

Abstract
Aims
Cardiac hypertrophy by activation of the β-adrenergic receptor (βAR) is mediated more efficiently by the β1-AR than by the β2-AR. We investigated the signalling mechanism by which the β1-AR mediates cardiac hypertrophy.
Methods and results
Experiments were performed in cultured neonatal rat cardiomyocytes. Hypertrophy was determined by the protein/DNA content and atrial natriuretic factor transcription. Phosphorylation of Akt and Src was assessed by immunoblotting. Isoproterenol (ISO, 10 µM), a non-selective β-AR agonist, caused selective downregulation of the β1-AR (control β1 vs. β2: 35 vs. 65%, Bmax 78 ± 4 fmol/mg; 4 h, 10 vs. 90%, 61 ± 5 fmol/mg). Concanavalin A (Con A, 0.5 µg/mL), an inhibitor of endocytosis, prevented downregulation of β1-ARs by ISO treatment (4 h, 35 vs. 65%, 73 ± 8 fmol/mg), suggesting that β1-ARs selectively undergo endocytosis. Interference with β1-AR endocytosis by Con A, carboxyl terminal peptide of β-AR kinase-1, dominant negative (DN) β-arrestin-1, or DN dynamin inhibited β-adrenergic hypertrophy, suggesting that the endocytosis machinery plays a key role in mediating β-adrenergic hypertrophy. Activation of Akt by the β1-AR was blocked by inhibition of the endocytosis machinery, suggesting that endocytosis mediates activation of Akt. Akt plays a critical role in β-adrenergic hypertrophy, since DN Akt blocked ISO-induced hypertrophy. β-Adrenergic activation of Akt is mediated by Src, which associates with the endocytosis machinery and is necessary and sufficient to mediate β-adrenergic hypertrophy.
Conclusion
Activation of the endocytosis machinery is required for activation of Akt, which, in turn, critically mediates β1-AR-induced cardiac hypertrophy.
Keywords: Myocytes, Cell culture, Protein kinases, Protein phosphorylation
1. Introduction
Several patho-physiological conditions in the heart elevate sympathetic nervous activity, which causes cardiac hypertrophy through stimulating the β-adrenergic receptor (β-AR) signalling.1,2 Sustained stimulation of β-ARs leads to reduced cardiac function and development of congestive heart failure.3 Conversely, treatment of heart failure patients with β-AR antagonists prevents progression of cardiac dysfunction and reduces mortality.2
We have shown that hypertrophy of cultured cardiac myocytes produced by stimulation of endogenous β-ARs is more efficiently mediated by β1-ARs compared with β2-ARs despite the fact that both β1- and β2-ARs are expressed in this cell type.4 Efficient coupling of the β1-AR with cardiac hypertrophy over the β2-AR has also been suggested in transgenic mice studies, which utilize overexpression of the β-AR subtypes in the heart.5–7 Although long-term (>1 year) overexpression of β2-ARs in the mouse heart eventually causes cardiomyopathy, the mechanisms by which β1- and β2-ARs mediate cardiomyopathy seem distinct: inhibition of p38-MAPK reduces cardiac dysfunction in β2-AR overexpressed mice but not in β1-AR overexpressed ones.8 At present, however, the mechanisms by which β1- and β2-ARs differentially mediate cardiac hypertrophy are not clearly understood. This issue is important, since modulating the function of β-AR subtypes has been considered as a possible treatment for congestive heart failure.9
The myocardial β1- and β2-ARs couple to substantially different signalling mechanisms.10 Ligand binding to the β1-AR causes activation of the α subunit of the heterotrimeric G protein, Gαs, while the β2-AR couples to both Gαs and Gαi.10 β1- and β2-ARs differentially affect contractile properties and intracellular Ca2+ transients in cardiac myocytes.10 However, intracellular mediators of cardiac hypertrophy stimulated by the β1-AR have not been fully understood.
The β-ARs are phosphorylated and internalized upon ligand binding. Multiple molecules, including Gβγ, β-AR kinase (β-ARK), β-arrestin-1, Src, dynamin, and clathrin, are involved in the initial process of β-AR internalization via clathrin-coated pits.11 Although receptor internalization has been traditionally portrayed as a mechanism to terminate receptor signalling, it could mediate activation of signalling molecules, such as MAP kinases,11 which in turn play a key role in mediating cell growth responses by G protein-coupled receptors (GPCRs). Whether or not such endocytosis-dependent signalling mechanisms mediate cardiac hypertrophy remains to be elucidated.
In this study, we examined the signalling mechanism by which the β1-AR stimulates cardiac hypertrophy. Since our initial observation indicated that the β1-AR is internalized after stimulation with agonists in a more prolonged fashion than the β2-AR in cardiac myocytes, we examined the role of receptor endocytosis in mediating β-AR-induced cardiac hypertrophy.
2. Experimental procedures
2.1. Materials
PP1 was obtained from Biomol (Plymouth Meeting, PA, USA).
2.2. Primary cultures of neonatal rat ventricular myocytes
Primary cultures of cardiac ventricular myocytes from 1-day-old Crl:(WI)BR-Wistar rats (Charles River Laboratories, Wilmington, MA, USA) were prepared as described.4 The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. Myocytes were cultured in serum-free conditions for 48 h before experiments.
