

Abstract
Factor XIa (FXIa) inhibition by protease nexin-2 (PN2KPI) was compared with trypsin inhibition by basic pancreatic trypsin inhibitor (BPTI). PN2KPI was a potent inhibitor of FXIa (Ki ∼ 0.81 nM) and trypsin (Ki ∼ 0.03 nM), but not of other coagulation proteases (thrombin, FVIIa, FIXa, FXa, FXIIa, plasmin, kallikrein, Ki > 185 nM). PN2KPI was ∼775-fold more potent than BPTI in FXIa inhibition, but both exhibited similar potencies against trypsin. Studies of FXIa and trypsin inhibition by PN2KPI and BPTI and P1 site swap mutants (PN2KPI-R15 K, BPTI-K15 R) demonstrated that FXIa inhibition by PN2KPI and P1 site swap mutants and trypsin inhibition by PN2KPI and BPTI conform to a single-step, slow equilibration inhibitory mechanism, whereas FXIa-inhibition by BPTI follows a classical, competitive inhibitory mechanism. Mutation of P1 impaired FXIa inhibition by PN2KPI-R15 K ∼14-fold, enhanced FXIa inhibition by BPTI-K15 R ∼150-fold, and had no effect on trypsin inhibition. Arginine at the P1 site of either PN2KPI or BPTI confers high affinity and specificity for FXIa, whereas either arginine or lysine suffices for trypsin inhibition. Thus, PN2KPI is a highly specific inhibitor of FXIa among coagulation enzymes, but the flexibility of trypsin renders it susceptible to inhibition by both wild-type and mutant forms of PN2KPI and BPTI.
Keywords: BPTI, FXIa, inhibition mechanism, protease nexin 2 Kunitz domain, trypsin
Coagulation factor XIa (FXIa), the activated form of the zymogen (FXI), other coagulation enzymes and trypsin are all members of a diverse family of serine peptidases that carry out disparate biological functions (1). FXI is a homodimeric 160-kDa glycoprotein, present in human plasma, which is essential for normal haemostasis, since its deficiency is associated with a hemorrhagic disorder termed haemophilia C (2). The zymogen is activated by coagulation proteases including thrombin, FXIIa and FXIa (3–6). The activation product of this event is FXIa, which in turn can activate FIX to FIXa to initiate the consolidation phase of blood coagulation resulting in fibrin formation at the site of blood vessel injury (2, 7–10). The 30-kDa light-chain region of FXIa is highly homologous to trypsin and harbours its serine protease catalytic activity.
Important control mechanisms exist for the regulation of coagulation protease activities. Serine protease inhibitors (SERPIN) have been proposed as physiological regulators of FXIa function in plasma, including protease nexin 1 (11), antithrombin III (12), C1 inhibitor (13, 14), α-1-protease inhibitor (15, 16) and α-2-antiplasmin (17). On platelet activation by physiological stimulators, protease nexin 2 (PN2) is secreted from α-granules into plasma and inhibits FXIa (18–20). PN2, a member of the class of Kunitz-type inhibitors, is a ∼120-kDa isoform of the Alzheimer’s β-amyloid protein precursor that has been shown to be a highly potent and physiologically relevant inhibitor of FXIa on the basis of detailed kinetic studies (18, 19, 21–24). PN2 is a slow, tight binding inhibitor of FXIa with a reported Ki of 290–450 pM (18, 21, 23). The KPI domain of PN2 (PN2KPI) is 57 residues in length (Glu289-Ile345 in the 751 amino acid isoform of PN2) and contains the entire FXIa inhibitory function of PN2 (22–25). A homologous protein belonging to the same Kunitz family called basic (or bovine) pancreatic trypsin inhibitor (BPTI; aprotinin; TrasylolR) is also an inhibitor of several human serine proteases such as trypsin, plasmin, kallikrein, activated protein C and FXIa (26, 27).
Although a great deal is known about the structural biology of the proteases, FXIa and trypsin, and their inhibitors, PN2KPI and BPTI, very little information is available concerning the mechanisms by which these two Kunitz-type inhibitors regulate the activities of the two serine proteases. The rationale for the present study, which focused on a comparison of the mechanisms of inhibition of human FXIa and bovine trypsin by the human PN2KPI and BPTI, is based on the striking structural homology between the human FXIa/PN2KPI complex (28) and the bovine trypsin/BPTI complex (29), in addition to the fact that BPTI (as TrasylolR) is frequently utilized therapeutically in human subjects in the treatment and prevention of thrombo-embolism, e.g. in patients undergoing cardiopulmonary bypass surgery (30, 31). Moreover, a large number of naturally occurring nonhuman serine proteinase inhibitors have been identified as potent inhibitors of human proteinases. This category of nonhuman inhibitors includes bovine BPTI (26, 27, 32, 33), tick anticoagulant peptide (34–36) and Boophilin from the cattle tick (37), which inhibit several human enzymes including proteinases in coagulation cascade. Human Kunitz-type inhibitors are also known to inhibit nonhuman proteinases. For example, bovine trypsin and bovine chymotrypsin are inhibited by human TFPI (38), TFPI 2 (39) and PN2KPI (21, 40), whereas mouse NGF-gamma is inhibited by human PN2KPI (21). In addition to these biochemical studies, three-dimensional structures of cross-species enzyme/inhibitor complexes have also been documented, including the crystal structures of human PN2KPI in complex with bovine trypsin, bovine chymotrypsin (41), and rat anionic trypsin (42) as well as BPTI in complex with human mesotrypsin and human cationic trypsin (33). To further understand these biochemically important interactions, we have studied the kinetic mechanisms of inhibition of the serine proteinases, FXIa and trypsin by the Kunitz inhibitors, PN2KPI and BPTI.
PN2KPI and BPTI are structurally very similar Kunitz inhibitory protein family members, each of which has six conserved cysteine residues making three intrachain disulphide bonds resulting in stable compact structures containing two loop regions that interact with the proteases. X-ray crystal structures of these inhibitory proteins have been solved in complex with several serine proteases such as trypsin, chymotrypsin and the catalytic domain of FXIa (28, 41, 42). Structural data from X-ray crystallography reveal similar superimposable backbone structures except for a small region from residues 39–41 (43). The current study compares these structurally homologous inhibitors, PN2KPI and BPTI, and contrasts their biochemical properties in the inhibition of the structurally similar serine proteinases, human FXIa and bovine trypsin. We further demonstrate, by using P1 site swap mutants, PN2KPI-R15 K and BPTI-K15 R, that the P1 site residues of the inhibitors are responsible in part for the functional differences in their enzyme specificities.
Experimental Procedures
Native and mutant inhibitor gene constructs for Pichia pastoris expression
The PN2KPI domain was amplified by polymerase chain reaction (PCR) from the full-length human PN2 gene as previously described (28). PCR-based site-directed mutagenesis (QuikChange, Strategene, USA) was utilized to introduce an Arg to Lys mutation at the P1 site and Ala mutations (PN2KPI-P13A, R15A, M17A, S19A, R20A and F34A) as explained in detail elsewhere (28). Utilizing a similar approach BPTI was obtained as a 206-bp PCR amplification product from bovine liver cDNA (Biochain Institute, Inc., USA) and ligated into a yeast expression vector, pPICZαA (Invitrogen, USA) and the BPTI P1 site was mutated from Lys to Arg (BPTI-K15 R) by inserting the codon preferentially used in yeast. The pPICZαA plasmids containing native and mutant inserts were sequenced from 5′ to the α-mating factor secretion signal using an AOX5 primer to confirm the mutations at the desired sites as well as the reading frame integrity.
Native and mutant protein expression in yeast cells
As explained in detail elsewhere for PN2KPI (28), the pPICZαA plasmid containing PN2KPI, BPTI and respective mutant gene constructs were incorporated into the methylotrophic yeast Pichia pastoris X33 competent cell genome (Invitrogen, USA) by recombinational cloning. Zeocin selected yeast clones were lysed either by lyticase (Sigma, USA) or by repeated freeze-thaw cycles. The lysates were checked by PCR using respective gene specific forward and reverse primers for correct insert size. Successful clones were propagated in buffered medium containing glycerol and yeast nitrogen base (BMGY) and expressed in buffered medium containing methanol and yeast nitrogen base (BMMY) for 96 h at 30°C under vigorous agitation, supplemented with 0.5% methanol every 24 h.
