

Abstract
The continuity of duplex DNA is generally considered a prerequisite for chromosome continuity. However, as previously shown in yeast as well as human cells, the introduction of a double-strand break (DSB) does not generate a chromosome break (CRB) in yeast or human cells. The transition from DSB to CRB was found to be under limited control by the tethering function of the RAD50/MRE11/XRS2 (MRX) complex. Using a system for differential fluorescent marking of both sides of an endonuclease-induced DSB in single cells, we found that nearly all DSBs are converted to CRBs in cells lacking both exonuclease 1 (EXO1) activity and MRX complex. Thus, it appears that some feature of exonuclease processing or resection at a DSB is critical for maintaining broken chromosome ends in close proximity. In addition, we discovered a thermal sensitive (cold) component to CRB formation in an MRX mutant that has implications for chromosome end mobility and/or end-processing.
Keywords: double-strand break repair, chromosome break, exonuclease 1, MRX, fluorescent imaging
INTRODUCTION
Chromosome breaks (CRBs) are associated with many diseases including cancer and are detected cytologically following treatment with a variety of clastogens [1] and [2]. It is therefore important to understand the mechanisms and events that control their appearance. Primary and secondary double-strand breaks (DSBs) are generally considered as precursors to CRBs. While chromosome continuity is expected to influence chromosome dynamics, previous findings in yeast and mammalian cells demonstrated that DNA continuity is not essential to maintaining chromosome continuity. A site-specific DSB is inefficient at generating a CRB based on single molecule studies in yeast [3], [4], and mammalian cells [5], even if the DSB fails to undergo repair.
How chromosomal proteins that prevent the transition from DSBs to CRBs at the site of breakage is poorly understood. Using approaches that combine genetics and single molecule chromosome imaging in yeast, we had examined [3] the consequences of a DSB induced by an I-SceI homing endonuclease at a site closely-flanked (4 and 9 kb) by bacterial operators that can bind lacI-GFP and tetR-CFP, respectively [6]. The generation of a CRB was identified by spatial separation of the fluorescent spots (see Fig. 1). Less than a few percent of the DSBs led to a CRB in wild type (WT) cells; however, in the absence of the Rad50/Mre11/Xrs2 (MRX) complex, 12% of cells with a DSB also experienced a CRB [3]. Protein tethering at the site of the DSB, not the DNA nuclease function of MRX was proposed to limit DSB to CRB transition. Similar results were obtained in a system employing the same fluorescent markers placed 50 kb [4] from both sides of a site-specific DSB. The apparent inefficiency of DSB induction of CRBs led us to ask whether additional genetic factors prevent CRBs. In particular we were interested in the role that end-processing, i.e., resection, might play, since this might influence the ability to hold ends together or allow recruitment of additional repair machinery. We found that a combination of exonuclease 1 (EXO1) and MRX mutations results in highly efficient generation of CRBs from I-SceI induced DSBs. In addition, there is a cold-sensitive component that contributes to CRB induction.
Fig. 1. Systems for the detection of a CRB induced by site-specific DSBs using nearby probes.

A) I-SceI system (adapted from [3]): A DSB is generated by I-SceI endonuclease which is under control of a GAL1 promoter. Multiple repeat sequences of lacO which binds lacI-GFP and tetO which binds tetR-CFP are 4.7 kb and 9.2 kb, respectively, from the DSB site. The spindle pole body is identified by Spc29-RFP chimeric protein. A CRB is identified by separation of > 0.8 μm separation of the GFP and CFP spots. B) HO-system (adapted from [4]): Two multiple repeat sequences of lacO, which binds lacI-GFP, are located 50 kb from an HO-endonuclease site. The endonuclease is under the control of a GAL1 promoter.
RESULTS
Systems detect a CRB induced by a site-specific DSB using fluorescent probes
Presented in Fig. 1 are the two previously reported [3] and [4] systems that have been employed to assess the ability of a DSB to alter chromosome continuity. In the first (Fig. 1A), lacI-GFP molecules and tetR-CFP proteins bind to multiple copies of their cognate sequences at 5 and 9 kb, respectively, from a sequence that is targeted by galactose-inducible I-SceI [3]. CRBs are identified by a greater than 0.8 micron separation of the markers in cells that are large-budded (due to DSB-induced arrest). Also included is a spindle marker Spc29-RFP that is used to ascertain cells arrested in G2. The second system (Figure 1B) described by [4], utilizes a single lacI-GFP probe to identify sites that are 50 kb from a sequence that is targeted by galactose-inducible HO-endonuclease. Again, CRBs are identified by spot separation of > 0.8 μm (the separation was > 0.5 μm in [4]) in cells that are large-budded. However, other factors might contribute to spot separation as discussed below. While both I-SceI and HO are efficient (Fig. 2 and Supplementary Fig. 1), the former is slower at inducing DSBs [7].
Fig. 2. A DSB to CRB transition is primarily determined by exonuclease 1 and MRX following induction of a DSB.



Cells were grown in YEPD overnight, transferred in YEP lactate overnight, and resuspended in synthetic medium with galactose at 30°C as described in the Material and Methods. A) I-SceI induced CRBs. Presented is the % of large budded cells with separated (> 0.8 μm) lacI-GFP and tetR-CFP spots after 8 h I-SceI induction. Previous results [3] are included (gray bars). The exo1Δ mutant exhibited a high frequency of CRBs after DSB induction, reaching ~40% in large budded cells. The frequency of CRBs was substantially greater in exo1Δ rad50Δ double mutant. Presented in parenthesis are the numbers of cells with CRBs/total cells examined. While the present rad50Δ vs WT results are not statistically different, the previous results [3] were at the p = 0.01 level. The combination of results from the two sets of data (indicated by brackets) are significant at the p = 0.01 level (see Material and Methods). The “**” indicates significant difference from the WT at p < 0.01 level. The “***” indicates significant difference from the single mutant at p < 0.01 level. B) I-SceI DSB cutting efficiency. DNA-plugs were prepared from imaging samples at 0, 4, and 8 h, and chromosomes were separated by PFGE. Chr II and digested fragments were detected by Southern blotting with probes as described in the Material and Methods. The intensity of bands was determined using ImageQuant software, and cutting efficiency was determined by the ratio of the centric and acentric fragments/total Chr II. C) HO-induced CRBs. After HO-induction for 6 h, the % of large budded cells with two lacI-GFP spots was determined. Assuming 100% cutting, the efficiency of DSB to CRB transition is 11% in the WT strain, 27% in rad50Δ, 36% in exo1Δ and 65% in exo1Δ rad50Δ. Because the HO system uses a single color, these results may include sister chromatid separation. Presented in parentheses are the numbers of cells with CRBs/total cells examined. The “*” indicates significant difference from the WT at p < 0.01 level. The “**” indicates significant difference from the single mutant at p < 0.01 level.
