

Abstract
In striated muscle, the basic contractile unit is the sarcomere, which comprises myosin-rich thick filaments intercalated with thin filaments made of actin, tropomyosin and troponin. Troponin is required to regulate Ca2+-dependent contraction, and mutant forms of troponins are associated with muscle diseases. We have disrupted several genes simultaneously in zebrafish embryos and have followed the progression of muscle degeneration in the absence of troponin. Complete loss of troponin T activity leads to loss of sarcomere structure, in part owing to the destructive nature of deregulated actin–myosin activity. When troponin T and myosin activity are simultaneously disrupted, immature sarcomeres are rescued. However, tropomyosin fails to localise to sarcomeres, and intercalating thin filaments are missing from electron microscopic cross-sections, indicating that loss of troponin T affects thin filament composition. If troponin activity is only partially disrupted, myofibrils are formed but eventually disintegrate owing to deregulated actin–myosin activity. We conclude that the troponin complex has at least two distinct activities: regulation of actin–myosin activity and, independently, a role in the proper assembly of thin filaments. Our results also indicate that sarcomere assembly can occur in the absence of normal thin filaments.
Keywords: Muscle, Sarcomere, Troponin, Zebrafish
Introduction
Muscle cells possess an elaborate cytoskeletal structure, which is essential for the coordination of contraction and the generation of force. The basic machinery of contraction is organised into bundles of long myofibrils, which are each made of stacks of sarcomeres. Sarcomeres consist of thin filaments, attached to Z discs, and thick filaments, attached to M lines. Filamentous actin associates with tropomyosin, troponin and other proteins to form thin filaments, which intercalate with thick filaments that are predominantly made of myosin. The contraction of striated muscle is regulated by the troponin protein complex, which mediates the Ca2+-dependent interaction of actin and myosin within the sarcomere. The troponin complex possesses three subunits: troponin C, troponin I and troponin T. Troponin T anchors the complex to the thin filament by binding to tropomyosin. Following stimulation, calcium ions released from the sarcoplasmic reticulum bind to troponin C and this leads to conformational changes of the complex. Troponin I consequently relieves its inhibition, which allows the binding of myosin to actin. Simultaneously, troponin T and tropomyosin alter conformation, allowing myosin-binding sites on actin to be exposed (Alberts et al., 2007; Galinska-Rakoczy et al., 2008; Phillips et al., 1986; Zot and Potter, 1987).
In humans, loss of function of the slow-twitch-muscle-specific troponin T isoform TNNT1 causes a recessive form of Nemaline myopathy (OMIM 605355), a genetically heterogeneous disease resulting from mutations in thin filament genes and characterised by muscle weakness and rod-like inclusions in muscle fibres (Johnston et al., 2000). Dominant mutations in the fast-twitch skeletal troponin T isoform TNNT3 or the troponin I isoform TNNI2 are responsible for a disorder called distal arthrogryposis type 2B (OMIM 601680) (Sung et al., 2003), characterised by clenched fist, overlapping fingers, ulnar deviation and positional foot deformities. Mutations affecting the three cardiac subunits of the complex (TNNC1, TNNT2 and TNNI3) are also associated with a variety of cardiomyopathies (Kimura et al., 1997; Landstrom et al., 2008; Mogensen et al., 2003; Mogensen et al., 2004; Murphy et al., 2004; Peddy et al., 2006; Thierfelder et al., 1994).
In addition to the known role in the regulation of contraction, there is some evidence that troponin is required for formation and maintenance of sarcomeres. In zebrafish, mutations of the silent heart locus, which encodes cardiac troponin T2, lead to sarcomere loss and myocyte disarray, attributed to coordinate reduction of thin filament gene expression (Sehnert et al., 2002). A similar disruption of sarcomere assembly was observed in Tnnt2−/− mice, which die during embryogenesis due to failure of cardiac contraction (Ahmad et al., 2008; Nishii et al., 2008). Body wall muscle contraction and sarcomere organisation are disrupted in Caenorhabditis elegans mutants defective for genes encoding troponin T or troponin I (Burkeen et al., 2004; Myers et al., 1996). In Drosophila melanogaster, severe mutant alleles of troponin T lead to a failure of sarcomere assembly, whereas hypomorphic troponin mutant alleles initially develop normal muscle and then undergo muscle damage (Fyrberg et al., 1990).
We have used zebrafish to investigate the role of thin filament proteins in the process of sarcomere assembly. Zebrafish development is rapid, with muscle fully differentiated by 48 hours post-fertilisation (hpf) (Kimmel et al., 1995). Zebrafish muscle has a similar composition to human muscle, in comparison with invertebrates (Bassett et al., 2003). Moreover, the optical properties of the embryo make it highly amenable to light microscopy. Like in mouse and human, the troponin genes in zebrafish exist in several isoforms, differentially expressed in cardiac, slow-twitch or fast-twitch skeletal muscle (Fu et al., 2009; Hsiao et al., 2003). Additionally, troponin genes have been duplicated in the zebrafish genome relative to mammalian genomes. The opportunity to simultaneously knockdown expression of different genes using combinations of antisense morpholino oligonucleotides (MOs) in zebrafish allows us to overcome this genetic redundancy (Nasevicius and Ekker, 2000).