Purification of recombinant Kunitz inhibitory domain proteins
Purification of recombinant PN2KPI and its mutant protein followed the protocol explained earlier (28). For BPTI and BPTI-K15 R purification certain modifications were made as explained below. Pichia pastoris BMMY cultures containing secreted proteins were centrifuged to remove yeast cells. The supernatant was precipitated using saturating concentrations of ammonium sulphate and centrifuged at 11,000g for 30 min. Pellets were resuspended in 50 mM Tris buffer, pH 7.8 and the proteins were purified using desalting columns (G25 16/60, Amersham Biosciences Corp., USA) followed by ion exchange chromatography (10–20 ml SP HP columns, Amersham Biosciences Corp., USA) in 50 mM Tris buffer pH 7.8 and eluted with sodium chloride. The eluted samples were concentrated in dialysis tubing (Float-a-lyser, Spectrum Laboratories, Inc., USA) using an external dehydrant, cross-linked sodium polyacrylate gel (Spectragel; Spectrum Laboratories, Inc., USA), at 4°C. Concentrated samples were size fractionated using size-exclusion columns (HiLoad 30, 16/60, Amersham Biosciences Corp., USA) in 150 mM NaCl, 50 mM Tris buffer, pH 7.8 (TBS).
The concentrations of purified proteins were estimated either by the bicinchoninic acid assay (Pierce Biotechnology, Inc., USA) or from their extinction coefficient, 5960 M−1 cm−1 at 280 nM, estimated from the protein sequence (ProtParam, ExPASy, Swiss Institute of Bioinformatics). The yields of various preparations of PN2KPI and mutants were 0.7–10 mg l−1, whereas those of BPTI and BPTI-K15 R were 1–3.5 mg l−1 of culture volume. On silver-stained SDS–PAGE gels under reducing conditions, the purified proteins were single, sharp bands at 6.3–6.5 kDa (data not shown) and probed by western blotting using rabbit anti-Alzheimer’s β-amyloid protein precursor polyclonal antibody (Chemicon International, USA) for PN2KPI.
Measurement of initial rates of S-2366 hydrolysis by FXIa
Increasing concentrations of S-2366 (l-pyro-Glu-Pro-Arg-p-nitroanilide-HCl; 0-2 mM) in 15 µl of 50 mM Tris, 150 mM NaCl, 5 mM CaCl2, 0.1% BSA, pH 7.6 (TBSB) was placed in the wells of a 96-well microplate to which 135 µl of premixed and preincubated (30 min) enzyme/inhibitor mixture in TBSB was added. The final concentration of the FXIa (Haematologic Technologies Inc., VT, USA) in the reaction mixture was 1 nM. Generation of the product pNA was monitored by measuring absorbance at 405 nM in a microplate reader for 10 min at 37°C. Initial velocity measurements were analyzed using KaleidaGraph v3.5 nonlinear regression software.
Determination of equilibrium inhibition constant (Ki)
To establish Ki values for the inhibition FVIIa, FIXa, FXa, plasmin (all from Haematologic Technologies Inc., VT, USA), thrombin, FXIIa, plasma kallikrein (all from Enzyme Research Laboratories, IN, USA) and trypsin by PN2KPI, BPTI and their mutants, assays were carried out in 50 mM Tris, 150 mM NaCl, 0.1% BSA, pH 7.5 buffer (TBSB) using appropriate chromogenic or fluorogenic substrates to measure residual activity after incubation with the Kunitz inhibitors. The final enzyme concentrations used were thrombin (1 nM), FVIIa (5 nM or 50 nM in the presence or absence, respectively, of 50 nM tissue factor, obtained from Calbiochem, CA,USA), FIXa (50 nM), FXa (1 nM), FXIIa (25 nM), plasmin (10 nM) or plasma kallikrein (1 nM) in 135 µl volume for 30 min at 37°C in a microtitre plate to establish equilibrium between the inhibitor and the enzyme. To this preincubation mixture, 15 µl of substrate was added to the final reaction volume of 150 µl. The substrates (all obtained from Chromogenix, DiaPharma Group, Inc., OH, unless otherwise indicated) and their final concentrations for the reactions were: 200 µM of S-2366 (l-pyroGlu-Pro-Arg-pNA-HCl) for thrombin; 2000 µM of Chromozyme t-PA (CH3-SO2-D-Phe-Gly-Arg-pNA; Roche Diagnostics, IN, USA) for FVIIa; 3000 µM of Spectrozyme-FIXa (MeSO2-D-CHG-Gly-Arg-pNA.AcOH; American Diagnostica Inc., CA, USA) for FIXa; 400 and 80 µM of S2765 (N-α-Z-D-Arg-Gly-Arg-pNA.2HCl) for FXa and trypsin, respectively; 250 or 300 µM of S2302 (Pro-Phe-Arg-pNA.2HCl) for FXIIa or plasma kallikrein, respectively; and 400 µM of S2403 (pyroGlu-Phe-Lys-pNA.HCl) for plasmin. The substrate concentrations used were at or above their determined Km values for their respective enzymes. The initial reaction velocities of chromogenic substrate cleavage, determined for 10–15 min at 37°C in a microplate reader at 37°C (Thermomax, Molecular Devices, USA) were converted to fraction of amidolytic activity remaining. For fluorogenic assays increasing concentrations of PN2KPI, BPTI and mutants in TBSB, were incubated with FXIa (25 pM) in 90 µl volumes for 30 min at room temperature in black polystyrene microtitre plates (Corning, NY, USA) to establish equilibrium between the inhibitor and the enzyme. To this preincubation mixture, 10 µl of the fluorogenic substrate, Boc-Glu(OBzl)-Ala-Arg-methylcoumarin acetate (Boc-EAR-MCA; Peptides International, Inc., Louisville, Kentucky) was added to a 275 µM final concentration in a total reaction volume of 100 µl. For studies with trypsin, increasing concentrations of PN2KPI (0–0.7 nM), or BPTI (0–2.8 nM) in TBSB were incubated with 100 pM trypsin (bovine source, Sigma, St Louis, Missouri) in 90 µl volume for 30 min at room temperature to establish equilibrium between the inhibitor and the enzyme. Ten microlitres of the fluorogenic substrate Boc-EAR-MCA was added to a final concentration of 6 µM in a total reaction volume of 100 µl. The progress of hydrolysis was monitored at an emission wavelength (Em440) of 440 nm after excitation at 350 nm (Ex350). The fluorimetric initial reaction velocity readings for 10–15 min in a microplate reader at room temperature (Spectramax M2, Molecular Devices, USA) were converted to fraction of amidolytic activity remaining. In both colorimetric and fluorescence assays, the IC50 values were determined by plotting residual enzyme activity against inhibitor concentration using KaleidaGraph v3.5 software and Ki eq were calculated using the equation explained in ‘Data analyses’ section.
Progress curve generation
Release of a highly fluorescent product from the peptidyl substrate Boc-EAR-MCA by FXIa or trypsin was monitored in the presence of varying concentrations of PN2KPI or BPTI. The values of Km for Boc-EAR-MCA hydrolysis by FXIa and trypsin were determined to be 275 and 1.2 µM, respectively. Substrate hydrolysis was initiated by the addition 10 µl of enzyme (25 pM FXIa or 1 nM trypsin, final concentrations) to 90 µl of a mixture of inhibitor (varying concentrations) and fluorogenic substrate in TBSB in 96-well black polystyrene microtitre plates. The progress of substrate hydrolysis was monitored for up to 50 min at 15- or 60 s intervals at room temperature (ranging from 24°C to 27°C) in a fluorescence plate reader as described earlier. In order to study whether trypsin inhibition by BPTI follows irreversible inhibition kinetics, progress curves of substrate hydrolysis were generated using final concentrations of trypsin (1 nM), BPTI (16 nM) and varying concentrations (1.2–12 µM) of fluorogenic substrate (44). The progress of fluorogenic substrate hydrolysis was monitored for 50 min at room temperature.
Data analyses
For other than the tight binding inhibitors, values of Ki eq were derived from their IC50 using the following equation:
| (1a) |
where, [S] is substrate concentration, Km is the Michaelis constant.
For tight-binding inhibitors the Ki eq were calculated using the following equation (45):
| (1b) |
where Vs is the velocity after a steady state has been reached between enzyme and inhibitor, Vo is the velocity of product formation at the start of the reaction, Ki eq represents the equilibrium inhibition constant, [S] is substrate concentration, Km is the Michaelis constant and I is the inhibitor concentration.
Inhibition by slow, tight-binding inhibitors can be described by two general mechanisms (46–49) as depicted in Fig. 1. In mechanism 1, the formation of the final tight enzyme–inhibitor complex occurs slowly but directly without any substantial accumulation of an initial loose complex. In this mechanism, k′1 and k′–1 are the second- and first-order kinetic rate constants. In mechanism 2, there is accumulation of an initial enzyme-inhibitor complex with an inhibition constant of K′i that isomerizes in a second step to form the tight complex and both k2 and k–2 are first-order kinetic constants.
Fig. 1.

Enzyme inhibition mechanisms. In mechanism 1, the binding of inhibitor and isomerization of the complex are simultaneous and not distinguishable and therefore, k1 and k–1 represent the overall on-rate and off-rate constants. In mechanism 2, on the other hand, inhibition of enzyme occurs in two steps, characterized by the formation of a loose EI complex described by rate constants k′1 and k′–1 and isomerization of EI–EI* (the stable complex) occurs with an on-rate of k2 and an off-rate of k–2.