The transition of most DSBs to CRBs is prevented by a combination of the exonuclease function of Exo1 and the MRX complex
As shown in Fig. 2A, an I-SceI induced DSB generates a CRB in 3–5% of large budded WT cells (also described previously [3]). Consistent with our earlier results [3], the frequency is increased to ~11% in the MRX deletion mutant rad50Δ (Fig. 2A). While the present results are not statistically significant, the previous results [3] for WT vs rad50Δ are significant as well as when the two sets of data are combined, as indicated in Fig. 2A.) This difference between WT and the rad50Δ mutant in the present experiments might be explained by differences in cutting efficiency since there was a decrease in cut molecules in the population at later times in the WT strain (Fig. 2B) (possibly some DSBs produced by the slow cutting I-SceI endonuclease can be repaired between sister chromatids in the WT cells). However, under conditions of comparable, rapid-cutting by HO (Supplementary Fig. 1) there is still a difference in CRB induction (Fig. 2C).
Surprisingly, the level of CRBs is dramatically increased to 46% in cells lacking exonuclease 1 (exo1Δ; Fig. 2A). As shown in Fig. 2B, nearly 80% of the molecules in the population are cut. Induction of CRBs by HO-endonuclease is also increased in an exo1Δ mutant (Fig. 2C). Although there is more rapid and efficient cutting by HO-endonuclease, the large chromosomal distance and use of single color fluorescent markers (Fig. 1B) may limit the detection of CRBs. There is a high level of spot separation in the No-DSB control (~10%; [4] and unpublished) that could also be due to sister chromatid separation. The CRB frequency is nearly the same (Fig. 2A) with an exo1 mutant specifically deficient in nuclease function (exo1-D173A) [8] and [9] as with the exo1Δ mutant. The high CRB frequency cannot be attribute to a lack of repair since exo1Δ mutants are efficient at DSB repair [10]. Thus, we conclude the Exo1 nuclease function is a major component in preventing a DSB transitioning to a CRB.
Remarkably, when the exo1Δ or the exo1-D173A nuclease mutations were combined with a rad50Δ mutation, the frequency of large budded cells with a CRB following I-SceI induction increased to~80% or ~65% with HO (Fig. 2C). Thus, the combination of deficiencies in both Exo1 activity and MRX results in nearly all DSBs transitioning to CRBs, given the efficiency of I-SceI cutting (Fig. 2B). It is not clear why the CRB frequency is less with HO. Possibly, the spatial distribution and even opportunities for identical sequences to interact may lower the opportunity to see separated spots. Overall, these results suggest that in the absence of MRX and its associated tethering function [11], the DSB to CRB transition is largely prevented by a mechanism that is specifically dependent upon the nuclease activity of Exo1.
The relationship between temperature and appearance of CRBs
If there is a structural basis to the prevention of CRBs then reducing the temperature might increase the likelihood of CRB formation from a DSB. This was not the case for either the WT or the exo1Δ mutant as shown in Fig. 3A. However, in the absence of MRX the appearance of CRBs is strongly influenced by temperature over the range 23° – 37° C. At 37°, ~6% of the cells contain a CRB. With reduction in temperature from 37° to 30° to 23°, we found a dramatic increase in the appearance of CRBs from 6 to 12 to 40%, respectively. Deletion of the Mre11 component of MRX also increased CRBs when the temperature was reduced from 30° (12%; [3]) to 23° (compare with Fig. 3B). Given an efficiency of DSB induction by I-SceI in the WT and rad50Δ strains of 61% and 90% at 23° (data not shown), respectively, the absence of MRX reveals a cold-sensitive component that can effectively prevent DSBs from transitioning to CRBs.
Fig. 3. Affect of temperature on the appearance of DSB-induced CRBs.



Cells were grown at 30°C, and shifted to 23°C, 30°C, or 37°C during I-SceI induction. A) Increased CRBs for rad50Δ mutants but not exo1Δ at reduced temperature. Transition from a DSB to a CRB was 5% in WT and 12% in rad50Δ at 30°C. The I-SceI induction was for 8 hr. The “*” indicates significant difference from the WT at p < 0.01 level. The “**” indicates significant difference from the single mutant at p < 0.01 level. Comparisons are for the 23°C and the 30°C sets of data. B) CRB induction at 23°C in various mutants after I-SceI induction for 8 hr. The “*” indicates significant difference from the WT at p < 0.01 level. For exo1Δ mre11-16A the single asterisk indicates significant difference from the WT and the mre11-16A mutant. The “**” indicates significant difference from the single mutant at p < 0.01 level (for sae2Δ rad50Δ, the comparison is with the sae2Δ mutant). C) HO-induced CRBs at 23°C. Cells were grown at 30°C and shifted to 23°C during HO induction. The cells with CRBs after HO induction for 6 h were identified as containing two or more GFP spots. The “*” indicates significant difference from the WT at p < 0.01 level. The “**” indicates significant difference from the single rad50Δ mutant at p < 0.01 level. Presented in parenthesis are the numbers of cells with CRBs/total cells examined.
We also examined the mre11-D16A mutant which increases sensitivity to radiation and lacks nuclease activity in vitro [12], [13], and [14]. Like Mre11, the mre11-D16A protein can complex with Rad50 in vitro and tether ends of DNA molecules [13]. As shown in Fig. 3B, the ability of a DSB to induce a CRB at 23° was similar to that in WT cells suggesting that the cold sensitivity in cells lacking MRX complex is not related to a deficiency in Mre11 nuclease mediated end-processing. Loss of both Exo1 and MRX resulted in efficient DSB to CRB transition at both temperatures for both the I-SceI and HO induced DSBs (Fig. 3B and 3C). Thus, the cold-sensitive component appears related to Exo1 associated nuclease functions and/or temperature dependent properties of the DNA/chromatin ends (see Discussion).