2.3. Receptor-binding assays
Receptor binding for intact cells was performed as described by Morisco et al.4 Myocytes were rinsed with a binding buffer containing Opti-MEM (Invitrogen, Carlsbad, CA, USA) supplemented with 0.1% bovine serum albumin, 0.1 mg/mL bacitracin, and incubated with (−)-[125I]iodocyanopindolol ([125I]CYP, 0.1 pM–3 nM, specific activity 2200 Ci/mmol, Perkin Elmer, Waltham, MA, USA) in the assay (0.3 mL) buffer for 1 h at 37°C. Myocytes were washed with the binding buffer. To determine the cell surface ligand binding, cells were exposed to 50 mM glycine–150 mM NaCl, pH 3, for 10 min at 4°C. The cell surface-bound radioactivity was collected by aspiration of the acid wash solution and three rinses with ice-cold Opti-MEM. The cell-associated radioactivity was measured by harvesting cells, and both fractions were quantitated by gamma counting. To determine the total cellular ligand binding, myocytes were incubated with 1 mL of 5% trichloroacetic acid (TCA) for 1 h at 4°C. Radioactivity released in TCA was counted. Specific binding of [125I]CYP was defined as total binding minus non-specific binding of [125I]CYP in the presence of 1 µM propranolol. Bmax and dissociation constant (Kd) were determined from saturation binding curves using non-linear-fitting curves (GraphPad, San Diego, CA, USA). The analysis of β-AR subtypes was performed as described previously.4
2.4. Transient transfection and reporter gene assays
Transfection was carried out using atrial natriuretic factor (ANF)-Luc (−638) (1 µg)12 or a plasmid containing the yeast GAL4-binding sites linked to firefly luciferase (pFR-Luc) together with a plasmid containing the activation domain of CREB fused with the GAL4 DNA-binding domain (GAL4-CREB). To correct for differences in the transfection efficiency, an SV40 driven β-galactosidase plasmid (SV40-βgal) was co-transfected.
2.5. Immunoblotting
Akt activity was determined using anti-phospho-Akt (Serine 473) antibody (Cell Signaling Technology, Danvers, MA, USA). For the analysis of Src, myocytes were lysed with lysis buffer containing 150 mM NaCl, 15 mM HEPES (pH 7.0), 0.1% SDS, 1% NP-40, 1% Na-deoxycholate, 2 mM EDTA, 50 mM NaF, 1 mM Na vanadate, 10 µg/mL aprotinin, 10 µg/mL trypsin inhibitor, and 10 µg/mL leupeptin. Src activity was determined using anti-phospho-Src (Tyrosine 416) antibody (Cell Signaling Technology).
2.6. Adenovirus transduction
Adenoviruses harbouring dominant negative (DN) Akt [Akt (T308A/S473A)], β-ARK carboxyl terminal peptide (β-ARK-CT), and DN dynamin [dynamin (K44A)] were provided by Drs G. Condorelli (University of California, San Diego, CA, USA), R.P. Xiao (National Institute of Aging, Baltimore, MD, USA), and J.E. Pessin (The University of Iowa, Iowa City, IA, USA), respectively.
2.7. Statistics
Data are given as mean ± SEM. Statistical analyses were performed using the analysis of variance. The post-test comparison was performed by the method of Tukey. Significance was accepted at the P < 0.05 level.
3. Results
3.1. The β1-AR is selectively internalized following isoproterenol stimulation
We initially hypothesized that differences in ligand binding-induced downregulation between β1- and β2-ARs may explain differences in the hypertrophic effects of β1- and β2-ARs.4 We examined downregulation of the β-ARs in response to isoproterenol (ISO), a non-selective agonist for the β-AR, using hydrophobic radioligand [125I]-CYP. Treatment of neonatal rat cardiac myocytes with ISO (10 µM) for 4 h caused downregulation of the β1-AR, but did not significantly reduce [125I]-CYP binding to β2-AR (control β1 vs. β2: 35 vs. 65%, Bmax 78 ± 4 fmol/mg; after 4 h, 10 vs. 90%, 61 ± 5 fmol/mg). Concanavalin A (Con A), an inhibitor of endocytosis,13 prevented ISO-induced downregulation of the β1-AR (at 4 h, 35 vs. 65%, 73 ± 8 fmol/mg). We also measured cell surface radioligand binding by the acid wash technique. The proportion of the β1-AR vs. β2-AR on the cell surface gradually decreased upon ISO stimulation and was 11 vs. 89% at 4 h. These results indicate that ISO causes selective endocytosis of the β1-AR over the β2-AR in cardiac myocytes. This result would seem to contradict the selective mediation of cardiac hypertrophy by the β1-AR4 if endocytosis only terminates the cell signalling through β-ARs. However, since the endocytosis machinery could actively mediate some signalling mechanisms,11 we hypothesized that the endocytosis machinery mediates ISO-induced cardiac hypertrophy.
3.2. Concanavalin A inhibits isoproterenol-induced increases in protein/DNA content and atrial natriuretic factor transcription
Since Con A prevented endocytosis of the β1-AR, we examined the effect of Con A on ISO-induced increases in the protein/DNA content (protein/DNA) and ANF transcription, major features of ISO-induced cardiac hypertrophy.4 Forty-eight-hour stimulation of myocytes with ISO significantly increased protein/DNA, consistent with our previous observation.4 Con A did not affect basal protein/DNA, but it significantly inhibited ISO-induced increases in protein/DNA (Figure 1A). In contrast, Con A did not significantly affect FBS-induced increases in protein/DNA (Figure 1A), suggesting that the effect of Con A on ISO-induced increases in protein/DNA is specific. Con A significantly inhibited ISO-induced increases in ANF transcription (Figure 1B). On the other hand, Con A affected neither ISO-induced increases in cAMP production (Figure 1C) nor ISO-induced increases in the GAL4-CREB activity (Figure 1D), suggesting that Con A does not globally affect β-AR-mediated signalling mechanisms. Taken together, Con A-sensitive mechanisms, such as internalization of the β1-AR, play an essential role in mediating ISO-induced cardiac hypertrophy.
Figure 1.