A general equation for progress curves for slow-binding inhibition is given by the following equation:
| (2) |
where Vo is the velocity of product formation at the start of the reaction, Vs is the velocity after a steady state has been reached between enzyme and inhibitor, kobs is the observed first order rate constant that characterizes the transition from initial velocity to steady state velocity and P is the concentration of product formed at time t.
Kinetic parameters Km and kcat for substrate hydrolysis in the presence of the inhibitor are related to Vo and kobs as follows:
Mechanism 1
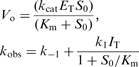 |
(2a) |
Thus, if enzyme inhibition occurs involving mechanism 1 the initial velocity would be independent of the inhibitor concentration and kobs versus I would be linear with an y-intercept of k–1.
Mechanism 2
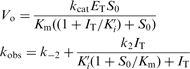 |
(2b) |
where, ET and IT are total concentrations of enzyme and inhibitor, respectively, and S0 is substrate concentration.
Thus, for inhibition by mechanism 2, Vo is inversely related to the inhibitor concentration and kobs versus I is hyperbolic. Therefore, by analyzing how Vo and kobs are related to the inhibitor concentration one may assess whether the enzyme inhibition is a single step (mechanism 1) or a two-step (mechanism 2) phenomenon.
KaleidaGraph (v3.5) was used for obtaining values of Vo, Vs and kobs. Values of k–1 (mechanism 1) or k–2 (mechanism 2) were derived from progress curves by using the following relationship (45, 49):
| (3) |
The second-order rate constant k1 (mechanism 1) was then obtained from the following linear equation:
|
(4) |
Calculated Ki (Kical) is thus given by k–1/k1.
To determine whether trypsin forms a non-dissociable complex with BPTI under our experimental conditions, we generated progress curves using fixed enzyme and inhibitor concentrations while varying the substrate concentration. Product formation approaches a finite value [P∞] with progress of time and this value decreases with decrease of substrate concentration represented by the following equation (44):
| (5) |
where V is the rate of hydrolysis of substrate in the absence of inhibitor, [S] is the substrate concentration, Km is the Michaelis constant, k1 app represents the apparent association rate constant of the enzyme–inhibitor complex in the presence of the substrate and I is the inhibitor concentration. From the values of k1 app at different substrate concentrations, the association rate constant k1 for formation of trypsin–BPTI complex was calculated using the following equation:
| (6) |
The intercept of the plot of 1/k1 app versus [S] represents 1/k1.
Results
Inhibition of coagulation proteases and trypsin by PN2KPI
Initial studies were aimed at determining the specificity of interactions of PN2KPI with a variety of coagulation enzymes and trypsin (Supplementary Fig. S1 and Table I). FXIa and trypsin were inhibited with Ki values of 0.81 nM and 0.03 nM, respectively, whereas no evidence of thrombin inhibition was observed, and the Ki values for FVIIa (in the absence and presence of tissue factor) FIXa, FXa, FXIIa and plasmin were all >400 nM, whereas kallikrein was inhibited by PN2KPI with a Ki value of 183 nM.
Structural comparisons of PN2KPI and BPTI
The primary structures of the highly homologous Kunitz inhibitors PN2KPI and BPTI are shown in Fig. 2A. For comparison of three-dimensional structures, we superimposed the PN2KPI X-ray crystal structure from the PN2KPI-FXIa catalytic domain complex (PDB:1ZJD) (28) and the BPTI structure from the BPTI–trypsin complex (PDB: 2FTL) (29). The crystal structures of these two proteins were superimposable, displaying a striking similarity of the main-chain conformations of PN2KPI and BPTI. The two subdomains within these inhibitors termed loops 1 and 2 (Fig. 2B) that interact most closely with their cognate proteinases are highly homologous sharing primary sequence identity of 70% and 83%, respectively between PN2KPI and BPTI (Fig. 2A). A short sequence of ascending residues, 39–41, adjacent to loop 2 displays some backbone deviation between the two inhibitors (Fig. 2B). However, this sequence of residues does not interact with the cognate proteinases as observed from the crystal structures of the enzyme–inhibitor complexes.
Fig. 2.

The primary and tertiary structures of the Kunitz-type inhibitors, PN2KPI and BPTI (A and B) and comparison of interactions of P1 site residues Arg15 and Lys15 (of PN2KPI and BPTI, respectively) with the Asp189 of FXIa catalytic domain and Trypsin (C and D). (A) The primary amino acid sequence of PN2KPI and BPTI. The P1 site residue is shown with an arrowhead. Underlined sequences are the enzyme interacting loops 1 and 2 of the inhibitor, which share a sequence identity of 70% and 83%, respectively. Cysteine residues that make intrachain disulfide bonds are shown in yellow and disulfide bonds are shown in solid lines. (B) Superimposed X-ray crystal structures of PN2KPI (in pink, PDB: 1ZJD) and BPTI (in green, PDB: 2FTL) shown with P1 site residues Arg15 and Lys15 side chains. Loops 1 and 2, and N and C terminals are indicated. (C) PN2KPI in complex with FXIa catalytic domain (dark blue; PDB:1ZJD) and trypsin (light blue; PDB:1TAW). Arg15 of PN2KPI (pink and red) protrudes into the S1 specificity pocket of the catalytic domain of FXIa and trypsin to establish a salt bridge with Asp189. (D) Lys15 of BPTI (grey) protrudes similarly into the S1 specificity pocket of trypsin (light blue; 2FTL) to establish a hydrogen bond with Asp189, with the intervention of a water molecule (orange).
Inhibition of FXIa, kallikrein, plasmin and trypsin by PN2KPI mutants
Based on an inspection of the X-ray crystal structure of the PN2KPI/FXIa complex (28), we have selected several key residues in PN2KPI for mutational analysis to determine the effects on inhibition of FXIa, plasmin, kallikrein and trypsin (Table II), the proteases most susceptible to inhibition by PN2KPI, according to our results (Table I). The PN2KPI-R15A lost virtually all its inhibitory activity against all four proteases, confirming the essential role of the P1 residue of the Kunitz inhibitor in protease inhibition. Interestingly, the PN2KPI-M17A mutant displayed ∼25-fold gain of function against kallikrein without any significant change in Ki values for FXIa, plasmin or trypsin. Whereas we observed a significant loss of activity for PN2KPI-P13A, PN2KPI-R20A and PN2KPI-F34A mutants in the inhibition of FXIa, kallikrein and plasmin, there was no significant effect of any of these mutants in trypsin inhibition.
Table II.
Inhibition of FXIa, plasma kallikrein, plasmin and trypsin by PN2KPI mutantsa.
| Ki [nM] | ||||
|---|---|---|---|---|
| Inhibitors | FXIa | Kallikrein | Plasmin | Trypsin |
| PN2KPI | 0.81 | 183 | 459 | 0.03 |
| PN2KPI-P13A | 2.96 | 1005 | 2,690 | 0.048 |
| PN2KPI-R15A | NIb | NIb | NIb | NIb |
| PN2KPI-M17A | 0.81 | 7.4 | 479 | 0.054 |
| PN2KPI-S19A | 0.93 | 253 | 615 | 0.024 |
| PN2KPI-R20A | 2.83 | 6,320 | 914 | 0.041 |
| PN2KPI-F34A | 4.92 | 398 | 2,640 | 0.088 |
aTrypsin data represent fluorogenic assay results, whereas the results for the rest of the enzymes are based on chromogenic assays.
bNo inhibition observed at inhibitor concentrations up to 4 µM.
Table I.
Inhibition of coagulation proteases by PN2KPI.
| Enzyme | FIIa | FVIIa | FVIIa+TF | FIXa | FXa | FXIa | FXIIa | Plasmin | Kallikrein | Trypsin |
|---|---|---|---|---|---|---|---|---|---|---|
| Ki [nM] | NIa | 3,500 | 756 | 5,500 | 1,150 | 0.81 | 7,610 | 459 | 183 | 0.03 |
aNo inhibition observed at the highest inhibitor concentration (8 µM).