Interestingly, the mre11-D16A mutation is reported to disrupt the MRX complex in vivo, based on the loss of ability to immunoprecipitate the complex [14]. The present results argue that the complex is functional, since there is no loss in ability to prevent the DSB→CRB transition at 23° or 30° [3], although the mutant exhibited the expected increase in sensitivity to both ionizing radiation and MMS (Supplementary Fig. 2). We also found that the mre11-D16A mutant had only a small effect on resection in vivo at the site-specific DSB (see below).
Other mutants were also examined for their impact on CRB formation at 23°. The Sae2 protein which is associated with MRX complex can affect exo- and endonuclease functions [15], [16], and [17]. The sae2Δ mutation had a relatively small effect on the appearance of CRBs compared to the rad50Δ and mre11Δmutants; the frequency of cells with a CRB was ~10% compared to ~3% in WT cells (Fig. 3B). Another protein factor that binds DSBs and affects end-processing is Ku70/80 [18]. Consistent with previous results at 30° [3] the level of CRBs in cells lacking Ku70 was comparable to that in WT cells.
Relationship between resection and appearance of CRBs
Since both the exonuclease 1 and MRX can participate in 5′ to 3′ resection of DSB ends in growing cells (summarized in [20] and [21]) and G2 arrested cells [10], we examined the relationship between changes in end-processing and the appearance of CRBs. To determine possible roles of end-processing in the increased frequencies of CRBs, we assessed resection at DSBs in the various mutants utilizing an approach based on pulse-field gel electrophoresis that we recently described (PFGE; [10]). The generation of single-strand tails of at least a few hundred bases at the ends of chromosomes or large chromosomal fragments by resection leads to a reduction in mobility (giving the appearance of larger molecular weight) under the standard conditions used for PFGE. This “PFGE-shift” assay provides an opportunity to address the fraction of broken molecules that undergo resection (i.e., shifted vs unshifted) as well as extent of resection [10]. For example, a unique cut site in a chromosome will generate two molecules of expected size. However, resection of the broken ends leads to an apparent increase in size (see Fig. 4 below); in the absence of resection, there is no change in mobility.
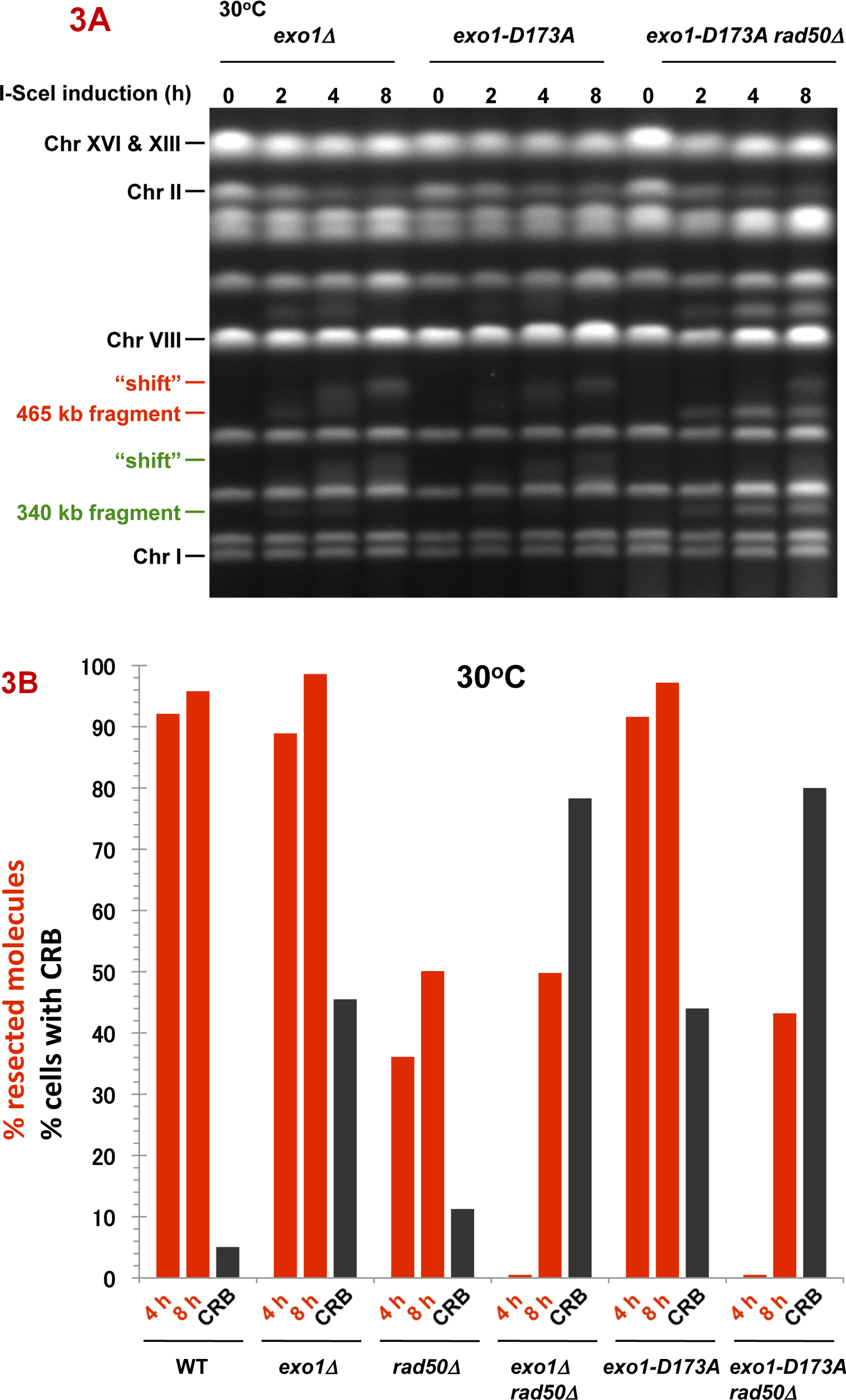
Fig. 4. DSB resection in MRX and exonuclease 1 mutants as determined by pulse-field shift.