Concanavalin A negatively affects isoproterenol-induced cardiac hypertrophy. (A) Cultured neonatal rat cardiac myocytes were treated with isoproterenol (10 µM) or foetal bovine serum (FBS, 20%) for 48 h in the presence or absence of concanavalin A (Con A, 0.5 µg/mL) and protein/DNA was determined. Experiments were performed in triplicate six times. **P < 0.01, ***P < 0.001 vs. control. (B) Cardiac myocytes were transfected with ANF-Luc and SV40-β-galactosidase. Twenty-four hours after transfection, myocytes were stimulated with isoproterenol (10 µM) in the presence or absence of concanavalin A (0.5 µg/mL) for an additional 24 h. Experiments were performed in triplicate six times. **P < 0.001 vs. respective control. (C) Myocytes were pre-treated with IBMX for 30 min and then stimulated with isoproterenol (10 μM) for 15 min in the presence or absence of concanavalin A (0.5 µg/mL). The level of cAMP was determined by RIA. The values of cAMP were adjusted by the mean protein content. Experiments were performed in triplicate three to five times. **P < 0.05 vs. control. (D) Myocytes were transfected with pFR-Luc (1 µg/mL), GAL4-CREB, and the SV40-β-galactosidase. After 24 h, cells were stimulated with isoproterenol for 24 h with or without concanavalin A. Luciferase activities were normalized by those of β-galactosidase. Experiments were performed in triplicate three times. **P < 0.05 vs. control, #P < 0.01 vs. isoproterenol.
3.3. Gβγ mediates isoproterenol-induced increases in atrial natriuretic factor transcription
Upon ligand binding to the β-ARs, Gβγ subunits are dissociated from Gsα. Gβγ physically interacts with β-ARK-1, thereby allowing β-ARK-1 to phosphorylate the β-ARs. Thus, Gβγ is critically involved in the endocytosis of the β-ARs.11 Co-expression of Gβ2 and Gγ2 dose-dependently stimulated ANF-Luc reporter gene activity in myocytes (Figure 2A). Thus, increased expression of the Gβγ subunit is sufficient to stimulate ANF transcription.
Figure 2.

Gβγ plays an essential role in isoproterenol-induced atrial natriuretic factor transcription. (A) Neonatal rat cardiac myocytes were transfected with atrial natriuretic factor-luciferase (ANF-Luc), expression plasmids encoding Gβ2 plus Gγ2 and the SV40-β-galactosidase. Experiments were performed in duplicate five times. *P < 0.05, **P < 0.001 vs. control. (B) Cardiac myocytes were transfected with ANF-Luc, an expression plasmid encoding β-ARK-CT, and the SV40-β-galactosidase. Some myocytes were stimulated with isoproterenol (10 µM) for 24 h. Experiments were performed in duplicate five times. **P < 0.001 vs. control. (C) Myocytes were transduced with control adenovirus or adenovirus harbouring β-ARK-CT. Myocytes were stimulated with isoproterenol (10 µM) for 48 h. Protein/DNA in control virus-transduced myocytes without isoproterenol stimulation was expressed as 1. Experiments were performed in triplicate three times. *P < 0.01 vs. control virus-transduced myocytes without isoproterenol stimulation.
β-ARK-CT inhibits the cell signalling mediated by Gβγ.14 Expression of β-ARK-CT significantly inhibited ISO-induced activation of ANF transcription (Figure 2B). Adenovirus-mediated transduction of β-ARK-CT, but not β-galactosidase, significantly inhibited ISO-induced increases in protein/DNA (Figure 2C). β-ARK-CT inhibited selective downregulation of β1-AR by ISO (β1 vs. β2 in the presence of β-ARK-CT at 4 h, 31 vs. 69%) in cardiac myocytes. These results suggest that Gβγ, another important component of the endocytosis machinery of the β-AR, is required for ISO-induced ANF transcription. The isoform-specific role of Gβ and Gγ in mediating ISO-induced ANF transcription remains to be elucidated using specific knock-down of Gβ and Gγ isoforms.
3.4. Dominant negative β-arrestin-1 and dominant negative dynamin block isoproterenol-induced cardiac hypertrophy
We further examined whether the endocytosis machinery plays a key role in mediating ISO-induced cardiac hypertrophy. DN β-arrestin-1 [β-arrestin-1 (319–418)] binds to clathrin cages but fails to interact with β-ARs, thereby preventing endocytosis of the β-ARs.15 Transfection of DN β-arrestin-1 inhibited ISO-induced increases in ANF transcription (Figure 3A). DN dynamin inhibits clathrin-coated vesicle-mediated endocytosis.16 Transduction of adenovirus vector harbouring DN dynamin significantly inhibited ISO-induced increases in protein/DNA (Figure 3B) and ANF transcription (Figure 3C). In contrast, control adenovirus did not affect ISO-induced increases in protein/DNA. These results are consistent with the notion that the endocytosis machinery plays an important role in mediating β-adrenergic cardiac hypertrophy.
Figure 3.

Dominant negative β-arrestin-1 and dominant negative dynamin inhibit isoproterenol-induced cardiac hypertrophy. (A) Cardiac myocytes were transfected with ANF-Luc and SV40-β-galactosidase, together with expression vector encoding dominant negative β-arrestin-1. Twenty-four hours after transfection, myocytes were stimulated with isoproterenol (10 µM) for an additional 24 h. Luciferase activities normalized by β-galactosidase activities were expressed as relative to unstimulated samples transfected with 0.5 µg of pcDNA3.1 (Cont). Experiments were performed in duplicate six times. **P < 0.001 vs. control. (B) Myocytes were transduced with either control virus or adenovirus harbouring dominant negative dynamin. Myocytes were then stimulated with isoproterenol (10 µM) for 48 h. Protein/DNA in control virus-transduced myocytes without isoproterenol stimulation was expressed as 1. **P < 0.01 vs. control virus-transduced myocytes without isoproterenol stimulation. Experiments were performed in duplicate six times. (C) Cardiac myocytes were transfected with ANF-Luc and SV40-β-galactosidase. Twenty-four hours after transfection, myocytes were stimulated with isoproterenol (10 µM). Cardiac myocytes were also transduced with either control virus or adenovirus harbouring dominant negative dynamin. Myocytes were then stimulated with isoproterenol (10 µM) for 24 h. Luciferase activities were normalized by those of β-galactosidase. Experiments were performed in duplicate six times. *P < 0.05, **P < 0.01 vs. control.