Equilibrium inhibition of FXIa and trypsin by PN2KPI and BPTI and P1 site mutants
To determine values of Ki for inhibition of FXIa and trypsin by PN2KPI and BPTI (Supplementary Fig. S2), the inhibitors were preincubated with the enzyme for 30 min at room temperature, and the residual enzyme activity was determined. The Ki value (Table III) for PN2KPI inhibition of FXIa was 0.81 ± 0.31 nM (Supplementary Fig. S2A), consistent with previous reports (18–24) indicating that PN2 is a tight-binding inhibitor of FXIa. In contrast, the Ki for inhibition of FXIa by BPTI determined by the equilibrium method was 627 nM (Supplementary Fig. S2B) demonstrating that in spite of the striking similarity in three-dimensional structure, the affinity of PN2KPI for FXIa is ∼775-fold tighter than that of BPTI for FXIa. To examine the contribution of the P1 residue of the two Kunitz- type inhibitors on the inhibition of FXIa, we prepared two mutant proteins PN2KPI-R15 K (mimicking BPTI at the P1 site) and BPTI-K15 R (mimicking PN2KPI at the P1 site). These P1 site mutants PN2KPI-R15 K (Supplementary Fig. S2C) and BPTI-K15 R (Supplementary Fig. S2D) were characterized by Ki values in FXIa inhibition of 11.3 nM and 4.1 nM, respectively (Table III). In marked contrast, the Ki values for trypsin inhibition by PN2KPI (Supplementary Fig. S2E), BPTI (Supplementary Fig. S2F), PN2KPI-R15 K (Supplementary Fig. S2G) and BPTI-K15 R (Fig. S2H), ranging between 0.016 and 0.076 nM (Table III), were not significantly different biologically.
Table III.
Kinetic studies for inhibition of FXIa and trypsin by Kunitz inhibitors.
| Enzyme | Inhibitor | Mechanism | Ki eq [nM] | k1 M−1 s−1 | k–1 s−1 | KI cal [nM] |
|---|---|---|---|---|---|---|
| FXIa | PN2KPI | 1 | 0.81 ± 0.3 | 0.24 × 106 | 3.2 × 10−4 | 1.3 |
| PN2KPI-R15 K | 1 | 11.3 ± 0.6 | 1.7 × 105 | 3.1 × 10−3 | 18.2 | |
| BPTI | Competitive | 627 ± 9.1 | 708 ± 81a | |||
| BPTI-K15 R | 1 | 4.1 ± 0.3 | 2.87 × 105 | 2.3 × 10−3 | 8.01 | |
| Trypsin | PN2KPI | 1 | 0.03 ± 0.007 | 3.4 × 106 | 3.7 × 10−4 | 0.049 |
| PN2KPI-R15 K | 0.047 ± 0.003 | |||||
| BPTI | 1 | 0.072 ± 0.001 | 4.4 × 105 | 3.2 × 10−5 | ||
| BPTI-K15 R | 0.016 ± 0.002 |
aCalculated from Michaelis–Menten plot represented in Fig. 5B.
Progress curve analysis of FXIa and trypsin inhibition
To investigate the mechanisms by which PN2KPI and BPTI inhibit FXIa and trypsin, progress curves generated in the absence and presence of various inhibitor concentrations were analysed. The progress curves for FXIa inhibition by PN2KPI (0–32 nM) and by BPTI (0–5 µM) were monitored at 15–60-s intervals for a maximum duration of 50 min. FXIa inhibition displayed distinct progress curve patterns for inhibition by PN2KPI (Fig. 3A) and BPTI (Fig. 5A). In the case of inhibition of FXIa by PN2KPI (Fig. 3A), PN2KPI-R15 K (Fig. 4A) and BPTI-K15 R (Fig. 6A) the initial rates of fluorogenic substrate hydrolysis were independent of inhibitor concentration, and the progress curves demonstrated progressive decreases with increasing inhibitor concentration. On the contrary, the initial rates of inhibition of FXIa by BPTI were linear at all inhibitor concentrations and the slopes were progressively decreased with increasing concentrations of BPTI (Fig. 5A). Similarly, the progress curves of trypsin inhibition by PN2KPI and BPTI (Figs 7A and 8A) demonstrated initial rates that were independent of inhibitor concentrations as were found for FXIa inhibition by PN2KPI. These data demonstrate that FXIa inhibition by PN2KPI and trypsin inhibition by BPTI both conform to mechanism 1 with slow, single-step equilibration between enzyme and inhibitor.
Fig. 3.

Progress curve of fluorogenic substrate (Boc-EAR-MCA) hydrolysis by FXIa in the presence of PN2KPI and the secondary plot. (A) Hydrolysis of Boc-EAR-MCA (275 µM) by FXIa (25 pM) in the presence of varying concentrations of PN2KPI (0–32 nM) were monitored for 3,000 s. Inhibitor concentrations used are shown on the right, adjacent to the respective curve. (B) Secondary plot of kobs values versus PN2KPI concentration. The values of kobs were obtained by fitting progress curve data shown in Figure (A) using equation (2). The kobs (s−1) values for each inhibitor concentration were calculated from the averaged values (± SEM) of three assays each done in duplicate. All assays were background subtracted with substrate autolysis for the entire assay duration.
Fig. 5.

Progress curve of fluorogenic substrate (Boc-EAR-MCA) hydrolysis by FXIa in the presence of BPTI. (A) Hydrolysis of Boc-EAR-MCA (275 µM) by FXIa (25 pM) in the presence of varying concentrations of BPTI (0–5 µM) were monitored for 3,000 s. Inhibitor concentrations used are shown on the right, adjacent to the respective curve. (B) S-2366 hydrolysis by FXIa (1 nM) in the presence of varying concentrations of BPTI. Michaelis–Menten titration curves of initial rates as a function of substrate concentration were generated as described in the ‘Experimental procedures’ section. Each data point represents the mean (± SD) of two to four assays each done in duplicate.
Fig. 4.

Progress curve of fluorogenic substrate (Boc-EAR-MCA) hydrolysis by FXIa in the presence of PN2KPI-R15 K and the secondary plot. (A) Hydrolysis of Boc-EAR-MCA (275 µM) by FXIa (25 pM) in the presence of varying concentrations of PN2KPI-R15 K (0–120 nM) were monitored for 3,000 s. Inhibitor concentrations used are shown on the right, adjacent to the respective curve. (B) Secondary plot of kobs values versus PN2KPI-R15 K concentration. The kobs (s−1) values for each inhibitor concentration were calculated from the averaged values (± SD) of three assays each done in duplicate. All assays were background subtracted with substrate autolysis for the entire assay duration.
Fig. 6.

Progress curve of fluorogenic substrate (Boc-EAR-MCA) hydrolysis by FXIa in the presence of BPTI-K15 R. (A) Hydrolysis of Boc-EAR-MCA (275 µM) by FXIa (25 pM) in the presence of varying concentrations of BPTI-K15 R (0–80 nM) were monitored for 3,000 s. Inhibitor concentrations used are shown on the right, adjacent to the respective curve. (B) Secondary plot of kobs values versus BPTI-K15 R concentration. The kobs (s−1) values for each inhibitor concentration were calculated from the averaged values (± SD) of three assays each done in duplicate.
Fig. 7.

Progress curve of fluorogenic substrate (Boc-EAR-MCA) hydrolysis by trypsin in the presence of PN2KPI and the secondary plot. (A) Hydrolysis of Boc-EAR-MCA (6 µM) by trypsin (0.5 nM) in the presence of varying concentrations of PN2KPI (0–20 nM) were monitored for 2,500 s. Inhibitor concentrations used are shown on the right, adjacent to the respective curve. (B) Secondary plot of kobs values versus PN2KPI concentration. The kobs (s−1) values for each inhibitor concentration were calculated from the averaged values (± SD) of five assays each done in duplicate.
Fig. 8.

Progress curves of fluorogenic substrate (Boc-EAR-MCA) hydrolysis by trypsin in the presence of BPTI. (A) Represents substrate (6 µM) hydrolysis by trypsin (1 nM) in the presence of varying concentrations of BPTI (0–16 nM). Inhibitor concentrations are shown on the right, adjacent to the respective curve. (B) Represents hydrolysis at varying concentrations of substrate (curves 1 and 2, 12 µM; 3, 6 µM; 4, 3 µM; 5, 1.2 µM) using trypsin (1 nM) and in the absence (curve 1) and in the presence (curves 2–5) of BPTI (16 nM). The apparent association rate constants (k1 app) for each substrate concentration are obtained from the respective curv es, using equation (5) (see ‘Data analyses’ section) and the value of k1 was then obtained from a secondary plot of 1/k1 app versus [S], (inset).
In secondary plots, values of kobs obtained from progress curves using equation (2) were plotted as a function of the corresponding inhibitor concentrations. A linear relationship of kobs versus inhibitor concentration was observed for PN2KPI (Fig. 3B), PN2KPI-R15 K (Fig. 4B) and BPTI-K15 R (Fig. 6B). In all these cases, inhibition of FXIa conforms to mechanism 1 (Fig. 1 and Table III), which takes place in a single step involving the gradual formation of a tight complex. In contrast, BPTI inhibition of FXIa follows simple steady-state kinetics with the rate of substrate hydrolysis remaining the same throughout the time period at each inhibitor concentration and progressively lower rates observed with progressive increases in inhibitor concentration (Fig. 5A). This is shown in a separate confirmatory assay (Fig. 5B) in which titration curves of chromogenic substrate (S-2366) hydrolysis by FXIa in the presence of varying concentrations of BPTI demonstrated increasing values of Km without any alteration of Vmax, indicative of pure competitive inhibition (Table I). Similar to FXIa inhibition, trypsin inhibition by PN2KPI also displays a single step inhibition process displaying a linear relationship of kobs versus inhibitor concentration (Fig. 7B).