DNA-plugs were prepared from samples used for imaging and chromosomes were separated by PFGE; gels were stained with SYBR Gold. I-SceI induction resulted in cutting of the 815 kb Chr II into 470 kb and 345 kb fragments. With time, resection leads to a shift in these bands to slower mobilities [10]. A) PFGE-shift following incubation at 30 °C. B) and C) PFGE-shift following incubation at 23 °C of various mutants. Most chromosome fragments were shifted in the mre11-D16A mutant and exo1Δ mre11-D16A double mutant.
Cutting of Chr II in the WT strain by I-SceI results in the appearance of two fragments (345 kb and 470 kb) within 2 h (not shown), and the DNA of most chromosomes II are broken by 4 h, as shown in Fig. 4A. In addition, nearly all broken molecules at 4 h (>90%) exhibit the PFGE-shift associated with resection (e.g., 470 kb fragment). Given the low frequency of CRBs, this suggests that factors that prevent the transition to CRBs can function efficiently even when ends are resected.
The resection in the WT strain differs from that observed in the rad50Δ mutant (Fig. 4A). Only ~35% of the broken molecules at 30° are shifted at 4 h increasing to ~50% at 8 h, as summarized in Fig. 5A. (Similar results were reported by Westmoreland et al. [10] for events at an HO-endonuclease induced DSB except that the time involved was shorter due to the more rapid cutting by HO-endonuclease.) The defect in resection at I-SceI induced DSBs is somewhat greater at 23° (Fig. 4B). Even though there is a marked defect in resection in the rad50δ, the small increase in CRBs at 30° (Fig. 2A and 5A) suggests that the resection associated with MRX has only a modest role in preventing CRBs and that another factor(s) maintains chromosome ends in close proximity at 30° although to a much lesser extent 23°. We also examined the mre11-D16A mutant. Although reported to be nuclease defective in vitro, resection was comparable to WT (Fig. 4C), supporting the view that MRX may not have a major direct role in resection of a DSB in vivo [20].
Fig. 5. Summary of resection and CRBs at 30°C and 23 °C.

The percentages of molecules that were resected were determined from PFGE images such as those described in Fig. 4 and are presented as red bars. The images were analyzed using KODAK MI software for the amount of broken 470 kb molecules that were unshifted or shifted (material above the 470 kb band). The black bars correspond to percentages of large budded cells that contain CRBs. The I-SceI induction was for 8 h.
Results with mutants lacking exonuclease 1 function (exo1Δ and exo1-D173A) contrast with those obtained with the rad50Δ strain. In spite of a defect in Exo1, the fraction of molecules resected is only slightly reduced from WT at 30° or 23° C (Fig. 5 and Supplementary Fig. 3), consistent with previous reports of compensating nucleases ([9]; also see [10]). Since the frequency of exo1Δ or exo1-D173A cells with a CRB is ~45% (Fig. 4, 5 and Supplementary Fig. 3A and 3B) there must be a specific role for exonuclease 1 activity in preventing a DSB from generating a CRB. The large increase at 30° in CRBs in the exo1 rad50 double mutant (~80%; Fig. 2 and 5 and Supplementary Fig. 2B) as compared to the single rad50Δ mutant (~11%; Fig. 5) as well as the cold-sensitive increase in CRBs in the single rad50Δ or mre11Δ mutants further suggests that Exo1 plays an important role in preventing CRBs when the MRX complex is inactivated. The effect of Exo1 is not simply explained by a reduction in resection, since the frequency of CRBs is much smaller in the rad50Δ mutant at 30° C, where there is a clear reduction in resection, than in the exo1Δ mutant. The combination of exo1 mutations with rad50Δ leads to a large reduction in the frequency of resected molecules after 4 h of I-SceI expression at 30° (35% vs <5%) but reaches that of the rad50Δ single mutant at 8 h. However, at 23° the frequency of resected molecules in the double mutant is 10% as compared to ~50% in the rad50Δ mutant following 8 h.
During these studies we observed that retention of both fluorescent markers GFP and CFP (5 kb and 9 kb away from the DSB, respectively, as described in Fig. 1) decreased upon I-SceI induction of a DSB. The frequency of large budded cells containing both markers went from >95% to ~20–25% in the WT strain (Fig. 6) suggesting that processing of ends could lead to reduced ability of the LacI and TetR repressor molecules to bind to their multicopy sequences. Surprisingly, there was much greater spot stability (both markers were detected in ~60–80% of cells) when exonuclease 1 activity was removed (exo1Δand exo1-D173A; Fig. 6A) although there was comparable cutting efficiency (Fig. 2B) and frequency of molecules with resected ends (Fig. 5). In the rad50Δ mutant there was also a large decrease in cells with both spots (~25%) which might be due to the exonuclease 1 acting on one or the other DSB ends even though resection is reduced in the rad50Δ mutant (Fig. 5). Similar results were found at 23° (Fig. 6B).
Fig. 6. Appearance of both CFP and GFP “spots” following DSB induction.


After I-SceI induction at 30°C or 23°C for 8 h, the percentage of large budded cells containing both lacI-GFP and tetR-CFP fluorescent markers was determined. The “WT no DSB” strain lacks an I-SceI recognition sequence on chromosome II. The “*” indicates significant difference from the WT and rad50Δ at p < 0.01 level. The “**” indicates significant difference from the exo1Δ and the exo1Δ rad50Δ mutants at the p < 0.01 level. Presented in parentheses are the numbers of cells with both spots/total cells examined.
These findings suggest that the DNA processing or resection carried out in the presence of Exo1 is efficient at preventing the DSB to CRB transition and that the exonuclease activity associated with exonuclease 1 plays a major role. Possibly in the absence of exonuclease 1, resection at DSBs is less processive as indicated in our previous studies with radiation-induced DSBs [10] and less able to displace associated proteins (see Discussion).
DISCUSSION
We previously identified a set of repair complexes that influence the transition from a DSB to a visually detectable CRB. Elimination of the MRX-mediated tethering resulted in a small but significant increase in CRBs. In the present study we found that the transition from DSB to chromosome discontinuity is greatly increased when Exo1 is defective. The removal of Exo1 or inactivation of Exo1 nuclease activity resulted in an approximate 10-fold increased likelihood of a CRB.