3.5. Isoproterenol-induced activation of Akt is mediated by β1-ARs and receptor endocytosis-dependent mechanisms
We next examined which endocytosis-dependent signalling mechanism mediates β-adrenergic cardiac hypertrophy. We have shown previously that members of the MAP kinase family do not mediate ISO-induced increases in ANF transcription, whereas Akt plays an important role in mediating β-AR-induced cardiac hypertrophy.12 ISO as low as 0.1 µM induced Akt phosphorylation in cardiac myocytes (Supplementary material online, Figure S1). We examined whether internalization of the β-AR is required for β-AR-induced activation of Akt. ISO-induced increases in Ser473 phosphorylation were inhibited by the β1-AR antagonist, betaxolol, but not by the β2-AR antagonist, ICI115,881 (Figure 4A) or the α1-AR antagonist, prazosin (Supplementary material online, Figure S1), suggesting that ISO-induced activation of Akt is mediated by the β1-ARs. Interestingly, inhibition of receptor endocytosis by Con A treatment blocked ISO-induced Ser473 phosphorylation of Akt (Figure 4B). Similarly, adenovirus harbouring β-ARK-CT inhibited ISO-induced phosphorylation of Akt (Figure 4C). These results suggest that the endocytosis machinery plays an important role in β-AR-induced activation of Akt.
Figure 4.
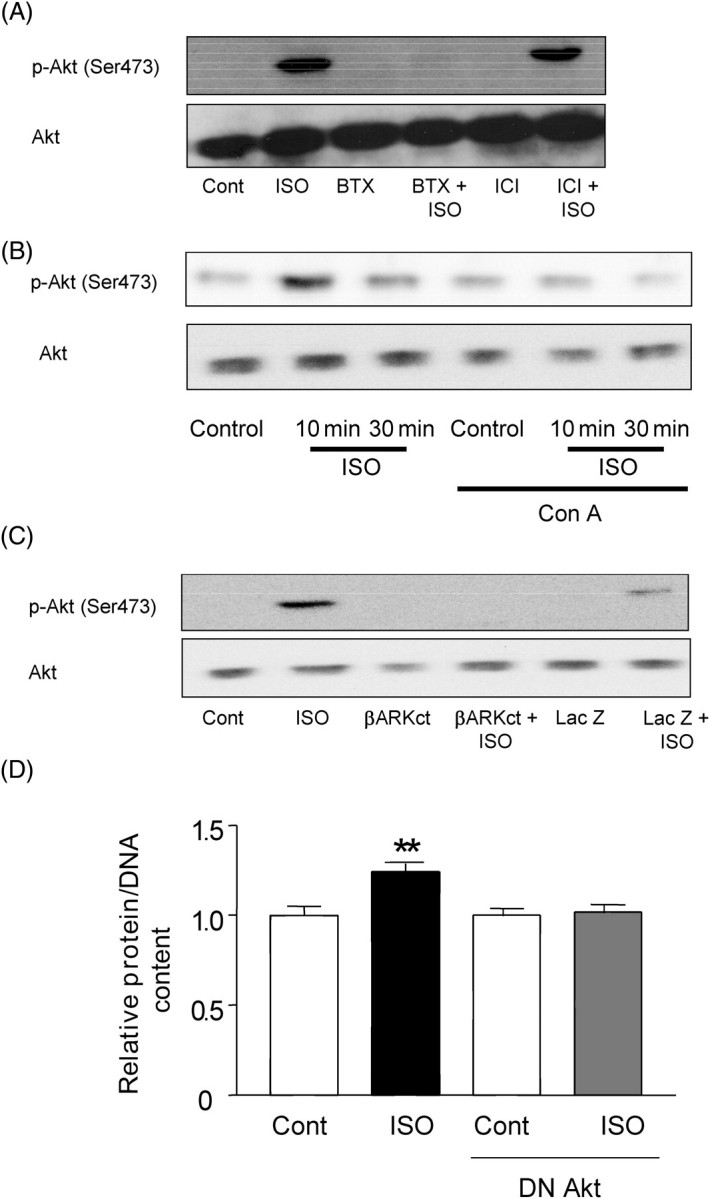
Isoproterenol-induced activation of Akt is mediated by β1-AR and concanavalin A-dependent mechanisms and plays an important role in mediating cardiac hypertrophy. (A) Cardiac myocytes were treated with isoproterenol (10 µM) for 30 min with or without betaxolol (10 µM) or ICI115,881 (10 µM), and activation of Akt was quantitated using an antibody against Serine 473-phosphorylated Akt. Lanes 3 and 4, betaxolol treated. Lanes 5 and 6, ICI115,881 treated. Lanes 2, 4, and 6, isoproterenol treated. (B) Cardiac myocytes were treated with isoproterenol (10 µM) for 10 or 30 min in the presence or absence of concanavalin A (Con A, 0.5 µg/mL), and the activated form of Akt was quantitated using an antibody against Serine 473-phosphorylated Akt. The results shown are representative of three experiments. (C) Myocytes were transduced with either control adenovirus (Lac Z) or adenovirus harbouring β-ARK-CT. Myocytes were stimulated with isoproterenol (10 µM) for 30 min, and activation of Akt was quantitated using phopho-specific antibody. In (A)–(C), the results shown are representative of three experiments. (D) Cardiac myocytes were transduced with control adenovirus or adenovirus harbouring dominant negative Akt (DN Akt). Myocytes were stimulated with isoproterenol (10 µM) for 48 h. Protein/DNA in control virus-transduced myocytes without isoproterenol stimulation was expressed as 1. Experiments were performed in duplicate six times. **P < 0.01 vs. control.