Progress curves of trypsin inhibition by BPTI (Fig. 8A) demonstrate unequivocally that the initial rates of trypsin inhibition were independent of BPTI concentration suggesting that the inhibition conforms to mechanism 1. However, we were unable to obtain reliable kobs values from these progress curves possibly because of the extremely slow rate of dissociation of the enzyme–inhibitor complex. Indeed Zhou et al. (44) demonstrated that trypsin inhibition by BPTI follows apparently irreversible kinetics with an association rate constant of 0.37 × 106 M−1 s−1. Vincent and Lazdunski (50) reported the first-order rate constant of dissociation of the same inhibition reaction to be 6.6 × 10−8 s−1. We attempted to generate progress curves for the dissociation of the complex, but even at the end of 60 min free trypsin liberated was <1% (data not shown). This fact together with the slow loss of activity of trypsin on standing in solution beyond 60 min as observed from chromogenic or fluorogenic substrate hydrolysis made it impossible to generate the data required for calculation of the relevant rate constants. We therefore aimed to determine whether trypsin forms a non-dissociable complex with BPTI under our experimental conditions, which are significantly different from those used previously (44). For this purpose, we generated progress curves using fixed enzyme and inhibitor concentrations while varying the substrate concentration (Fig. 8B). It is apparent from these results that product formation approaches a finite value [P∞] with progress of time and that this value decreases with decrease of substrate concentration. These results confirm the previous conclusion that trypsin inhibition by BPTI is non-dissociable (44). From progress curves shown in Fig. 8B we could obtain the values of k1 app (the apparent association rate constant of the enzyme–inhibitor complex in the presence of the substrate) using equation (6). The value of the association rate constant k1 (4.4 × 105 M−1 s−1) of trypsin-BPTI complex formation was then obtained from the intercept of the secondary plot of 1/k1app versus [S] shown as an inset in Fig. 8B. The dissociation rate constant k–1 (3.2 × 10−5 s−1) shown in Table III was obtained assuming Ki eq = k–1/k1.
Discussion
Factor XI (FXI) is the precursor of a plasma serine protease formed by factor XIIa or thrombin that participates in the consolidation phase of blood coagulation and is implicated in pathological thrombosis, rendering it a potential target for the development of antithrombotic agents. As the Kunitz-type inhibitor, protease nexin-2 (PN2-KPI) is a potent inhibitor of FXIa, the major question addressed in the present study is the physiological relevance, specificity and mechanism of action of the platelet α-granular protein, PN2/AβPP. Previous studies from our laboratory indicate that the major interactions required for FXIa inhibition by PN2 are localized to the catalytic domain of FXIa and the KPI domain of PN2 (24). We have also found that secretion of PN2 from activated platelets limits the lifetime of FXIa activity within the locus of activated platelets. However, in the presence of activated platelets, HK and Zn2+ ions, FXIa bound to the surface of activated platelets is protected from inactivation by PN2KPI (23, 51).
It has been demonstrated (52) that over-expression of PN2/AβPP in circulating platelets of transgenic mice was associated with inhibition of thrombosis in vivo, whereas mice with a genetic knockout of the gene for PN2/AβPP demonstrated a significant increase in thrombosis. Moreover, platelet PN2/AβPP transgenic mice developed larger haematomas in an experimental intracerebral haemorrhage model, whereas AβPP gene knockout mice exhibited reduced haemorrhage size. The question raised by these elegant studies relates to the mechanism by which over-expression of PN2/AβPP can inhibit thrombosis and enhance haemorrhage and deletion of this Kunitz inhibitor can promote thrombosis and inhibit haemorrhage. The present studies strongly suggest that the only plasma coagulation enzyme inhibited by physiological concentrations of PN2KPI achievable in plasma is FXIa. Thus, as shown in Table I, FXIa is inhibited by PN2KPI with a Ki ∼ 0.81 nM, a concentration considerably higher than the virtually undetectable level of PN2/AβPP in plasma (19, 53), but well below the concentrations (3–5 nM) that can be achieved at physiologic concentrations of platelets after secretion from α-granules (19, 23). Thus, in the vicinity of a platelet thrombus, the concentration of PN2/AβPP is likely to be sufficient to regulate FXIa activity, but insufficient to have any significant effect on other plasma serine proteases, Ki values (183–5500 nM) for which are 36-fold to 1,100-fold higher than the inhibitor concentration (3–5 nM) achieved after platelet secretion.
In reported specificity studies of target plasma proteases inhibited by PN2/AβPP and by PN2KPI, there is a general consensus that neither thrombin (21, 54) nor FXIIa (21, 40) is inhibited by PN2KPI, in agreement with the present studies which show no inhibition by thrombin and a Ki ∼ 7610 nM for FXIIa (Table I). There is also a consensus that both FXIa (18, 21, 40, 55) and trypsin (18, 21, 40) are potently inhibited by PN2KPI with Ki values of 0.25–2.7 nM and 0.02–0.42 nM, respectively, i.e. similar values to those reported herein (0.81 nM and 0.03 nM, respectively, Table I). In contrast, the highly homologous plasma protease, kallikrein is inhibited with a Ki (183 nM, Table I) >35-fold higher than the concentration of PN2/AβPP (3–5 nM) achieved after platelet secretion, compared with a value of 515 nM obtained by Dennis et al. (55) and no inhibition observed by Van Nostrand et al. (21), suggesting that kallikrein is not a target for inhibition by PN2/AβPP under physiological conditions. In contrast, widely divergent results have been reported for FIXa inhibition, as Schmaier et al. (56) have reported a Ki value of 190 nM for PN2KPI, and have shown that the presence of phospholipid (PS/PC) vesicles and FVIIIa protect FIXa from inhibition (57), whereas, in contrast, Neuenschwander et al. (45) have reported a Ki value of 9,500 nM for FIXa inhibition by PN2KPI, in reasonably good agreement with our Ki value of 5,500 nM (Table I). Similar discrepancies exist for inhibition of FXa by PN2KPI because Mahdi et al. (58) have reported Ki values of 33–72 nM, whereas Van Nostrand et al. (21) initially reported no inhibition and subsequently reported a Ki value of 59 nM (40), whereas Dennis et al. (55) reported a Ki value of 240 nM, compared with our value of 1,150 nM (Table I). Finally, Mahdi et al. (59) found that PN2KPI inhibited FVIIa with Ki values of 110–150 nM in the absence of tissue factor, compared with >1,000 nM reported by Dennis et al. (55) and 3,500 nM in the present study (Table I). In the presence of tissue factor, PN2KPI is a slightly more effective inhibitor of FVIIa, with Ki values of 68–78 nM reported by Schmaier et al. (59), compared with 300 nM reported by Dennis et al. (55) and 756 nM in the present study (Table I). The conclusion to be drawn from all these studies and the data presented herein is that, among all the plasma coagulation proteases examined for inhibition by PN2KPI, FXIa is the only physiologically relevant target, considering the concentrations of this Kunitz inhibitor achievable in the vicinity of a haemostatic thrombus.
In addition to determining the specificity PN2KPI for FXIa and trypsin the present work focuses on elucidating the mechanisms of inhibition of FXIa and trypsin, by the two structurally similar Kunitz-type inhibitors, PN2KPI and BPTI. The inhibition of FXIa by PN2KPI was studied in detail previously (23, 24), and trypsin inhibition by BPTI has been characterized as a slow, tight-binding interaction (44). However, the precise mechanism of FXIa inhibition by either PN2KPI or BPTI and the mechanism of trypsin inhibition by PN2KPI are unknown. It is particularly interesting to determine these kinetic mechanisms and the functional roles of the P1 residues in these two inhibitors because of the high affinity and specificity of FXIa inhibition by PN2KPI, the marked difference in Ki for FXIa inhibition by PN2KPI and BPTI and the striking structural similarity between these two inhibitors in complex with their cognate proteases (Fig. 2). The present investigations demonstrate marked differences in equilibrium inhibition constants for FXIa inhibition by PN2KPI (Ki 0.81 nM) and BPTI (Ki 627 nM) as shown in Table III. Moreover, progress curve analyses demonstrated characteristic differences in the mechanisms of FXIa inhibition by PN2KPI and BPTI. Thus, the inhibition of FXIa by PN2KPI, by PN2KPI-R15 K, and by BPTI-K15 R were all demonstrated to conform to mechanism 1 (Fig. 1), in which a slow equilibration occurs between the enzyme and the inhibitor. In contrast, FXIa inhibition by BPTI conforms to a simple steady-sate kinetic mechanism (Fig. 5A), and analysis of the Michaelis–Menten plot (Fig. 5B) demonstrated competitive inhibitory mechanism, from which it can be calculated that the value of Ki cal is 708 nM, which compares favourably with the value of Ki eq (627 nM), determined from equilibrium inhibition experiments (Table III). Although there are reports on the inhibition of serine proteases by BPTI (26, 27) this is the first report on the mechanism of FXIa inhibition by BPTI that is best described as classical competitive inhibition. These results are interesting in the context of recent studies by Neuenschwander et al. (45), who have demonstrated that the inhibition of FIXa by PN2KPI (Ki ∼ 20 µM) proceeds by mechanism 2 (45), in contrast to our results for FXIa inhibition by PN2KPI (Ki 0.81 nM), which are consistent with mechanism 1.