The Exo1 5′ to 3′ single strand exonuclease activity is involved in several DNA metabolic functions including removal of mismatches during replication [22], processing of lagging strand intermediates [23] and [9], and participating in recombination and resection of uncapped telomeres [24], [25], and [26]). In human cells exonuclease 1 contains exonuclease and flap endonuclease activities and interacts with mismatch repair (MMR) proteins [27], [28], and [29]. Human Exo1 also interacts with Werner syndrome (WRN) protein, and WRN stimulates the Exo1 flap endonuclease activity [30]. In yeast, Exo1 has a modest effect on resection of a site-specific DSB or radiation-induced DSBs in logarithmically-growing and G2 arrested yeast [10]. In logarithmically growing yeast, factors that affect resection include Exo1, the helicase/endonuclease SGS1 along with DNA2 and MRX [20]; deletion of any one of these can affect the pattern of end-processing [31], [32], and [20]. Thus, it is surprising that loss of Exo1 activity would have such a large impact on chromosome continuity. However, as shown previously and summarized in Fig. 5 and Supplementary Fig. 3B, Exo1 accounts for much of the remaining resection in a rad50Δ mutant.
We propose an additional role for Exo1, that of maintaining chromosome continuity upon introduction of a DSB. Since the prevention of CRBs specifically requires Exo1 nuclease activity, we suggest that this nuclease-mediated end-processing assures that ends are maintained in close proximity even though, as shown in Figure 4, most ends are resected. Although structural functions have been ascribed to Exo1 [33], the large effect of the D173A mutant strongly argues for an important nuclease specific role in preventing CRBs. Surprisingly, not only does Exo1 play a major role in preventing CRBs, there was a dramatic increase in CRBs when the Exo1 function was inactivated in cells lacking the MRX complex (mrx−) resulting in nearly all chromosomes with a DSB target site exhibiting a CRB (within the limits of detection). While there is a small but significant increase [11]in CRBs in an MRX defective background, the Exo1-mediated function is over 80-90% efficient at preventing a DSB→CRB transition in mrx− cells. On the other hand, while about 30–40% of DSBs can lead to CRBs in the Exo1 mutants, the generation of even more CRBs is preventable by the tethering function of MRX and not its nuclease activity (Fig. 4C).
The finding that the Exo1 activity itself plays a major role in preventing DSB generation of CRBs is unexpected in light of its primary function as an exonuclease, especially since most molecules are resected in mutants lacking Exo1 activity. It is interesting that the stability of the CFP and GFP fluorescent “spots” is greatly increased in Exo1 mutants. The resection characteristics of Exo1 differ from those of the other nucleases that affect DSB end-processing and is consistent with a slower or less processive resection in exo1 mutants [10] and [20]. Possibly Exo1 is more efficient at resecting protein-associated DNA (i.e., chromatin) or DNA with modified protein, such as stretches of phosphorylated H2A (which can extend to hundreds of kb)[34], [35], and [36] following DSB induction.
While we have established a prominent role for Exo1, the mechanism of Exo1 prevention of CRB formation, even in the absence of MRX, remains to be determined. Possibly other proteins that associate with single-strand DNA generated at DSB ends can interact to prevent separation of ends. It is interesting that the nearly 4-fold increase in CRBs with reduction in temperature (30° to 23°C) that is specific to the MRX mutants suggests a cold sensitive component in the Exo1 mediated prevention of DSB→CRB transitions. However, it is also possible that the physical status of the chromosome ends generated after end-processing might impact the thermodynamic properties of ends thereby affecting the detection of CRBs. The elasticity of a polymer network such as DNA/chromatin is expected to be influenced by entropic recoil of the polymer chain [37] and [38]. In well-defined in vitro systems the fluctuation of monomeric chains of a polymer network is temperature sensitive, resulting in chain shortening with increased temperature due to the increased number of states each monomer in the chain can occupy [39]. When translated to a chromosome polymer that is broken, this would lead to the appearance of chromosome breaks. Such an inverse relationship between CRB frequency and temperature would be opposite to the expected effect of temperature on an enzymatic reaction.
It is interesting that translocations arising by endjoining of HO-induced broken ends on different chromosomes are prevented by factors such as Mre11 and Sae2 [19] that also reduce the likelihood of DSB to CRB transitions. Importantly, translocations are also prevented by the associated nuclease activities. The Exo1 nuclease activity may function similarly if it influences opportunities to resect chromatin associated DNA. While Exo1 can resect “dirty” ended radiation-induced as well “clean” site-specific DSBs [10], the loss of Exo1 does not greatly affect DSB repair or radiation sensitivity implying overlapping repair functions with other exonucleases. In light of the present results, Exo1 also might function to prevent the transition of randomly induced DSBs to CRBs. If this is the case, Exo1 could influence the likelihood of chromosomal rearrangements which are induced by radiation in wild type yeast [40]
Material and Methods
Strains
The following strain KS406 which contained galactose-inducible I-SceI (Gal1/10-I-SceI) was developed previously, as described in [3], and has the following genotype:
MATα ade5-1 trp1-289 ura3-Δ leu2-3,112 lacO::ChrII tetO::LEU2
thr1::HISpLacI-GFP-Nat ade1::URAptetR-ECFP Hyg SPC29-RFP-Bsd
-
arg4::hisG-Gal1/10-I-SceI lys2::I-SceI.
lacO::ChrII corresponds to the lacO array that binds the LacI-GFP fluorescent probe;
tetO::LEU2 corresponds to the tetO array that binds the tetR-ECFP fluorescent probe.
thr1::HISpLacI-GFP-Nat corresponds to LacI-GFP integrated at THR1 locus using the marker Nat [nourseothricin] where LacI-GFP is driven by the HIS promoter.
ade1::URAptetR-ECFP corresponds to tetR-ECFP integrated at the ADE1 locus using the Hyg (hygromycin) marker.
SPC29-RFP-Bsd corresponds to the SPC29-RFP fluorescent probe integrated at SPC29 using the Bsd (blasticidin) marker.
arg4::hisG-Gal1/10-I-SceI corresponds to the galactose inducible I-SceI integrated at the ARG4 locus. lys2::I-SceI corresponds to the I-SceI target sequence at the LYS2 locus.