We disrupted expression of different members of the troponin T family. In fast-twitch muscle, redundant expression of three different troponin T genes (tnnt2c, tnnt3a and tnnt3b) guarantees normal assembly in single loss-of-function experiments, but the concomitant knockdown of all three genes leads to a block in normal myofibrillogenesis. We demonstrate that it is the loss of regulation of the actin–myosin activity that leads to failure of sarcomere assembly. When we block myosin function with the myosin inhibitor blebbistatin, striations are recovered but an additional effect of the loss of troponin T function on thin filament assembly is unmasked. Finally, we have identified a second class of phenotype that results when only single troponin genes are disrupted. Depletion of tnnt3a or tnnt3b by MOs, or loss of tnni4a.2 through mutation, allows assembly but leads to sarcomere disintegration, which is completely suppressed by inhibition of myosin activity.
Results
Depletion of troponin T activity in fast-twitch muscle disrupts sarcomere assembly
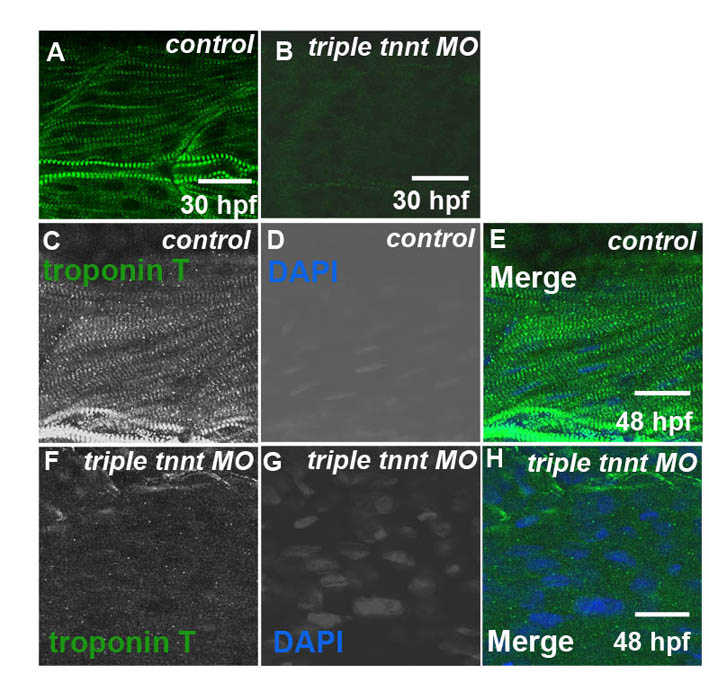
In the zebrafish trunk skeletal musculature, slow-twitch fibres and fast-twitch fibres are separated. Mononucleate slow-twitch fibres are superficially located, immediately under the skin, and are parallel to the anteroposterior axis, whereas multinucleated fast-twitch fibres are located deeper in the trunk, are more abundant and have an oblique orientation (Bassett et al., 2003; Devoto et al., 1996). Three troponin T genes, tnnt2c, tnnt3a and tnnt3b, are expressed in fast-twitch muscle during zebrafish development (Table 1; supplementary material Fig. S1). The tnnt2c gene is strongly expressed in myotomes from the beginning of somitogenesis and is downregulated from 36 hpf, whereas the tnnt3a and the tnnt3b genes are expressed from 18 hpf and 24 hpf, respectively (Hsiao et al., 2003; Thisse and Thisse, 2005). We knocked down expression of all three genes simultaneously with translation-blocking MOs. Embryos injected with triple tnnt MOs were shorter than embryos injected with a single MO and were occasionally bent, with abnormally shaped somites and pericardial oedema but with beating hearts, which indicated that the cardiac muscle in these embryos was functional. At 28 hpf, embryos were immotile, and myofibrils in fast-twitch muscle depleted of tnnt2c, tnnt3a and tnnt3b were markedly disorganised and very rarely possessed any striations (Fig. 1A,B), indicating that functional troponin T is required for sarcomere assembly in fast-twitch muscle. When analysed at 48 hpf, fast-twitch muscle in the triple MO-injected embryos showed a reduced level of filamentous actin, which was distributed in small foci. This was coupled with a loss of striation, as indicated by the anti-titin antibody T12, which recognises an epitope in proximity of the Z line (Fig. 1C,D). Phalloidin staining for actin, coupled with antibody staining for α-actinin, showed colocalisation of the two proteins in foci, although actin also appeared in aggregates without α-actinin (Fig. 2C; supplementary material Fig. S2C). Interestingly, other markers indicated that, although myofibrils were substantially disorganised, not all sarcomeric components were completely delocalised. We used a second anti-titin antibody T11, which recognises an epitope at the boundary between the I-band and A-band (Trombitas and Pollack, 1993), and detected some regions with short stretches of periodic T11 staining in the embryos injected with triple tnnt MOs (Fig. 1E,F). Similarly, an antibody directed against myomesin, a major component of M lines, showed that M lines also partially maintained regular spaced distribution in some regions (Fig. 1G,H). Tropomyosin failed to appear in striations but showed normal levels of expression in troponin-T-depleted muscles (Fig. 1I,J). Because the tropomyosin antibody that we had used in single staining did not work well in combination with phalloidin, we cloned tropomyosin 3 in frame with the red fluorescent protein mCherry and injected the mRNA encoding for this fusion protein in wild-type and triple tnnt MO-injected embryos. We then fixed the embryos and stained them with phalloidin. Our results show that tropomyosin and actin colocalised in the controls but did not overlap in the triple tnnt MO-injected embryos (supplementary material Fig. S3). Transmission electron microscope (TEM) analysis confirmed these results, showing that a large part of troponin-T-depleted muscle was filled with dispersed thick filaments (Fig. 1M). We also observed areas with disorganised and expanded Z lines (Fig. 1N), areas with thin-filament-free sarcomeres with no Z lines (Fig. 1O), and areas with accumulations of material, which, on the basis of light microscope analysis, we believe to be actin (Fig. 1P).