In order to test whether Akt plays an essential role in ISO-induced cardiac hypertrophy, we examined the effect of adenovirus-mediated transduction of DN Akt on ISO-induced increases in protein/DNA in cardiac myocytes. DN Akt significantly suppressed ISO-induced increases in the protein/DNA (Figure 4D). Transduction of control adenovirus did not affect ISO-induced increases in protein/DNA. These results suggest that activation of the endocytosis machinery by the β-AR causes activation of Akt, which in turn mediates β-adrenergic cardiac hypertrophy.
3.6. Src plays an essential role in isoproterenol-induced Akt activation and cardiac hypertrophy
Recruitment of c-Src to the receptor is involved in the mechanism leading to receptor internalization and activation of downstream signalling molecules, such as MAP kinases, in response to β-AR stimulation.17 We therefore examined whether Src is activated by ISO in cardiac myocytes. ISO stimulation caused increases in the tyrosine kinase activity as well as Tyr 416 phosphorylation of Src (Figure 5A and Supplementary material online, Figure S1). Interestingly, ISO-induced activation of Akt was blocked in the presence of the Src-specific inhibitor PP1 (Figure 5B), suggesting that ISO-induced activation of Akt is mediated by Src.
Figure 5.

Src mediates isoproterenol-induced activation of Akt and cardiac hypertrophy. (A) Cardiac myocytes were stimulated with isoproterenol (10 µM), and activation of Src was determined by anti-Tyrosine 418 phospho-specific Src antibody. Levels of Src phosphorylation were expressed relative to unstimulated samples. Experiments were performed in triplicate three times. (B) Cardiac myocytes were stimulated with isoproterenol (10 µM) with or without PP1 (1 µM) for 10 or 30 min. Activated forms of Akt were quantitated using an antibody against phospho-Serine 473 Akt. The result shown is representative of three experiments. (C) and (D) Cardiac myocytes were treated with or without PP1 (10 nM) for 30 min and then stimulated with isoproterenol (10 µM) in the presence or absence of PP1 for an additional 48 h (C) or 1 h (D). (C) Protein/DNA in control myocytes without PP1 treatment or isoproterenol stimulation was expressed as 1. Experiments were performed in duplicate six times. (D) Re-organization of polymerized actin was detected by phalloidin staining. The result shown is representative of three experiments. (E) and (F) Cardiac myocytes were transfected with ANF-Luc and SV40-β-galactosidase, together with expression vector encoding dominant negative Src (E) or constitutively active Src (F). The total amount of plasmid was adjusted to 0.5 µg by adding pcDNA3.1. Twenty-four hours after transfection, some myocytes were stimulated with isoproterenol (10 µM) for 24 h. Luciferase activities normalized by β-galactosidase activities were expressed relative to unstimulated samples with 0.5 µg of pcDNA3.1 (Control). Experiments were performed in duplicate six to nine times.
To determine whether activation of Src is required for β-AR-induced cardiac hypertrophy, we examined the effect of inhibition of Src on ISO-induced cardiac hypertrophy. ISO-induced increases in the protein/DNA were significantly inhibited by PP1 (Figure 5C). PP1 also significantly inhibited ISO-induced increases in actin reorganization, as determined by FITC-phalloidin staining (Figure 5D). DN Src dose-dependently inhibited ISO-induced increases in ANF transcription (Figure 5E). These results suggest that Src plays an essential role in mediating β-AR-induced cardiac hypertrophy. Although constitutively active Src stimulated ANF transcription, it did not do so as strongly as ISO (Figure 5F). Additional signalling mechanisms may be required for full activation of ANF transcription by ISO.
4. Discussion
In the present investigation, we studied the signalling mechanism by which the β1-AR mediates cardiac hypertrophy in response to β-AR stimulation (Figure 6). Our results suggest that the β1-AR undergoes prolonged endocytosis (>4 h) after ISO stimulation and that activation of the endocytosis machinery, instead of turning off the signal through the β-AR, positively mediates β-AR-induced cardiac hypertrophy. Furthermore, the endocytosis machinery is required for Akt activation, which in turn mediates cardiac hypertrophy in response to β-AR stimulation. Internalization-evoked signalling of GPCRs is required for cytoprotection in ischaemic pre-conditioning18 and impairment of cardiac function following mechanical stress.19 However, the requirement of the endocytosis machinery in cardiac hypertrophy has not been previously demonstrated.
Figure 6.

A hypothesis regarding the role of the endocytosis machinery in β-adrenergic cardiac hypertrophy. Ligand binding to the β1-AR leads to activation of Src and the endocytosis machinery. Activation of the endocytosis machinery causes activation of Akt, which in turn mediates cardiac hypertrophy. It is also possible that endocytosis and hypertrophy are regulated in parallel by common upstream signalling mechanisms. A, betaxolol; B, β-ARK-CT; C, dominant negative β-arrestin-1; D, dominant negative Src and PP1; E, concanavalin A and dominant negative dynamin.