The following mutant derivatives were created in this strain [3]: mre11Δ, xrs2Δ, mre11-D16A, and yku70Δ mutants. The RAD50 and SAE2 genes were deleted by PCR-based amplified TRP1 fragments of RAD50 as follows:
5′-GTGGTAGCAACCATTGAGAGGCAAAAACAAGGGAACGACGGAAA GCAGGCATGTCTGTTATTAATTTCAC-3′
and
5′-TCTAATTAATCAATCAAAGTCTATCCCTTCGTAGATATTATGG GGTCTTTCTATTTCTTAGCATTTTTGA-3′;
SAE2: 5′-ATACCTGCATTTCCATCCATGCTGTAAGCCATTAGGTGTTTGT CATGTCTGTTATTAATTTCAC-3′
and
5′-CAAAAAAAATGTATTTGAAGTAATGAATAAAGAATGATGA TCGCTGGCGTCTATTTCTTAGCATTTTTGA-3′.
The EXO1 gene was deleted using a kanMX module amplified from a kanMX deletion library. The exo1-D173A mutant was created using the YIp-yExo-D173A plasmid (a gift from Dr. Thomas A. Kunkel, NIEHS/NIH) that was inserted into EXO1. The exo1-D173A mutation was created by a “popout” using 5-FOA selection. A “No I-SceI” strain was generated by replacement of the I-SceI recognition site at the lys2 locus with the functional LYS2 gene. HO strains were derived from JK40.6 (a gift from Dr. David P. Toczyski, UCSF Cancer Center). Deletion of exo1 gene was made by one-step replacement through a PCR disruption technique using a kanMX module.
Detection of CRBs
I-SceI strains were grown overnight at 30°C in YPDA medifum (2% Bacto-Peptone, 1% yeast extract, 2% dextrose, 60 μg/mL adenine sulfate), resuspended in YEP lactate medium (2% Bacto-Peptone, 1% yeast extract, 3.15% lactic acid [pH 5.5]) and grown for an additional 18 h at 30°C. The cells were then transferred to synthetic lactate medium (3.15% lactic acid [pH 5.5]) lacking histidine containing 2% galactose and 20 mM 3-aminotriazole (3-AT) (Sigma, St. Louis, MO) for 8 h at 23°, 30°, or 37°C. After I-SceI induction for 8 h, over 80% of cells were large budded. Cells were harvested and concentrated by centrifuge. Cells were put on ice until they were imaged. Then large budded cells were examined at room temperature. G2/M arrest was also monitored by the distance between the Spc29-RFP spots. The microscopic imaging procedure was described previously [3]. HO strains were grown overnight at 30°C in YPDA medium, resuspended in YEP lactate medium, and transferred to synthetic lactate medium containing 2% galactose and 50 mM CuSO4 as described in [4]. The subsequent procedures were the same as used for the I-SceI strains.
Assay to detect loss of GFP or CFP spots
To detect loss of fluorescent spots, cells were prepared as described above. After I-SceI induction for 8 h, large budded cells were observed using fluorescent microscopy and the ratio of cells with both CFP and GFP spots versus total large budded cells was determined.
Pulse Field Gel Electrophoresis
Strains were grown under the same procedures used in the imaging experiment. After I-SceI induction, cells were harvested at 0, 4, and 8 h, and PFGE plugs were prepared as previously described [10] and [41]. Chromosomes were separated with Transverse Alternating Field Electrophoresis (TAFE) using a GeneLineII (Beckman Instruments, Fullerton, CA). The electrophoresis was performed at 350mA with 30 – 100 s pulse switch for 23 h 24 min. Following separation of chromosomes, gels were stained with SYBR Gold (Invitrogen, San Diego, CA), and the gel image was photographed using a GelLogic200 imaging system (Eastman Kodak, Rochester, NY). To calculate the ratio of resected molecules, the intensity of the broken Chr II 470 kb fragment was analyzed using KODAK Molecular Imaging software (Rochester, NY). The ratio of shifted molecule versus both shifted and un-shifted molecules was determined.
Efficiency of I-SceI cutting or HO cutting
Pulsed field gels were blotted to Hybond N+ (GE Healthcare, Buckinghamshire, England), and chromosome II or chromosome VII was detected by Southern blotting. Cutting efficiency was measured by Southern, and resection efficiency was measured by stained gel. The probes were amplified with the following primers. Chr II probe 1:
5′-GTTCCTGTTAACAACTAGTAG-3′ and 5′-TACCCATTTAACACCTGCCATG-3′; Chr II probe 2:
5′-TCAGTGTCAAGAGTAGAAGTG-3′ and 5′-TGAAGAAGCTGCATTTGCAAG-3′; Chr VII probe 1:
5′-CTACACCCTAATCACAATAG-3′ and 5′-GTTAGCGGTATTGAGGAATC-3′; Chr VII probe 2:
5′-AAGAGCCAAGCCAGGTGACC-3′ and 5′-TCTTGGTATACATTATTAGC-3′;
The PCR amplified probes were labeled with 32P-dCTP. The probes were used to hybridize both the centric and acentric fragment. Autoradiographs were examined with a Typhoon 9400 variable mode imager (GE Healthcare) and band intensities were analyzed using ImageQuant Version 5.2 (Molecular Dynamics, Sunnyvale, CA).
Statistical analysis
Differences between pairs of genotypes in the proportion of cells with CRBs were evaluated using likelihood ratio statistics (likelihood ratio exact test) and p-values were evaluated via permutation techniques. Permutation-based p-values were used because of the small counts for some genotypes. The Cochran-MantelHaenszel test was used to calculate a single p value in Fig. 2A using combined data from two experiments involving WT and rad50Δ (present results and [3]). All statistical calculations were done using StatXact 8 (Cytl Inc., Cambridge, MA).
Supplementary Material
DNA-plugs were prepared from imaging samples at 0, 3, and 6 h; chromosomes were separated by PFGE. Chr VII and digested fragments were detected by Southern blotting with probes for the centric and acentric fragment described in the Material and Methods. The intensity of bands was measured using ImageQuant software. Cutting efficiency was determined by the ratio of the centric and acentric fragment/total Chr VII.