Table 1.
Troponin genes described in this study

Fig. 1.

Disruption of sarcomere assembly of fast-twitch muscle of embryos simultaneously depleted of tnnt2c, tnnt3a and tnnt3b. (A,B) Phalloidin staining shows forming and mature striated myofibrils in control embryos at 28 hpf (A) but completely disrupted myofibrils in triple tnnt MO-injected embryos at the same stage (B). MJ, myotendinous junctions; HM, horizontal myoseptum. (C,D) At 48 hpf, the titin epitope T12 (C′,D′; green) localises to normal Z lines, and actin (C″,D″; red) appears in striations in control embryos (C), whereas the two proteins are randomly dispersed in the cytoplasm in triple tnnt MO-injected embryos (D). DAPI stains the nuclei (C‴,D‴; blue). Single channels are shown in grey. (E–H) The titin epitope T11 localises to the I-band–A-band boundaries and myomesin to the M lines in control embryos (E,G). They are still seen in discrete structures, although these are scattered around in the cytoplasm, in triple tnnt MO-injected embryos (F,H). (I,J) Tropomyosin localisation to thin filaments observed in the control (I) is lost in the triple tnnt MO-injected embryos (J). Scale bars: 25 μm, insets are 2× greater magnifications of corresponding panels. (A,B) Lateral views of rostral somites of 28 hpf embryos. (C–J) Lateral views of a single myotome. Confocal images were obtained from planes deep in the trunk musculature. (K–P) TEM images of fast-twitch muscles in 48 hpf control (K,L) and in triple tnnt MO-injected embryos (M–P). White arrowheads indicate Z lines and black arrows indicate M lines. In M, clusters of thick filaments can be observed. Accumulations of Z line proteins can be seen in N; in O, thin filament-free sarcomeres can be observed. The white asterisk marks the area where the Z line should have been. Accumulations of actin can be observed in P (black asterisk). Scale bars: 500 nm.
Fig. 2.

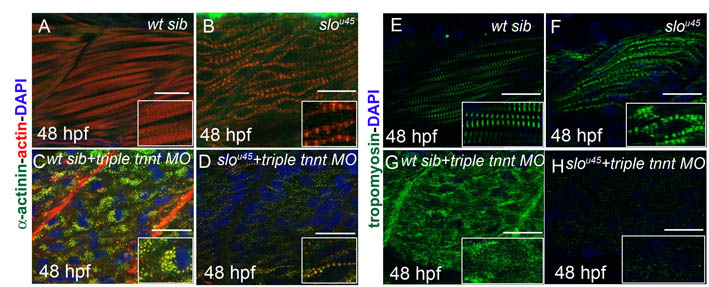
Recovery of striated myofibrils in triple tnnt-depleted embryos upon inhibition of myosin activity. (A–L) Phalloidin (red in A–D), α-actinin (green in A–D), fast myosin (white in E–H) and myomesin (white in I–L) staining of embryos incubated with DMSO reveals normal myofibrils in a control embryo (A,E,I) and the expected disruption in distribution of actin, Z lines, thick filaments and M lines in triple tnnt MO-injected embryos (C,G,K). When embryos are treated with blebbistatin, less compact myofibrils are observed in controls (B,F,J) and striated myofibrils can be seen in triple tnnt MO-injected embryos (D,H,L). (M–P) Tropomyosin does not recover its localisation to thin filaments in tnnt MO-injected embryos treated with blebbistatin (P). DAPI (blue) stains the nuclei. Scale bars: 25 μm, insets are 2× magnifications of corresponding panels.
To prove that the observed phenotype was specific, we cloned one of the three troponins, tnnt2c, in frame with mCherry. A DNA construct encoding the fusion protein downstream of the cytomegalovirus (CMV) promoter was injected into one-cell wild-type embryos. The exogenous DNA was expressed mosaically in a small subset of cells. At 28 and 48 hpf, isolated muscle cells producing the red fluorescent troponin T could be observed. In these cells, the protein localised correctly to thin filaments (supplementary material Fig. S4D–F). When this DNA construct was co-injected with the triple tnnt MOs, those cells producing the fusion protein recovered striations, whereas surrounding muscle cells not expressing the troponin-T–mCherry fusion protein showed disorganised distribution of actin and lacked striations (supplementary material Fig. S4G–I).
To determine the extent of troponin T downregulation, we used two different anti-troponin T antibodies (Ct3 and JLT-12) in whole-mount immunofluorescence assays. Both antibodies recognised troponin T on thin filaments in control embryos (supplementary material Fig. S5A,C and not shown) but did not react in triple tnnt MO-injected embryos (supplementary material Fig. S5B,F and not shown), indicating that troponin T expression was abolished by the tnnt MOs.