The results of FXIa inhibition by the two Kunitz inhibitors stand in marked contrast to those describing trypsin inhibition by PN2KPI and BPTI. Thus, the equilibrium inhibition constants for trypsin inhibition by PN2KPI (Ki 0.026 nM) and BPTI (Ki 0.072 nM) are only ∼3-fold different, compared with the ∼775-fold difference when FXIa is the protease (Table III). Progress curve analyses demonstrate that the formation of the trypsin–PN2KPI complex and the trypsin–BPTI complex, like FXIa inhibition by PN2KPI, conform to mechanism 1. However, as mentioned in the ‘Results’ section, we were unable to obtain kinetic constants of trypsin inhibition by BPTI from these progress curves since reliable kobs values could not be generated. Instead, the association rate constant (k1) was obtained from separate sets of experiments designed to obtain kinetic parameters for irreversible kinetics (44). As shown in Fig. 8B, product formation [P∞] approaches a constant value with time. Using this approach, we were able to obtain the association rate constant of trypsin inhibition by BPTI (Table III). These results do not exclude the possibility of reversible enzyme–inhibitor binding with a dissociation rate too slow to be measured during the time interval studied. Indeed the value of the dissociation rate constant k–1 (3.2 × 10−5 s−1) shown in Table III for trypsin-BPTI complex was found to be 10-fold slower (3.2 × 10−4 s−1) than that of FXIa-PN2KPI (Table III).
In addition to establishing the mechanistic characteristics describing the two Kunitz inhibitors, PN2KPI and BPTI, in FXIa and trypsin inhibition, we further investigated the importance of P1 site residues, i.e. Arg15 in PN2KPI and Lys15 in BPTI, in FXIa inhibition. The essential role of the P1 residue of PN2KPI was previously demonstrated in our laboratory by using a mutant protein, PN2KPI-R15A, which completely loses its inhibitory activity against FXIa (28). In addition, it has been demonstrated that the BPTI (K15A) mutant largely lost its trypsin inhibitory activity (60). As shown in Fig. 2, loop 1 of the enzyme-interacting double-loop region contains the P1 residue and is stabilized by a disulfide bond. This loop structure maintains similar backbone orientations in several Kunitz inhibitors, including PN2KPI and BPTI (Fig. 2B). The P1 residue of inhibitors in general is considered to be the most energetically important residue for high-affinity binding to serine proteases and therefore the primary determinant of specificity of an inhibitor for a given protease (61, 62). The guanidinium group of Arg15 in PN2KPI interacts directly with the carboxylate group of Asp189 in trypsin (PDB: 1TAW) (41) and of Asp189 in the catalytic domain of FXIa (PDB:1ZJD) (28) by forming a salt bridge in the S1 specificity pocket (Fig. 2C). Similarly, Lys15 in BPTI protrudes into the S1 specificity pocket of trypsin, while maintaining the same side chain orientation (similar to Arg15) and interacts with Asp189. However, the shorter Lys side chain (compared to Arg) relies on an ordered water molecule to establish the Lys15-Asp189 interaction in the BPTI–trypsin complex (Fig. 2D), (PDB:2FTL) (29). Thus, whereas PN2KPI and BPTI have similar backbone structures and P1 residue side chain orientations, differences in side chain bulkiness, length and consequent charge distribution, result in significant differences in local intermolecular interactions with FXIa. We hypothesized that these fine structural differences between PN2KPI and BPTI might have functional consequences.
To determine whether the presence of Arg15 or Lys15 at the P1 site of the inhibitor interacting with Asp189 of the protease translates into functional differences in their capacity to inhibit FXIa, P1 site PN2KPI and BPTI swap mutants along with their native parent molecules were examined for their inhibitory properties. Unlike the complete or near complete loss of enzyme inhibitory activity observed with alanine mutants of these inhibitors (28, 60), the mutant, PN2KPI-R15 K displayed partial loss of function, as previously observed (40). BPTI-K15 R on the other hand displayed >150-fold gain of function (Ki 4.1 nM) in FXIa inhibition, compared to its native counterpart BPTI (Ki 627 nM). The inhibitory activities for FXIa of the Arg15-containing PN2KPI and the BPTI-K15 R mutant are ∼14-fold and ∼150-fold more potent than those of the corresponding Lys15-containing forms. Thus, Arg15 is an important determinant of high-affinity binding and specificity for FXIa inhibition. In contrast, the presence of arginine or lysine at the P1 position had very little influence on the trypsin inhibitory activities of either PN2KPI or BPTI. Thus, the presence of arginine at the P1 position in BPTI or PN2KPI only slightly improves (5-fold and 2-fold, respectively) their affinity for trypsin. In an earlier study of the crystal structure of uncomplexed trypsin-D189 S (PDB: 1AMH) a single amino acid mutation in the substrate-binding pocket resulted in extensive structural changes around the mutated residue (63). However, a structural comparison of native trypsin with that of BPTI complexed to trypsin-D189 S revealed striking structural similarities (64), indicating the plasticity of trypsin accommodating BPTI and reverting back to its native structure. Thus, the structural plasticity of trypsin may explain why similar inhibition constants are seen with arginine or lysine containing Kunitz inhibitors. In contrast, in the case of FXIa the presence of Arg or Lys at the P1 site of the inhibitor determines both proteinase specificity and the kinetic mechanisms of proteinase interaction. The results of our mutational analysis of key residues in PN2KPI, based on the crystal structures of PN2KPI and BPTI in complex with their cognate proteinases (Table II) are particularly relevant in the context of the additional studies summarized in Table III, suggesting that it is the plasticity of trypsin that renders it particularly susceptible to inhibition by Kunitz inhibitors. Thus, it would appear that PN2/AβPP, contained within the α-granules of platelets and secreted into the vicinity of the haemostatic thrombus, is an exquisitely specific inhibitor of FXIa among all other plasma coagulation proteases since, within the vascular compartment, it would not encounter trypsin, a digestive enzyme secreted into the gastrointestinal tract.
Supplementary Data
Supplementary Data are available at JB online.
Funding
Supported by research grants from the National Institutes of Health (HL74124 and HL46213 to P.N.W.).
Conflict of interest
No conflict of interest.
Acknowledgements
We are grateful to Sriram Krishnaswamy (Children’s Hospital of the University of Pennsylvania), Paul Bock (Vanderbilt University School of Medicine) and Barrie Ashby (Temple University School of Medicine) for helpful discussions concerning the design and kinetic analysis of data. We thank Patricia Pileggi for help in manuscript preparation.