After ISO stimulation, the number of β1-ARs on the cell surface was decreased while that of β2-ARs was preserved. According to the traditional point of view, selective disappearance (endocytosis) of β1-ARs may not allow β1-ARs to cause sustained activation of signalling molecules for induction of cardiac hypertrophy. Our results indicated, however, that Con A treatment not only inhibited endocytosis of the β1-AR, but also blocked ISO-induced cardiac hypertrophy. Furthermore, β-ARK-CT, DN β-arrestin-1, DN dynamin, and the Src inhibitor, all of which prevent endocytosis of the β-AR,17,20,21 inhibited ISO-induced increases in ANF transcription and myocyte hypertrophy. These results are consistent with the notion that endocytosis of the β1-AR itself or activation of the endocytosis machinery is required for ISO-induced cardiac hypertrophy.
Internalization of the β-AR and/or accompanying clathrin-coated vesicle endocytosis plays a critical role in activation of downstream MAP kinases.11 Our results suggest that the endocytosis machinery is required for ISO-induced activation of Akt. The requirement of endocytosis for agonist-induced Akt activation has not been demonstrated in cardiac myocytes. We believe that Akt is the key signalling molecule which connects the endocytosis machinery and β-adrenergic hypertophy, because activation of Akt is necessary, whereas MAP kinases are not critically required, for β-adrenergic cardiac hypertrophy.12 Although activation of Akt could cause pathological hypertrophy,22 it may not mediate every aspect of hypertrophy by stimulation of the β1-AR. Thus, a possibility remains that other signalling molecules regulated by the endocytosis machinery may also participate in the pathogenesis of β1-AR-mediated cardiac hypertrophy. Whether the endocytosis machinery and subsequent activation of Akt are protective or detrimental for the heart in the long term remains to be elucidated.
In response to the stimulation of the β2-AR receptor, Src is recruited to the membrane through interaction with β-arrestin-1 and phosphorylates proteins involved in clathrin-mediated endocytosis, such as dynamin and clathrin heavy chain.17,23 We have shown that both activation of Src and receptor endocytosis are essential for β-adrenergic activation of Akt. That Src might directly regulate Akt seems unlikely because we could not obtain evidence that Src and Akt physically interact with one another (our unpublished observation). Alternatively, Src may regulate phosphatidylinositol 3′ kinase or Ca2+/calmodulin-dependent kinase, since both of these molecules are required for ISO-induced Akt activation in cardiac myocytes.12 Phosphatidylinositol 3′ kinase is recruited to the membrane in response to the stimulation of the β-AR in the heart.24 At present, we do not know yet whether Ca2+-dependent mechanisms are involved upstream or downstream of the endocytosis machinery.
Norepinephrine (NE), a physiological ligand for the β-AR, mediates hypertrophy also through α1-AR. Phenylephrine (PE), an agonist for the α1-AR, modestly activated Akt (Supplementary material online, Figure S2), and PE-induced activation of Akt was inhibited by Con A (Supplementary material online, Figure S3). Although the effect of PE may be partly mediated by the β-AR, these results suggest a possibility that the endocytosis machinery may be involved in activation of Akt by α1-ARs as well. Interestingly, activation of Src by PE is only modest. Thus, mechanisms mediating activation of the endocytosis machinery may not be identical between α1-ARs and β1-ARs (Supplementary material online, Figure S2). Stimulation of α1- or β-ARs induces hypertrophy in cultured adult cardiac myocytes.25 Whether or not the endocytosis-dependent signalling mechanism is critical for NE-induced hypertrophy in adult myocytes remains to be tested.
β1- and β2-ARs internalize with distinct time courses in fibroblasts.26 The time course and the extent of internalization of the β-AR subtypes substantially differ among various cell types. Differences in the expression level of intermediates required for endocytosis and distinct affinities of the β-AR subtypes for those intermediates may contribute to the distinct properties of receptor endocytosis among β-AR subtypes in various cell types.27 Although the involvement of β-ARKs and β-arrestin-1, a well-established mechanism for endocytosis of the β2-AR, has been suggested as a mechanism for endocytosis of the β1-AR,21,28 other mechanisms may also mediate internalization of the β1-AR. For example, endophilins specifically interact with the intracellular third loop of the β1-AR.29 Whether or not such a unique intermediate is responsible for the selective hypertrophic effects of the β1-AR is unknown. Alternatively, localization of β-ARs on the cell surface, and their association with caveolae, another important mechanism mediating endocytosis, may differ depending upon cell type.30 β2-ARs are predominantly localized to the caveolar space, whereas β1-ARs distribute between caveolae and other cell fractions in neonatal rat cardiac myocytes.31
Another interesting finding in this study was that overexpression of Gβγ stimulated ANF transcription and that expression of β-ARK-CT, a molecule which inhibits Gβγ-mediated signalling,14 significantly inhibited β-adrenergic cardiac hypertrophy. These results suggest that Gβγ plays an important role in β-adrenergic cardiac hypertrophy. Since β-ARK-1 is targeted to the β-ARs via its affinity for Gβγ,28 and because Gβγ binding is necessary for β-ARK-1 activation,32 Gβγ may play a critical role in mediating β-AR-induced cardiac hypertrophy through its effects on β-AR internalization. Alternatively, signalling molecules directly activated by a Gβγ-dependent mechanism, including phosphatidylinositol 3′-kinases,33 may also be involved in β-adrenergic cardiac hypertrophy. Overexpression of β-ARK-CT in transgenic mice failed to prevent pressure overload-induced cardiac hypertrophy,34 but did decrease hypertrophy caused by the myosin heavy chain missense mutation.28 Whether or not cardiac hypertrophy in vivo caused by agonists for GPCR is mediated by Gβγ-dependent mechanisms has not been elucidated.