Because expression of thin filament genes has been reported to be reduced in troponin-T-depleted cardiac muscle (Sehnert et al., 2002), we stained triple tnnt MO-injected embryos for expression of tropomyosin 3, troponin C, troponin I2 and troponin T3b by in situ hybridisation. We did not detect any change in the level or pattern of expression of the first two, but found tnni2 to be slightly upregulated and tnnt3b, one of the genes targeted with the MO, to be downregulated (data not shown).
Inhibition of myosin function suppresses loss of striated organisation but does not restore proper thin filament assembly
To assess whether the effect of troponin T depletion on myofibrils was due to deregulation of actin–myosin activity, we analysed the phenotype of embryos injected with tnnt2c, tnnt3a and tnnt3b MOs while simultaneously inhibiting myosin function. We used blebbistatin, a myosin inhibitor (Straight et al., 2003) known to inhibit contraction in skeletal muscle and cardiomyocytes (Skwarek-Maruszewska et al., 2009). We added blebbistatin to control MO-injected and triple tnnt MO-injected embryos at 26 hpf, and analysed embryos at 48 hpf. At this stage, both control MO- and triple tnnt MO-injected embryos were immotile and displayed heart oedema. Myofibrils in control MO-injected embryos treated with blebbistatin had become less compact, although sarcomere markers had not lost their striated appearance (Fig. 2B,F,J,N). We observed a recovery in myofibril structure in blebbistatin-treated triple tnnt MO-injected embryos, with immature sarcomeres that were, however, regularly aligned along myofibrils that had recovered periodicity of Z lines, thick filaments and M lines (Fig. 2D,H,L; supplementary material Fig. S2) but that showed no recovery of tropomyosin localisation to sarcomeres (Fig. 2P).
In electron micrographs, we observed misaligned myofibrils and sarcomeres with less-defined Z and M lines in blebbistatin-treated control embryos (Fig. 3B). The loss of sarcomeric integrity observed in DMSO-treated triple tnnt MO-injected embryos (Fig. 3C) was partially restored in blebbistatin-treated triple tnnt MO-injected embryos, where regularly spaced, enlarged Z lines with intervening thick filaments could be observed (Fig. 3D). In cross-sections, control muscles displayed regular, hexagonal arrangement of thick filaments surrounded by thin filaments (Fig. 3E), whereas the triple tnnt MO-injected embryos showed bundles of thick filaments with no intercalating thin filament, regardless of the blebbistatin treatment (Fig. 3F,G).
Fig. 3.

Blebbistatin treatment does not rescue the loss of normal thin filaments in troponin-T-depleted sarcomeres. (A–G) Electron microscopy images of 48 hpf embryos, in tangential (A–D) and cross-sections (E–G). Control embryos (A,E) show normal sarcomeres with well-defined Z lines (white arrowheads) and M lines (black arrows) and regular hexagonal arrangement of thick filaments with intercalating thin filaments (circle in E). In blebbistatin-treated control embryos (B), Z lines are less sharp and M lines are not clearly defined. In tnnt-depleted muscles (C,F), sarcomeric components are disorganised (C) and bundles of thick filaments in cross-sections do not appear to be intercalating with thin filaments (F). Upon blebbistatin treatment, wider and regularly spaced Z lines can be observed in triple tnnt MO-injected embryos (D) and virtually no thin filaments are visible inside thick filaments bundles (circle in G). Scale bars: 1 μm (A–D); 150 nm (E–G).
To examine the role of unregulated actin–myosin activity, we independently disrupted myosin assembly using sloth mutants. In sloth mutants, the chaperone protein Hsp90a.1 is defective, preventing efficient myosin folding and thick filament assembly (Granato et al., 1996; Hawkins et al., 2008). We injected the triple tnnt MOs into slou45 embryos. Wild-type sibling embryos injected with triple tnnt MOs displayed the expected phenotype with disrupted sarcomeres (supplementary material Fig. S6C). By contrast, slou45 mutant embryos had wavy myofibrils with I–Z–I brushes (supplementary material Fig. S6B). Injection of triple tnnt MOs in slou45 embryos did not lead to the formation of actin aggregates, and myofibrils recovered periodic distribution of actin and α-actinin (supplementary material Fig. S6D), confirming that rudimentary sarcomeres can form in the absence of myosin activity. In these embryos, however, tropomyosin did not recover regular distribution and appeared to be less abundant and unlocalised throughout the cytoplasm (supplementary material Fig. S6H).