Glossary
Abbreviations
- BMMY
buffered medium containing methanol and yeast nitrogen base
- BMGY
buffered medium containing glycerol and yeast nitrogen base
- BPTI
basic (or bovine) pancreatic trypsin inhibitor
- PN2KPI
protease nexin 2 Kunitz protease inhibitory domain
- PN2
protease nexin 2
- KPI
Kunitz protease inhibitor
- FXIIa
factor XIIa
- FXIa
factor XIa
- FXa
factor Xa
- FIXa
factor IXa
- FVIIa
factor VIIa
- FIIa
factor IIa (thrombin)
References
- 1.Page MJ, Di Cera E. Serine peptidases: classification, structure and function. Cell. Mol. Life Sci. 2008;65:1220–1236. doi: 10.1007/s00018-008-7565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh PN. Factor XI, in. In: Colman RW, Hirsh J, Marder VJ, Clowes AW, George JN, editors. Hemostasis and Thrombosis: Basic Principles & Clinical Practice. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 191–202. [Google Scholar]
- 3.Bouma BN, Griffin JH. Human blood coagulation factor XI. Purification, properties, and mechanism of activation by activated factor XII. J. Biol. Chem. 1977;252:6432–6437. [PubMed] [Google Scholar]
- 4.Naito K, Fujikawa K. Activation of human blood coagulation factor XI independent of factor XII. Factor XI is activated by thrombin and factor XIa in the presence of negatively charged surfaces. J. Biol. Chem. 1991;266:7353–7358. [PubMed] [Google Scholar]
- 5.Gailani D, Broze G.J., Jr Factor XI activation in a revised model of blood coagulation. Science. 1991;253:909–912. doi: 10.1126/science.1652157. [DOI] [PubMed] [Google Scholar]
- 6.Walsh PN, Griffin JH. Contributions of human platelets to the proteolytic activation of blood coagulation factors XII and XI. Blood. 1981;57:106–118. [PubMed] [Google Scholar]
- 7.Fujikawa K, Legaz ME, Kato H, Davie EW. The mechanism of activation of bovine factor IX (Christmas factor) by bovine factor XIa (activated plasma thromboplastin antecedent) Biochemistry. 1974;13:4508–4516. doi: 10.1021/bi00719a006. [DOI] [PubMed] [Google Scholar]
- 8.Di Scipio RG, Kurachi K, Davie EW. Activation of human factor IX (Christmas factor) J. Clin. Invest. 1978;61:1528–1538. doi: 10.1172/JCI109073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Osterud B, Bouma BN, Griffin JH. Human blood coagulation factor IX. Purification, properties, and mechanism of activation by activated factor XI. J. Biol. Chem. 1978;253:5946–5951. [PubMed] [Google Scholar]
- 10.Sinha D, Seaman FS, Walsh PN. Role of calcium ions and the heavy chain of factor XIa in the activation of human coagulation factor IX. Biochemistry. 1987;26:3768–3775. doi: 10.1021/bi00387a005. [DOI] [PubMed] [Google Scholar]
- 11.Knauer DJ, Majumdar D, Fong PC, Knauer MF. SERPIN regulation of factor XIa. The novel observation that protease nexin 1 in the presence of heparin is a more potent inhibitor of factor XIa than C1 inhibitor. J. Biol. Chem. 2000;275:37340–37346. doi: 10.1074/jbc.M003909200. [DOI] [PubMed] [Google Scholar]
- 12.Damus PS, Hicks M, Rosenberg RD. Anticoagulant action of heparin. Nature. 1973;246:355–357. doi: 10.1038/246355a0. [DOI] [PubMed] [Google Scholar]
- 13.Wuillemin WA, Eldering E, Citarella F, de Ruig CP, ten Cate H, Hack CE. Modulation of contact system proteases by glycosaminoglycans. Selective enhancement of the inhibition of factor XIa. J. Biol. Chem. 1996;271:12913–12918. doi: 10.1074/jbc.271.22.12913. [DOI] [PubMed] [Google Scholar]
- 14.Forbes CD, Pensky J, Ratnoff OD. Inhibition of activated Hageman factor and activated plasma thromboplastin antecedent by purified serum C1 inactivator. J. Lab. Clin. Med. 1970;76:809–815. [PubMed] [Google Scholar]
- 15.Scott CF, Schapira M, James HL, Cohen AB, Colman RW. Inactivation of factor XIa by plasma protease inhibitors: predominant role of alpha 1-protease inhibitor and protective effect of high molecular weight kininogen. J. Clin. Invest. 1982;69:844–852. doi: 10.1172/JCI110524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heck LW, Kaplan AP. Substrates of Hageman factor. I. Isolation and characterization of human factor XI (PTA) and inhibition of the activated enzyme by alpha 1-antitrypsin. J. Exp. Med. 1974;140:1615–1630. doi: 10.1084/jem.140.6.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saito H, Goldsmith GH, Moroi M, Aoki N. Inhibitory spectrum of alpha 2-plasmin inhibitor. Proc. Natl Acad. Sci. USA. 1979;76:2013–2017. doi: 10.1073/pnas.76.4.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith RP, Higuchi DA, Broze G.J., Jr Platelet coagulation factor XIa-inhibitor, a form of Alzheimer amyloid precursor protein. Science. 1990;248:1126–1128. doi: 10.1126/science.2111585. [DOI] [PubMed] [Google Scholar]
- 19.Van Nostrand WE, Schmaier AH, Farrow JS, Cunningham DD. Protease nexin-II (amyloid beta-protein precursor): a platelet alpha-granule protein. Science. 1990;248:745–748. doi: 10.1126/science.2110384. [DOI] [PubMed] [Google Scholar]
- 20.Bush AI, Martins RN, Rumble B, Moir R, Fuller S, Milward E, Currie J, Ames D, Weidemann A, Fischer P, Multhaup G, Beyreuther K, Masters CL. The amyloid precursor protein of Alzheimer's disease is released by human platelets. J. Biol. Chem. 1990;265:15977–15983. [PubMed] [Google Scholar]
- 21.Van Nostrand WE, Wagner SL, Farrow JS, Cunningham DD. Immunopurification and protease inhibitory properties of protease nexin- 2/amyloid beta-protein precursor. J. Biol. Chem. 1990;265:9591–9594. [PubMed] [Google Scholar]
- 22.Zhang Y, Scandura JM, Van Nostrand WE, Walsh PN. The mechanism by which heparin promotes the inhibition of coagulation factor XIa by protease nexin-2. J. Biol. Chem. 1997;272:26139–26144. doi: 10.1074/jbc.272.42.26139. [DOI] [PubMed] [Google Scholar]
- 23.Scandura JM, Zhang Y, Van Nostrand WE, Walsh PN. Progress curve analysis of the kinetics with which blood coagulation factor XIa is inhibited by protease nexin-2. Biochemistry. 1997;36:412–420. doi: 10.1021/bi9612576. [DOI] [PubMed] [Google Scholar]
- 24.Badellino KO, Walsh PN. Protease nexin II interactions with coagulation factor XIa are contained within the Kunitz protease inhibitor domain of protease nexin II and the factor XIa catalytic domain. Biochemistry. 2000;39:4769–4777. doi: 10.1021/bi9925468. [DOI] [PubMed] [Google Scholar]
- 25.Badellino KO, Walsh PN. Localization of a heparin binding site in the catalytic domain of factor XIa. Biochemistry. 2001;40:7569–7580. doi: 10.1021/bi0027433. [DOI] [PubMed] [Google Scholar]
- 26.Grzesiak A, Krokoszynska I, Krowarsch D, Buczek O, Dadlez M, Otlewski J. Inhibition of six serine proteinases of the human coagulation system by mutants of bovine pancreatic trypsin inhibitor. J. Biol. Chem. 2000;275:33346–33352. doi: 10.1074/jbc.M006085200. [DOI] [PubMed] [Google Scholar]
- 27.Stassen JM, Lambeir AM, Matthyssens G, Ripka WC, Nystrom A, Sixma JJ, Vermylen J. Characterisation of a novel series of aprotinin-derived anticoagulants. I. In vitro and pharmacological properties. Thromb. Haemost. 1995;74:646–654. [PubMed] [Google Scholar]
- 28.Navaneetham D, Jin L, Pandey P, Strickler JE, Babine RE, Abdel-Meguid SS, Walsh PN. Structural and mutational analyses of the molecular interactions between the catalytic domain of factor XIa and the Kunitz protease inhibitor domain of protease nexin 2. J. Biol. Chem. 2005;280:36165–36175. doi: 10.1074/jbc.M504990200. [DOI] [PubMed] [Google Scholar]
- 29.Hanson WM, Domek GJ, Horvath MP, Goldenberg DP. Rigidification of a flexible protease inhibitor variant upon binding to trypsin. J. Mol. Biol. 2007;366:230–243. doi: 10.1016/j.jmb.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McEvoy MD, Reeves ST, Reves JG, Spinale FG. Aprotinin in cardiac surgery: a review of conventional and novel mechanisms of action. Anesth. Analg. 2007;105:949–962. doi: 10.1213/01.ane.0000281936.04102.9f. [DOI] [PubMed] [Google Scholar]
- 31.Henry D, Carless P, Fergusson D, Laupacis A. The safety of aprotinin and lysine-derived antifibrinolytic drugs in cardiac surgery: a meta-analysis. CMAJ. 2009;180:183–193. doi: 10.1503/cmaj.081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scott CF, Wenzel HR, Tschesche HR, Colman RW. Kinetics of inhibition of human plasma kallikrein by a site-specific modified inhibitor Arg15-aprotinin: evaluation using a microplate system and comparison with other proteases. Blood. 1987;69:1431–1436. [PubMed] [Google Scholar]