Based on dilution pronging and exposure to ionizing radiation or growth in the presence of MMS, the two different MRE11-D16A isolates are more sensitive than the wild type, but less sensitive than a mutant completely lacking the RMX complex.
A) Resection at 30°C. DNA-plugs were prepared from samples used for imaging and chromosomes were separated by PFGE; gels were stained with SYBR Gold. I-SceI induction resulted in cutting of the 815 kb Chr II into 470 kb and 345 kb fragments. With time, resection leads to a shift in these bands to slower mobilities [10]. The additional band which is seen above ChrVIII following I-SceI induction appears to be due to inefficient cutting at a cryptic site in the genome in a chromosome larger than ChrVIII. Given the comparability in results obtained with HO and I-SceI for DSB to CRB transitions, we consider this additional, occasionally observed DSB irrelevant to the findings in this paper. B) Summary of resection at 30°C vs CRBs in various mutants. The percentages of molecules that were resected were determined from PFGE images such as those described in Fig. 4 and Supplementary Figure 1, and are presented as red bars. The images were analyzed using KODAK MI software for the amount of broken 470 kb molecules that were unshifted or shifted (material above the 470 kb band). The black bars correspond to percentages of large budded cells that contain CRBs.
{kind=link}
Acknowledgments
We greatly appreciate the statistical analysis provided by Dr. David Umbach as well imaging assistance provided by Dr. Masaki Akita. We thank the Resnick lab for critical comments and suggestions, especially Drs. Shay Covo, Kin Chan, Steven Roberts and Dmitry Gordenin. This work was supported by intramural research funds from NIEHS to MAR, project Z01-ES021016.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mills KD, Ferguson DO, Alt FW. The role of DNA breaks in genomic instability and tumorigenesis. Immunol Rev. 2003;194:77–95. doi: 10.1034/j.1600-065x.2003.00060.x. [DOI] [PubMed] [Google Scholar]
- 2.Albertson DG, Collins C, McCormick F, Gray JW. Chromosome aberrations in solid tumors. Nat Genet. 2003;34:369–376. doi: 10.1038/ng1215. [DOI] [PubMed] [Google Scholar]
- 3.Lobachev K, Vitriol E, Stemple J, Resnick MA, Bloom K. Chromosome fragmentation after induction of a double-strand break is an active process prevented by the RMX repair complex. Curr Biol. 2004;14:2107–2112. doi: 10.1016/j.cub.2004.11.051. [DOI] [PubMed] [Google Scholar]
- 4.Kaye JA, Melo JA, Cheung SK, Vaze MB, Haber JE, Toczyski DP. DNA breaks promote genomic instability by impeding proper chromosome segregation. Curr Biol. 2004;14:2096–2106. doi: 10.1016/j.cub.2004.10.051. [DOI] [PubMed] [Google Scholar]
- 5.Soutoglou E, Dorn JF, Sengupta K, Jasin M, Nussenzweig A, Ried T, Danuser G, Misteli T. Positional stability of single double-strand breaks in mammalian cells. Nat Cell Biol. 2007;9:675–682. doi: 10.1038/ncb1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robinett CC, Straight A, Li G, Willhelm C, Sudlow G, Murray A, Belmont AS. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J Cell Biol. 1996;135:1685–1700. doi: 10.1083/jcb.135.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sokolsky T, Alani E. EXO1 and MSH6 are high-copy suppressors of conditional mutations in the MSH2 mismatch repair gene of Saccharomyces cerevisiae. Genetics. 2000;155:589–599. doi: 10.1093/genetics/155.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tran PT, Erdeniz N, Dudley S, Liskay RM. Characterization of nuclease-dependent functions of Exo1p in Saccharomyces cerevisiae. DNA Repair (Amst) 2002;1:895–912. doi: 10.1016/s1568-7864(02)00114-3. [DOI] [PubMed] [Google Scholar]
- 10.Westmoreland J, Ma W, Yan Y, Van Hulle K, Malkova A, Resnick MA. RAD50 is required for efficient initiation of resection and recombinational repair at random, gamma-induced double-strand break ends. PLoS Genet. 2009;5:e1000656. doi: 10.1371/journal.pgen.1000656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell. 2001;8:1129–1135. doi: 10.1016/s1097-2765(01)00381-1. [DOI] [PubMed] [Google Scholar]
- 12.Furuse M, Nagase Y, Tsubouchi H, Murakami-Murofushi K, Shibata T, Ohta K. Distinct roles of two separable in vitro activities of yeast Mre11 in mitotic and meiotic recombination. EMBO J. 1998;17:6412–6425. doi: 10.1093/emboj/17.21.6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lewis LK, Storici F, Van Komen S, Calero S, Sung P, Resnick MA. Role of the nuclease activity of Saccharomyces cerevisiae Mre11 in repair of DNA double-strand breaks in mitotic cells. Genetics. 2004;166:1701–1713. doi: 10.1534/genetics.166.4.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krogh BO, Llorente B, Lam A, Symington LS. Mutations in Mre11 phosphoesterase motif I that impair Saccharomyces cerevisiae Mre11-Rad50-Xrs2 complex stability in addition to nuclease activity. Genetics. 2005;171:1561–1570. doi: 10.1534/genetics.105.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lobachev KS, Gordenin DA, Resnick MA. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell. 2002;108:183–193. doi: 10.1016/s0092-8674(02)00614-1. [DOI] [PubMed] [Google Scholar]
- 16.Clerici M, Mantiero D, Lucchini G, Longhese MP. The Saccharomyces cerevisiae Sae2 protein promotes resection and bridging of double strand break ends. J Biol Chem. 2005;280:38631–38638. doi: 10.1074/jbc.M508339200. [DOI] [PubMed] [Google Scholar]
- 17.Lengsfeld BM, Rattray AJ, Bhaskara V, Ghirlando R, Paull TT. Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol Cell. 2007;28:638–651. doi: 10.1016/j.molcel.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin SG, Laroche T, Suka N, Grunstein M, Gasser SM. Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell. 1999;97:621–633. doi: 10.1016/s0092-8674(00)80773-4. [DOI] [PubMed] [Google Scholar]
- 19.Lee K, Zhang Y, Lee SE. Saccharomyces cerevisiae ATM orthologue suppresses break-induced chromosome translocations. Nature. 2008;454:543–546. doi: 10.1038/nature07054. [DOI] [PubMed] [Google Scholar]