Partial loss of troponin activity in fast-twitch muscle leads to degradation of myofibrils
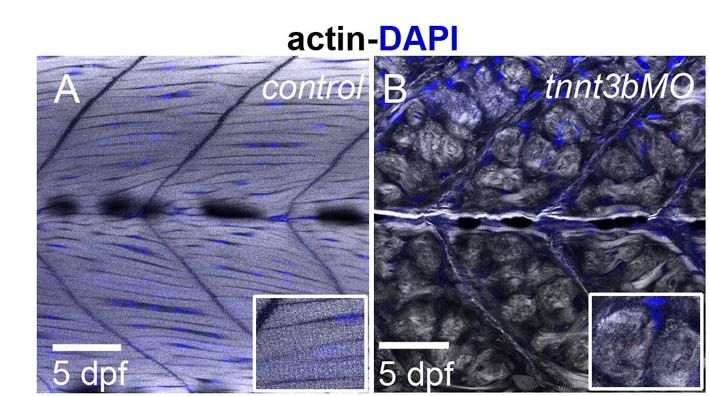
Single injection of tnnt3a or tnnt3b MOs impaired motility of embryos at 3 days post-fertilisation (dpf) and did not cause other major disruptions of embryonic development (Fig. 4A–C). Striated muscles possess a property known as form birefringence, which arises as a result of the parallel packing of long fibres, giving them the ability to polarise light. Larvae at 3 dpf showed complete (tnnt3a) or partial (tnnt3b) loss of muscle birefringence (Fig. 4D–F), indicating loss of myofibrillar organisation. Phalloidin staining showed a distinctive distribution of actin in round aggregates in tnnt3a MO-injected embryos (Fig. 4H). In tnnt3b MO-injected embryos, we found partial disruption, with striations preserved in the central part of myotubes but lost at the myotendinous junctions (Fig. 4I), indicating that at this stage tnnt3a can partially compensate for tnnt3b and that myotube ends are the first areas to be affected by a diminished availability of troponin T. At 5 dpf, however, myofibril disintegration progressed in tnnt3b MO-injected embryos and diffuse aggregates of actin could also be observed in the fast-twitch muscle of these embryos (supplementary material Fig. S7). At the ultrastructural level, we observed sarcomeres with several defects, including wider and wavier Z lines, and areas with disintegrated sarcomeres, dispersed thick filaments and accumulations of Z line proteins (Fig. 4J–L).
Fig. 4.

Myofibrils degrade in fast-twitch muscles of tnnt3-depleted embryos. (A–C) 3 dpf control (A), tnnt3a MO-injected (B) and tnnt3b MO-injected (C) embryos. (D–F) When trunks of the same embryos are observed under polarised light, embryos injected with tnnt3a MO (E) and tnnt3b MO (F) show total and partial loss of birefringence, respectively. (G–I) Lateral views of rostral somites in 3 dpf embryos. Confocal images from a plane deep in the trunk musculature show that actin is highly organised in fast muscle of control embryos (G), but appears completely disorganised in tnnt3a MO-injected embryos (H) and partially disorganised in tnnt3b MO-injected embryos (I; the inset shows a magnification of the myotendinous junction where actin localised in vertical Z lines or in horizontal stretched, non-striated filaments gives rise to a characteristic chequered appearance). Phalloidin is white and nuclei are counterstained with DAPI (blue). Scale bars: 50 μm; 10 μm in insets. (J–L) TEM images of fast-twitch muscles in 40 hpf control (J), tnnt3a MO- (K) and tnnt3b MO-injected embryos (L) reveal similar loss of sarcomeric integrity in the MO-injected embryos, with stacks of thick filaments and accumulations of Z line proteins (white arrowheads). Scale bars: 1 μm. (M,N) Forming fast-twitch fibres appear striated in both in control (M) and tnnt3a MO-injected (N) embryos at 28 hpf. Fast myosin is white and nuclei are blue. Scale bars: 20 μm. (O,P) At 48 hpf, M lines stained by myomesin (green) are regularly intercalated with thin filaments (phalloidin, red) in control embryos (O), whereas they cluster in groups and are excluded from actin-rich areas in tnnt3a MO-injected embryos (P, inset shows clusters). Scale bars: 15 μm; 10 μm in inset. M–P are lateral views of a single myotome, nuclei are stained with DAPI (blue).
In fast-twitch muscle, striations first become visible at 28 hpf. Phalloidin and myosin staining at this stage showed that sarcomeres initially form normally in tnnt3 MO-injected embryos (Fig. 4N and not shown) and that double knockdown of tnnt3a and tnnt3b had the same phenotype as single tnnt3a disruption (data not shown). In tnnt3a MO-injected embryos, sarcomeric proteins later became abnormally localised and separated into distinct cellular domains. For example, in muscle co-stained for actin and myomesin, groups of discrete and evenly spaced M lines could be seen distinctly separated from the actin aggregates (Fig. 4P).
We obtained independent confirmation of the effect of partial disruption of troponin function by analysing the knockout of a troponin I isoform in the same complex. A nonsense allele of tnni2a.4, a fast-twitch troponin I gene, was isolated in collaboration with the Zebrafish Mutation Resource at the Sanger Institute (http://www.sanger.ac.uk/cgi-bin/Projects/D_rerio/mutres/mutation.pl?project_id=966&mutation_id=170). The mutation is predicted to truncate the 176 amino acid protein after the 66th amino acid (Q67X) (Fig. 5B), eliminating more than half of the protein, including the C-terminus, which is thought to interact with actin monomers and tropomyosin on the thin filament (Galinska-Rakoczy et al., 2008) to inhibit actin–myosin interactions.
Fig. 5.

Progressive loss of fast-twitch fibre myofibrillar integrity in tnni2sa0058 mutant embryos. (A,B) Sequence traces from a wild-type fish (A) and a tnni2a.4sa0058 carrier fish (B) show the nucleotide change (asterisk) that converts C to T, generating a stop codon. (C–F) In a 5-day-old wild-type sibling observed under polarised light (C,E, at two different light intensities) trunk muscles are birefringent, whereas in a mutant embryo they are not (D,F). (G,H) Lateral views of single myotomes. Confocal images were obtained from planes deep in the trunk musculature. At 48 hpf, tnni2a.4 mutant embryos show myofibrils that are losing their integrity, as highlighted by phalloidin staining for thin filaments (G″,H″; red) and α-actinin staining for Z lines (G′,H′; green). DAPI stains the nuclei (G‴,H‴; blue). Single channels are shown in grey; overlap of signals shows in yellow in the merged panels (G,H). Scale bars: 10 μm. (I,J) At 3 dpf, fast myosin (white) is completely delocalised in tnni2a.4 mutants (J) whereas wild-type siblings show normal fast muscle (I). Nuclei are stained in blue. Scale bars: 20 μm. Insets are 2× magnifications of corresponding panels.