In summary, our results indicate that β1-ARs, compared with β2-ARs, undergo prolonged endocytosis upon ligand binding in cardiac myocytes and that activation of the endocytosis machinery is required for activation of Akt, which in turn mediates cardiac hypertrophy (Figure 6). Many GPCRs mediate cardiac hypertrophy, which generally requires long-term stimulation with agonists, despite the fact that they undergo internalization. If endocytosis of other GPCRs mediates cell-signalling mechanisms leading to hypertrophy, this could be an important mechanism by which the GPCR is able to generate sustained signals for cardiac hypertrophy. Our results suggest that the clathrin-mediated endocytosis machinery may be an important target for the treatment of cardiac hypertrophy because inhibition of β-AR endocytosis may selectively uncouple cardiac hypertrophy while preserving coupling between β-AR and cardiac contractility.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by National Institutes of Health grants HL67724, HL69020, HL59139, AG023039, and AG028787 and by American Heart Association (AHA) grant 0340123N. C.M. was supported by a fellowship from AHA Pennsylvania/Delaware Affiliate.
Acknowledgement
We thank Daniela Zablocki for critical reading of the manuscript.
Conflict of interest: none declared.
References
- 1.Vatner SF, Vatner DE, Homcy CJ. Beta-adrenergic receptor signaling: an acute compensatory adjustment—inappropriate for the chronic stress of heart failure? Insights from Gsalpha overexpression and other genetically engineered animal models. Circ Res. 2000;86:502–506. doi: 10.1161/01.res.86.5.502. [DOI] [PubMed] [Google Scholar]
- 2.Port JD, Bristow MR. Altered beta-adrenergic receptor gene regulation and signaling in chronic heart failure. J Mol Cell Cardiol. 2001;33:887–905. doi: 10.1006/jmcc.2001.1358. [DOI] [PubMed] [Google Scholar]
- 3.Limbird LE, Vaughan DE. Augmenting beta receptors in the heart: short-term gains offset by long-term pains? Proc Natl Acad Sci USA. 1999;96:7125–7127. doi: 10.1073/pnas.96.13.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morisco C, Zebrowski DC, Vatner DE, Vatner SF, Sadoshima J. Beta-adrenergic cardiac hypertrophy is mediated primarily by the beta(1)-subtype in the rat heart. J Mol Cell Cardiol. 2001;33:561–573. doi: 10.1006/jmcc.2000.1332. [DOI] [PubMed] [Google Scholar]
- 5.Engelhardt S, Hein L, Wiesmann F, Lohse MJ. Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc Natl Acad Sci USA. 1999;96:7059–7064. doi: 10.1073/pnas.96.12.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milano CA, Allen LF, Rockman HA, Dolber PC, McMinn TR, Chien KR, et al. Enhanced myocardial function in transgenic mice overexpressing the beta 2-adrenergic receptor. Science. 1994;264:582–586. doi: 10.1126/science.8160017. [DOI] [PubMed] [Google Scholar]
- 7.Dorn GW, II, Tepe NM, Lorenz JN, Koch WJ, Liggett SB. Low- and high-level transgenic expression of beta2-adrenergic receptors differentially affect cardiac hypertrophy and function in Galphaq-overexpressing mice. Proc Natl Acad Sci USA. 1999;96:6400–6405. doi: 10.1073/pnas.96.11.6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peter PS, Brady JE, Yan L, Chen W, Engelhardt S, Wang Y, et al. Inhibition of p38 alpha MAPK rescues cardiomyopathy induced by overexpressed beta 2-adrenergic receptor, but not beta 1-adrenergic receptor. J Clin Invest. 2007;117:1335–1343. doi: 10.1172/JCI29576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bristow MR. Beta-adrenergic receptor blockade in chronic heart failure. Circulation. 2000;101:558–569. doi: 10.1161/01.cir.101.5.558. [DOI] [PubMed] [Google Scholar]
- 10.Xiao RP. Beta-adrenergic signaling in the heart: dual coupling of the beta2-adrenergic receptor to G(s) and G(i) proteins. Sci STKE. 2001;2001:RE15. doi: 10.1126/stke.2001.104.re15. [DOI] [PubMed] [Google Scholar]
- 11.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 12.Morisco C, Zebrowski D, Condorelli G, Tsichlis P, Vatner SF, Sadoshima J. The Akt-glycogen synthase kinase 3 beta pathway regulates transcription of atrial natriuretic factor induced by beta-adrenergic receptor stimulation in cardiac myocytes. J Biol Chem. 2000;275:14466–14475. doi: 10.1074/jbc.275.19.14466. [DOI] [PubMed] [Google Scholar]
- 13.Rocha-Singh KJ, Hines DK, Honbo NY, Karliner JS. Concanavalin A amplifies both beta-adrenergic and muscarinic cholinergic receptor-adenylate cyclase-linked pathways in cardiac myocytes. J Clin Invest. 1991;88:760–766. doi: 10.1172/JCI115374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koch WJ, Hawes BE, Inglese J, Luttrell LM, Lefkowitz RJ. Cellular expression of the carboxyl terminus of a G protein-coupled receptor kinase attenuates G beta gamma-mediated signaling. J Biol Chem. 1994;269:6193–6197. [PubMed] [Google Scholar]
- 15.Krupnick JG, Santini F, Gagnon AW, Keen JH, Benovic JL. Modulation of the arrestin–clathrin interaction in cells. Characterization of beta-arrestin dominant-negative mutants. J Biol Chem. 1997;272:32507–32512. doi: 10.1074/jbc.272.51.32507. [DOI] [PubMed] [Google Scholar]