- 33.Salameh MA, Soares AS, Hockla A, Radisky ES. Structural basis for accelerated cleavage of bovine pancreatic trypsin inhibitor (BPTI) by human mesotrypsin. J. Biol. Chem. 2008;283:4115–4123. doi: 10.1074/jbc.M708268200. [DOI] [PubMed] [Google Scholar]
- 34.Waxman L, Smith DE, Arcuri KE, Vlasuk GP. Tick anticoagulant peptide (TAP) is a novel inhibitor of blood coagulation factor Xa. Science. 1990;248:593–596. doi: 10.1126/science.2333510. [DOI] [PubMed] [Google Scholar]
- 35.Krishnaswamy S, Vlasuk GP, Bergum PW. Assembly of the prothrombinase complex enhances the inhibition of bovine factor Xa by tick anticoagulant peptide. Biochemistry. 1994;33:7897–7907. doi: 10.1021/bi00191a017. [DOI] [PubMed] [Google Scholar]
- 36.Rezaie AR. Kinetics of factor Xa inhibition by recombinant tick anticoagulant peptide: both active site and exosite interactions are required for a slow- and tight-binding inhibition mechanism. Biochemistry. 2004;43:3368–3375. doi: 10.1021/bi036177y. [DOI] [PubMed] [Google Scholar]
- 37.Macedo-Ribeiro S, Almeida C, Calisto BM, Friedrich T, Mentele R, Sturzebecher J, Fuentes-Prior P, Pereira PJ. Isolation, cloning and structural characterisation of boophilin, a multifunctional Kunitz-type proteinase inhibitor from the cattle tick. PLoS ONE. 2008;3:e1624. doi: 10.1371/journal.pone.0001624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petersen LC, Bjorn SE, Olsen OH, Nordfang O, Norris F, Norris K. Inhibitory properties of separate recombinant Kunitz-type-protease-inhibitor domains from tissue-factor-pathway inhibitor. Eur. J. Biochem. 1996;235:310–316. doi: 10.1111/j.1432-1033.1996.0310f.x. [DOI] [PubMed] [Google Scholar]
- 39.Sierko E, Wojtukiewicz MZ, Kisiel W. The role of tissue factor pathway inhibitor-2 in cancer biology. Semin. Thromb. Hemost. 2007;33:653–659. doi: 10.1055/s-2007-991532. [DOI] [PubMed] [Google Scholar]
- 40.Van Nostrand WE, Schmaier AH, Siegel RS, Wagner SL, Raschke WC. Enhanced plasmin inhibition by a reactive center lysine mutant of the Kunitz-type protease inhibitor domain of the amyloid beta-protein precursor. J. Biol. Chem. 1995;270:22827–22830. doi: 10.1074/jbc.270.39.22827. [DOI] [PubMed] [Google Scholar]
- 41.Scheidig AJ, Hynes TR, Pelletier LA, Wells JA, Kossiakoff AA. Crystal structures of bovine chymotrypsin and trypsin complexed to the inhibitor domain of Alzheimer's amyloid beta-protein precursor (APPI) and basic pancreatic trypsin inhibitor (BPTI): engineering of inhibitors with altered specificities. Protein Sci. 1997;6:1806–1824. doi: 10.1002/pro.5560060902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perona JJ, Tsu CA, Craik CS, Fletterick RJ. Crystal structures of rat anionic trypsin complexed with the protein inhibitors APPI and BPTI. J. Mol. Biol. 1993;230:919–933. doi: 10.1006/jmbi.1993.1210. [DOI] [PubMed] [Google Scholar]
- 43.Hynes TR, Randal M, Kennedy LA, Eigenbrot C, Kossiakoff AA. X-ray crystal structure of the protease inhibitor domain of Alzheimer's amyloid beta-protein precursor. Biochemistry. 1990;29:10018–10022. doi: 10.1021/bi00495a002. [DOI] [PubMed] [Google Scholar]
- 44.Zhou JM, Liu C, Tsou CL. Kinetics of trypsin inhibition by its specific inhibitors. Biochemistry. 1989;28:1070–1076. doi: 10.1021/bi00429a022. [DOI] [PubMed] [Google Scholar]
- 45.Neuenschwander PF, Williamson SR, Nalian A, Baker-Deadmond KJ. Heparin modulates the 99-loop of factor IXa: effects on reactivity with isolated Kunitz-type inhibitor domains. J. Biol. Chem. 2006;281:23066–23074. doi: 10.1074/jbc.M603743200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cha S. Tight-binding inhibitors-I. Kinetic behavior. Biochem. Pharmacol. 1975;24:2177–2185. doi: 10.1016/0006-2952(75)90050-7. [DOI] [PubMed] [Google Scholar]
- 47.Morrison JF. The slow-binding and slow, tight-binding inhibition of enzyme-catalysed reactions. Trends Biochem. Sci. 1982;7:102–105. [Google Scholar]
- 48.Morrison JF, Walsh CT. The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 49.Huang ZF, Wun TC, Broze G.J., Jr Kinetics of factor Xa inhibition by tissue factor pathway inhibitor. J. Biol. Chem. 1993;268:26950–26955. [PubMed] [Google Scholar]
- 50.Vincent JP, Lazdunski M. Trypsin-pancreatic trypsin inhibitor association. Dynamics of the interaction and role of disulfide bridges. Biochemistry. 1972;11:2967–2977. doi: 10.1021/bi00766a007. [DOI] [PubMed] [Google Scholar]
- 51.Baird TR, Walsh PN. The interaction of factor XIa with activated platelets but not endothelial cells promotes the activation of factor IX in the consolidation phase of blood coagulation. J. Biol. Chem. 2002;277:38462–38467. doi: 10.1074/jbc.M205902200. [DOI] [PubMed] [Google Scholar]
- 52.Xu F, Davis J, Miao J, Previti ML, Romanov G, Ziegler K, Van Nostrand WE. Protease nexin-2/amyloid beta-protein precursor limits cerebral thrombosis. Proc. Natl. Acad. Sci. USA. 2005;102:18135–18140. doi: 10.1073/pnas.0507798102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Komiyama Y, Murakami T, Egawa H, Okubo S, Yasunaga K, Murata K. Purification of factor XIa inhibitor from human platelets. Thromb. Res. 1992;66:397–408. doi: 10.1016/0049-3848(92)90289-m. [DOI] [PubMed] [Google Scholar]
- 54.Sinha S, Dovey HF, Seubert P, Ward PJ, Blacher RW, Blaber M, Bradshaw RA, Arici M, Mobley WC, Lieberburg I. The protease inhibitory properties of the Alzheimer's beta-amyloid precursor protein. J. Biol. Chem. 1990;265:8983–8985. [PubMed] [Google Scholar]
- 55.Dennis MS, Lazarus RA. Kunitz domain inhibitors of tissue factor-factor VIIa. II. Potent and specific inhibitors by competitive phage selection. J. Biol. Chem. 1994;269:22137–22144. [PubMed] [Google Scholar]
- 56.Schmaier AH, Dahl LD, Rozemuller AJ, Roos RA, Wagner SL, Chung R, Van Nostrand WE. Protease nexin-2/amyloid beta protein precursor. A tight-binding inhibitor of coagulation factor IXa. J. Clin. Invest. 1993;92:2540–2545. doi: 10.1172/JCI116863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmaier AH, Dahl LD, Hasan AA, Cines DB, Bauer KA, Van Nostrand WE. Factor IXa inhibition by protease nexin-2/amyloid beta-protein precursor on phospholipid vesicles and cell membranes. Biochemistry. 1995;34:1171–1178. doi: 10.1021/bi00004a010. [DOI] [PubMed] [Google Scholar]
- 58.Mahdi F, Van Nostrand WE, Schmaier AH. Protease nexin-2/amyloid beta-protein precursor inhibits factor Xa in the prothrombinase complex. J. Biol. Chem. 1995;270:23468–23474. doi: 10.1074/jbc.270.40.23468. [DOI] [PubMed] [Google Scholar]
- 59.Mahdi F, Rehemtulla A, Van Nostrand WE, Bajaj SP, Schmaier AH. Protease nexin-2/Amyloid beta-protein precursor regulates factor VIIa and the factor VIIa-tissue factor complex. Thromb. Res. 2000;99:267–276. doi: 10.1016/s0049-3848(00)00245-0. [DOI] [PubMed] [Google Scholar]
- 60.Castro MJ, Anderson S. Alanine point-mutations in the reactive region of bovine pancreatic trypsin inhibitor: effects on the kinetics and thermodynamics of binding to beta-trypsin and alpha-chymotrypsin. Biochemistry. 1996;35:11435–11446. doi: 10.1021/bi960515w. [DOI] [PubMed] [Google Scholar]
- 61.Bode W, Huber R. Natural protein proteinase inhibitors and their interaction with proteinases. Eur. J. Biochem. 1992;204:433–451. doi: 10.1111/j.1432-1033.1992.tb16654.x. [DOI] [PubMed] [Google Scholar]
- 62.Laskowski M., Jr, Kato I. Protein inhibitors of proteinases. Annu. Rev. Biochem. 1980;49:593–626. doi: 10.1146/annurev.bi.49.070180.003113. [DOI] [PubMed] [Google Scholar]
- 63.Szabo E, Bocskei Z, Naray-Szabo G, Graf L. The three-dimensional structure of Asp189Ser trypsin provides evidence for an inherent structural plasticity of the protease. Eur. J. Biochem. 1999;263:20–26. doi: 10.1046/j.1432-1327.1999.00452.x. [DOI] [PubMed] [Google Scholar]
- 64.Perona JJ, Hedstrom L, Wagner RL, Rutter WJ, Craik CS, Fletterick RJ. Exogenous acetate reconstitutes the enzymatic activity of trypsin Asp189Ser. Biochemistry. 1994;33:3252–3259. doi: 10.1021/bi00177a016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.