Starting from 3 dpf, tnni2a.4sa0058 mutants were unable to swim properly and at 5 dpf failed to inflate the swim bladder. At this stage, trunk muscles showed no birefringence (Fig. 5C–F). As with tnnt3a MO-injected embryos, fast-twitch myofibrils showed a loss of striation and diffuse actin and α-actinin staining by 48 hpf (Fig. 5G,H). Myosin staining at 3 dpf also revealed severe disruption of thick filaments (Fig. 5I,J). When analysed at 28 hpf, mutants could not be distinguished from wild-type siblings, indicating that myofibrils had formed normally and therefore that the fast-twitch muscle disintegration was progressive.
The partial loss of troponin phenotype is suppressed by loss of myosin activity
We tested whether the phenotype is due to unregulated actin–myosin activity. In tnnt3 MO-injected embryos, myofibril degradation proceeds through a first stage in which actin is distributed in sharp vertical bands and stretched horizontal fibres in enlarged areas of the myotubes, to a second stage in which actin becomes localised to round aggregates (Fig. 4H,I; supplementary material Fig. S7B), again suggesting that deregulated actin–myosin activity might have a role in the genesis of this phenotype.
We added blebbistatin to control MO- and tnnt3a MO-injected embryos at 26 hpf and analysed embryos at 48 hpf. Untreated tnnt3a MO-injected embryos showed the expected disrupted actin localisation with no striations (Fig. 6C), whereas blebbistatin-treated embryos had wavy but clearly striated myofibrils (Fig. 6D). This indicated that, in the absence of actin–myosin contraction, the lack of troponin T function does not lead to sarcomere disintegration. Because the tnni2a.4 mutant phenotype is the same as the tnnt3a MO phenotype, we used blebbistatin to verify whether inhibition of actin–myosin contraction could suppress the disruption of sarcomeres in the tnni2a.4 mutants. We found that four out of 20 control DMSO-treated offspring from heterozygous parents displayed the expected disrupted actin phenotype at 48 hpf, whereas none of the 20 observed blebbistatin-treated embryos from the same clutch displayed obvious actin accumulations.
Fig. 6.

Suppression of the tnnt3a progressive myofibrillar disorganisation phenotype in the absence of functional thick filaments. (A–D) Incubation with the myosin inhibitor blebbistatin: phalloidin staining of embryos incubated with DMSO reveals normal myofibrils in a control embryo (A) and the expected disruption in actin distribution in a tnnt3a MO-injected embryo (C). When embryos are treated with blebbistatin, loose myofibrils are observed in a control embryo (B) and wavy but striated myofibrils can be seen in a tnnt3a MO-injected embryo (D). Scale bars: 25 μm, insets are 2× magnifications of corresponding panels. (E–H) Analysis in the sloth mutant: a 48 hpf wild-type sibling displays normally striated fast fibres (E). In a wild-type sibling injected with the tnnt3a MO (F), myofibrils undergo degradation and actin becomes diffuse. In a sloth mutant embryo (G), actin is distributed in thick filament-free sarcomere-like blocks along myofibrils. In a sloth mutant injected with tnnt3a MO (H), actin remains in blocks rather than dispersed in the cytoplasm. Scale bars: 10 μm, insets are 2× magnifications of corresponding panels. All images are lateral views of a myotome. Confocal images were obtained from planes deep in the trunk musculature. Phalloidin staining is white and DAPI for nuclei is blue.
We confirmed these results independently using sloth mutants. We injected the tnnt3a MO into slou45 embryos. Wild-type sibling embryos injected with tnnt3a MO displayed the expected phenotype with degrading myofibrils (Fig. 6F), whereas slou45 mutant embryos had wavy myofibrils with I–Z–I brushes (Fig. 6G). Injection of tnnt3a MO in slou45 embryos did not lead to the formation of actin aggregates, and myofibrils were indistinguishable from uninjected slou45 embryos of the same stage (Fig. 6H), confirming that normal interactions between myosin and actin are required for the Tnnt3 loss of function to affect sarcomere maintenance.
Disruption of tnnt2c abolishes sarcomere assembly in slow-twitch muscle
The tnnt2c gene is expressed in fast-twitch but also in slow-twitch muscle (http://zfin.org/cgi-bin/webdriver?MIval=aa-markerview.apg&OID=ZDB-GENE-030520-1). Embryos injected with tnnt2c MO alone developed normally and did not show any morphological abnormalities, but they were completely immotile at 24 hpf and lacked the normal twitching movements typical of this stage. We first observed spontaneous twitching at around 28 hpf, when fast-twitch muscle becomes functional. By 48 hpf, we found that embryos had recovered their ability to move, hatch and swim normally. Slow-twitch myofibrils of tnnt2c MO-injected embryos at 24 hpf were thin, the filamentous actin was completely dispersed or distributed in regularly spaced foci but not in properly formed sarcomeres (Fig. 7A,B), and myosin was found in blocks but also dispersed in the cytoplasm or in thin, loose myofibrils (Fig. 7C,D). Electron microscopic analysis of slow-twitch muscle ultrastructure confirmed the absence of normal sarcomeres at 24 hpf in tnnt2c MO-injected embryos and showed accumulation of Z line proteins and dispersed thick filaments (Fig. 7F, arrowheads and arrow, respectively). At 30 hpf, actin staining in tnnt2c MO-injected embryos showed sarcomeres that had grown laterally and were decorated with titin T12 (Fig. 7G,H), although myofibrils were still wavy. Fast-twitch muscle at this stage was normal. At 5 dpf, phalloidin staining showed a normal distribution of filamentous actin in the slow-twitch muscle of tnnt2c MO-injected embryos (Fig. 7J).