- 20.Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huertas P. DNA resection in eukaryotes: deciding how to fix the break. Nat Struct Mol Biol. 2010;17:11–16. doi: 10.1038/nsmb.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tran HT, Gordenin DA, Resnick MA. The 3′-->5′ exonucleases of DNA polymerases delta and epsilon and the 5′-->3′ exonuclease Exo1 have major roles in postreplication mutation avoidance in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:2000–2007. doi: 10.1128/mcb.19.3.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tishkoff DX, Boerger AL, Bertrand P, Filosi N, Gaida GM, Kane MF, Kolodner RD. Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proc Natl Acad Sci U S A. 1997;94:7487–7492. doi: 10.1073/pnas.94.14.7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maringele L, Lydall D. EXO1-dependent single-stranded DNA at telomeres activates subsets of DNA damage and spindle checkpoint pathways in budding yeast yku70Δ mutants. Genes Dev. 2002;16:1919–1933. doi: 10.1101/gad.225102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bertuch AA, Lundblad V. EXO1 contributes to telomere maintenance in both telomerase-proficient and telomerase-deficient Saccharomyces cerevisiae. Genetics. 2004;166:1651–1659. doi: 10.1534/genetics.166.4.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zubko MK, Guillard S, Lydall D. Exo1 and Rad24 differentially regulate generation of ssDNA at telomeres of Saccharomyces cerevisiae cdc13-1 mutants. Genetics. 2004;168:103–115. doi: 10.1534/genetics.104.027904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiu J, Qian Y, Chen V, Guan MX, Shen B. Human exonuclease 1 functionally complements its yeast homologues in DNA recombination, RNA primer removal, and mutation avoidance. J Biol Chem. 1999;274:17893–17900. doi: 10.1074/jbc.274.25.17893. [DOI] [PubMed] [Google Scholar]
- 28.Genschel J, Bazemore LR, Modrich P. Human exonuclease I is required for 5′ and 3′ mismatch repair. J Biol Chem. 2002;277:13302–13311. doi: 10.1074/jbc.M111854200. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Yuan F, Presnell SR, Tian K, Gao Y, Tomkinson AE, Gu L, Li GM. Reconstitution of 5′-directed human mismatch repair in a purified system. Cell. 2005;122:693–705. doi: 10.1016/j.cell.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 30.Sharma S, Sommers JA, Driscoll HC, Uzdilla L, Wilson TM, Brosh RM., Jr The exonucleolytic and endonucleolytic cleavage activities of human exonuclease 1 are stimulated by an interaction with the carboxyl-terminal region of the Werner syndrome protein. J Biol Chem. 2003;278:23487–23496. doi: 10.1074/jbc.M212798200. [DOI] [PubMed] [Google Scholar]
- 31.Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–2772. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tran PT, Fey JP, Erdeniz N, Gellon L, Boiteux S, Liskay RM. A mutation in EXO1 defines separable roles in DNA mismatch repair and post-replication repair. DNA Repair (Amst) 2007;6:1572–1583. doi: 10.1016/j.dnarep.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Downs JA, Allard S, Jobin-Robitaille O, Javaheri A, Auger A, Bouchard N, Kron SJ, Jackson SP, Cote J. Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol Cell. 2004;16:979–990. doi: 10.1016/j.molcel.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 36.Shroff R, Arbel-Eden A, Pilch D, Ira G, Bonner WM, Petrini JH, Haber JE, Lichten M. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol. 2004;14:1703–1711. doi: 10.1016/j.cub.2004.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bloom KS. Beyond the code: the mechanical properties of DNA as they relate to mitosis. Chromosoma. 2008;117:103–110. doi: 10.1007/s00412-007-0138-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bloom K, Joglekar A. Towards building a chromosome segregation machine. Nature. 2010;463:446–456. doi: 10.1038/nature08912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grosberg AY, Khokhlov AR. Giant Molecules: Here, There, and Everywhere. Academic Press; 1997. [Google Scholar]
- 40.Argueso JL, Westmoreland J, Mieczkowski PA, Gawel M, Petes TD, Resnick MA. Double-strand breaks associated with repetitive DNA can reshape the genome. Proc Natl Acad Sci U S A. 2008;105:11845–11850. doi: 10.1073/pnas.0804529105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma W, Westmoreland J, Nakai W, Malkova A, Resnick MA. Characterizing resection at random and unique chromosome and telomere ends. Methods and Protocols. doi: 10.1007/978-1-61779-129-1_2. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DNA-plugs were prepared from imaging samples at 0, 3, and 6 h; chromosomes were separated by PFGE. Chr VII and digested fragments were detected by Southern blotting with probes for the centric and acentric fragment described in the Material and Methods. The intensity of bands was measured using ImageQuant software. Cutting efficiency was determined by the ratio of the centric and acentric fragment/total Chr VII.
Based on dilution pronging and exposure to ionizing radiation or growth in the presence of MMS, the two different MRE11-D16A isolates are more sensitive than the wild type, but less sensitive than a mutant completely lacking the RMX complex.
A) Resection at 30°C. DNA-plugs were prepared from samples used for imaging and chromosomes were separated by PFGE; gels were stained with SYBR Gold. I-SceI induction resulted in cutting of the 815 kb Chr II into 470 kb and 345 kb fragments. With time, resection leads to a shift in these bands to slower mobilities [10]. The additional band which is seen above ChrVIII following I-SceI induction appears to be due to inefficient cutting at a cryptic site in the genome in a chromosome larger than ChrVIII. Given the comparability in results obtained with HO and I-SceI for DSB to CRB transitions, we consider this additional, occasionally observed DSB irrelevant to the findings in this paper. B) Summary of resection at 30°C vs CRBs in various mutants. The percentages of molecules that were resected were determined from PFGE images such as those described in Fig. 4 and Supplementary Figure 1, and are presented as red bars. The images were analyzed using KODAK MI software for the amount of broken 470 kb molecules that were unshifted or shifted (material above the 470 kb band). The black bars correspond to percentages of large budded cells that contain CRBs.