- 16.Kao AW, Ceresa BP, Santeler SR, Pessin JE. Expression of a dominant interfering dynamin mutant in 3T3L1 adipocytes inhibits GLUT4 endocytosis without affecting insulin signaling. J Biol Chem. 1998;273:25450–25457. doi: 10.1074/jbc.273.39.25450. [DOI] [PubMed] [Google Scholar]
- 17.Miller WE, Maudsley S, Ahn S, Khan KD, Luttrell LM, Lefkowitz RJ. Beta-arrestin1 interacts with the catalytic domain of the tyrosine kinase c-SRC. Role of beta-arrestin1-dependent targeting of c-SRC in receptor endocytosis. J Biol Chem. 2000;275:11312–11319. doi: 10.1074/jbc.275.15.11312. [DOI] [PubMed] [Google Scholar]
- 18.Tong H, Rockman HA, Koch WJ, Steenbergen C, Murphy E. G protein-coupled receptor internalization signaling is required for cardioprotection in ischemic preconditioning. Circ Res. 2004;94:1133–1141. doi: 10.1161/01.RES.0000126048.32383.6B. [DOI] [PubMed] [Google Scholar]
- 19.Tachibana H, Naga Prasad SV, Lefkowitz RJ, Koch WJ, Rockman HA. Level of beta-adrenergic receptor kinase 1 inhibition determines degree of cardiac dysfunction after chronic pressure overload-induced heart failure. Circulation. 2005;111:591–597. doi: 10.1161/01.CIR.0000142291.70954.DF. [DOI] [PubMed] [Google Scholar]
- 20.Lin FT, Krueger KM, Kendall HE, Daaka Y, Fredericks ZL, Pitcher JA, et al. Clathrin-mediated endocytosis of the beta-adrenergic receptor is regulated by phosphorylation/dephosphorylation of beta-arrestin1. J Biol Chem. 1997;272:31051–31057. doi: 10.1074/jbc.272.49.31051. [DOI] [PubMed] [Google Scholar]
- 21.Koch WJ, Rockman HA, Samama P, Hamilton RA, Bond RA, Milano CA, et al. Cardiac function in mice overexpressing the beta-adrenergic receptor kinase or a beta ARK inhibitor. Science. 1995;268:1350–1353. doi: 10.1126/science.7761854. [DOI] [PubMed] [Google Scholar]
- 22.Shioi T, McMullen JR, Kang PM, Douglas PS, Obata T, Franke TF, et al. Akt/protein kinase B promotes organ growth in transgenic mice. Mol Cell Biol. 2002;22:2799–2809. doi: 10.1128/MCB.22.8.2799-2809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilde A, Beattie EC, Lem L, Riethof DA, Liu SH, Mobley WC, et al. EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell. 1999;96:677–687. doi: 10.1016/s0092-8674(00)80578-4. [DOI] [PubMed] [Google Scholar]
- 24.Naga Prasad SV, Barak LS, Rapacciuolo A, Caron MG, Rockman HA. Agonist-dependent recruitment of phosphoinositide 3-kinase to the membrane by beta-adrenergic receptor kinase 1. A role in receptor sequestration. J Biol Chem. 2001;276:18953–18959. doi: 10.1074/jbc.M102376200. [DOI] [PubMed] [Google Scholar]
- 25.Pinson A, Schluter KD, Zhou XJ, Schwartz P, Kessler-Icekson G, Piper HM. Alpha- and beta-adrenergic stimulation of protein synthesis in cultured adult ventricular cardiomyocytes. J Mol Cell Cardiol. 1993;25:477–490. doi: 10.1006/jmcc.1993.1053. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki T, Nguyen CT, Nantel F, Bonin H, Valiquette M, Frielle T, et al. Distinct regulation of beta 1- and beta 2-adrenergic receptors in Chinese hamster fibroblasts. Mol Pharmacol. 1992;41:542–548. [PubMed] [Google Scholar]
- 27.Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- 28.Freedman NJ, Liggett SB, Drachman DE, Pei G, Caron MG, Lefkowitz RJ. Phosphorylation and desensitization of the human beta 1-adrenergic receptor. Involvement of G protein-coupled receptor kinases and cAMP-dependent protein kinase. J Biol Chem. 1995;270:17953–17961. doi: 10.1074/jbc.270.30.17953. [DOI] [PubMed] [Google Scholar]
- 29.Tang Y, Hu LA, Miller WE, Ringstad N, Hall RA, Pitcher JA, et al. Identification of the endophilins (SH3p4/p8/p13) as novel binding partners for the beta1-adrenergic receptor. Proc Natl Acad Sci USA. 1999;96:12559–12564. doi: 10.1073/pnas.96.22.12559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ostrom RS, Gregorian C, Drenan RM, Xiang Y, Regan JW, Insel PA. Receptor number and caveolar co-localization determine receptor coupling efficiency to adenylyl cyclase. J Biol Chem. 2001;276:42063–42069. doi: 10.1074/jbc.M105348200. [DOI] [PubMed] [Google Scholar]
- 31.Rybin VO, Xu X, Lisanti MP, Steinberg SF. Differential targeting of beta-adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. A mechanism to functionally regulate the cAMP signaling pathway. J Biol Chem. 2000;275:41447–41457. doi: 10.1074/jbc.M006951200. [DOI] [PubMed] [Google Scholar]
- 32.Pitcher JA, Touhara K, Payne ES, Lefkowitz RJ. Pleckstrin homology domain-mediated membrane association and activation of the beta-adrenergic receptor kinase requires coordinate interaction with G beta gamma subunits and lipid. J Biol Chem. 1995;270:11707–11710. doi: 10.1074/jbc.270.20.11707. [DOI] [PubMed] [Google Scholar]
- 33.Stephens L, Smrcka A, Cooke FT, Jackson TR, Sternweis PC, Hawkins PT. A novel phosphoinositide 3 kinase activity in myeloid-derived cells is activated by G protein beta gamma subunits. Cell. 1994;77:83–93. doi: 10.1016/0092-8674(94)90237-2. [DOI] [PubMed] [Google Scholar]
- 34.Rockman HA, Chien KR, Choi DJ, Iaccarino G, Hunter JJ, Ross J, Jr, et al. Expression of a beta-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci USA. 1998;95:7000–7005. doi: 10.1073/pnas.95.12.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.