Fig. 7.

Sarcomere assembly is delayed in slow-twitch muscle depleted of tnnt2c. (A–D) Mature sarcomeres can be seen in 24 hpf control embryos (A,C), but not in stage matched tnnt2c MO-injected embryos (B,D) stained for filamentous actin (A,B) and slow myosin (C,D). Scale bars: 25 μm, insets are 2× magnifications of corresponding panels. (E,F) Dysmorphology at the ultrastructural level is revealed by TEM images of slow muscle in 24 hpf control (E) and tnnt2c MO-injected embryos (F). Accumulations of Z line proteins (black arrowheads) and dispersed thick filaments (black arrow) can be found in the MO-injected embryos (F). Scale bars: 500 nm. (G,H) 30 hpf control embryos stained with an anti-titin antibody (G′,H′; green, T12), and phalloidin (G″,H″; red), show normal sarcomeres (G), whereas tnnt2c MO-injected embryos do not (H). Nuclei are counterstained with DAPI (G‴,H‴; blue). Single channels are shown in grey. The asterisk in H marks clustering nuclei in a lateral line rosette. Scale bars: 30 μm, insets are 2× magnifications of corresponding panels. (I,J) Slow-twitch muscle appeared normal at 5 dpf in control (I) and tnnt2c MO-injected (J) embryos. Scale bars: 50 μm.
We found that the recovery of striations in myofibrils of tnnt2c MO-injected embryos after 30 hpf (supplementary material Fig. S8) is due to the expression of another slow-twitch-muscle-specific troponin T encoded by a second, slow-twitch-muscle-specific troponin T gene, tnnt2d (http://zfin.org/cgi-bin/webdriver?MIval=aa-markerview.apg&OID=ZDB-GENE-050626-97) (for nomenclature see Materials and Methods, Table 1 and supplementary material Fig. S1). We designed a translation-blocking MO for tnnt2d and found that disruption of this gene on its own did not lead to any gross morphological abnormalities or developmental defect (data not shown). Immunostaining for myosin, and phalloidin staining for actin at 48 hpf in tnnt2c MO-injected embryos showed that slow-twitch muscle had almost completely recovered striations along their length, although myofibrils were thinner than in controls (Fig. 8B,B′,F). Depletion of tnnt2d led to mild alterations of muscle cell morphology, with some small myofibrils detaching from the main bundles (Fig. 8C,C′,G; also observed at 5 dpf, Fig. 8J). When tnnt2c and tnnt2d were simultaneously disrupted, we found that slow-twitch muscle did not recover at 48 hpf (compare Fig. 8D,D′,H with 8B,B′,F), demonstrating that from 30 hpf tnnt2d is able to provide enough troponin T to the slow-twitch muscle to compensate for tnnt2c loss of function.
Fig. 8.

tnnt genes are required for sarcomere assembly in slow-twitch muscle. (A–D) Slow myosin staining (white) in 48 hpf control embryos (A,A′) and in embryos injected with tnnt2c MO (B,B′), tnnt2d MO (C,C′) or co-injected with tnnt2d and tnnt2c MOs (D,D′) shows that myofibrillar organisation is lost in the double-injected embryos. Nuclei are counterstained with DAPI (blue). A–D are lateral views of rostral somites and A′–D′ are higher magnification images showing muscle cells. In both cases, confocal images are from the most superficial layer of trunk muscles. Scale bars: 25 μm (A–D); 10 μm (A′–D′). (E–H) Phalloidin staining (white) in 48 hpf control embryos (E) and in embryos injected with tnnt2c MO (F), tnnt2d MO (G) or co-injected with tnnt2d and tnnt2c MOs (H) confirms loss of sarcomeres in double-injected embryos. Nuclei are counterstained with DAPI (blue). Scale bars: 10 μm. (I–J) Slow myosin staining (white) of control (I) and tnnt2d MO-injected (J) embryos shows fairly normal slow fibres at 5 dpf in the MO-injected embryo, with some myofibrils detached from the main bundles (white arrowheads). Scale bars: 30 μm, insets are 2× magnifications of corresponding panels.
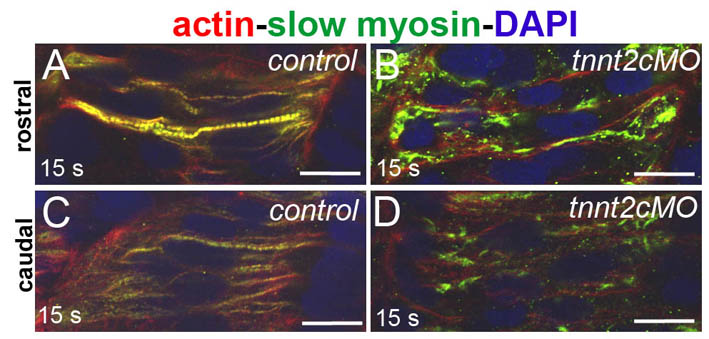
We examined the earlier steps of sarcomere formation in slow-twitch muscle at 14–16 somite stages. In control embryos, a few myofibrils could be detected in the anterior somites, whereas in caudal somites rare striations were just becoming detectable (supplementary material Fig. S9A,C). In tnnt2c MO-injected embryos we never observed similar structures, myosin appeared to aggregate in rods or foci, actin appeared more diffuse, and overlap of the signal was less frequent (supplementary material Fig. S9B,D), indicating that knockdown of tnnt2c impairs sarcomere assembly from the start.
Discussion
In this study, we have demonstrated that troponin is required for normal myofibrillogenesis. First, it is required to regulate actin–myosin interactions, and second it affects proper thin filament assembly. We also show that in the absence of normal thin filaments, assembly of rudimentary sarcomeres can occur.
Sarcomeres do not form in the absence of functional troponins
Despite the observation that sarcomeric substructures can form and align in the absence of main sarcomere components, such as thick filaments (Hawkins et al., 2008; Wohlgemuth et al., 2007), we did not find normal sarcomeres in muscles of embryos depleted of troponin T, instead aggregates of Z line proteins and dispersed thick filaments were observed from the very beginning of muscle differentiation. In silent heart mutants, defective for the cardiac isoform troponin T2a, sarcomere assembly is impaired and dispersed thick filaments were reported (Sehnert et al., 2002). In that study, it was proposed that a common regulation of thin filament genes would be altered in the absence of troponin T and that lack of assembly is due to downregulation of thin filament genes and the consequent dearth of thin filament proteins (Huang et al., 2009; Sehnert et al., 2002). Similar observations, however, have not been reported for mouse troponin mutants (Nishii et al., 2008). In D. melanogaster, downregulation of troponin I but not of other major thin filaments genes has been reported in flight muscles of a troponin T mutant (Nongthomba et al., 2007).
The consequences of tnnt2a disruption in the zebrafish heart have been further investigated with confocal microscopy (Huang et al., 2009). These authors reported loss of periodicity for thin and thick filaments and for Z and M lines, similar to that observed in skeletal muscle. They concluded that tnnt2a is required for the striations of thin filaments, which in turn affects the periodicity of Z bodies, M lines and thick filaments.
We show that the failure of assembly is principally due to the deregulated actin–myosin activity rather than to changes in the abundance of thin filament components or to lack of thin filament striations, because the regular distribution of major sarcomere components, lost in embryos depleted of the three troponin T proteins, is restored by blocking myosin activity.
In the absence of troponin, actin and myosin are not found together because their unregulated interaction is destructive. Instead, actin accumulates in aggregates and in abnormal Z lines, and myosin remains as dispersed thick filaments or appears in stacks of thick filaments in which myomesin can localise. Altered actin dynamics in this context probably lead to abnormal accumulation of Z line proteins in substantially enlarged Z lines (Fig. 9B).
Fig. 9.

Consequences of the loss of troponin function. (A) Schematic depiction of sarcomeres in the wild-type condition in which troponin (blue ovals), on thin filaments (thick blue lines) together with tropomyosin, regulates actin–myosin interactions. Thick filaments are shown in green. (B) In the absence of troponin, sarcomeres do not form, instead stacks of thick filaments, expanded Z lines (black ovals) and aggregates of filamentous actin (blue clouds) can be observed. (C) If in the absence of troponin, the actin–myosin activity is blocked, then sarcomeres form although filamentous actin (thin blue lines) does not associate with tropomyosin and Z lines are expanded.
Additionally, our data indicate that the loss of a functional troponin complex independently affects proper thin filament assembly (Fig. 9C). Although we found that the periodicity of filamentous actin is restored upon blocking actin–myosin activity and that actin filaments extend from expanded Z lines towards thick filaments, importantly we also found that filamentous actin is not associated with tropomyosin in the absence of troponin activity. Probably, actin filaments that are not decorated with troponin and tropomyosin cannot be detected in our electron microscopy preparations (Fig. 3). The association of tropomyosin with actin is stabilised by the binding of troponin T (Hitchcock-DeGregori and Heald, 1987), and it is likely that association of tropomyosin with actin becomes unstable in the troponin-T-depleted muscle. Our interpretation is that overall thin filament assembly is impaired in the absence of troponin; however, further analysis of the distribution of other proteins associated with thin filaments (e.g. nebulin) will be needed to further support this conclusion. Our in situ hybridisation experiments did not reveal major changes in tropomyosin expression in the triple tnnt MO-injected embryos and the troponin gene tnni2 was only slightly upregulated while tnnt3b was slightly downregulated. Therefore, we suggest that transcriptional changes are not major determinants of the phenotype arising from the loss of troponin activity.
Disruption of single fast-twitch muscle tnnt genes causes only partial loss of troponin activity because they are partially redundant. When we disrupt expression of a gene that is expressed early, such as tnnt2c in slow-twitch muscle, then sarcomere assembly is impaired. When we disrupt expression of genes that become functional later, after tissue differentiation is initiated, then the first steps of myofibril formation occur but deregulation of actin–myosin activity eventually leads to sarcomere disintegration, with sarcomeric components becoming abnormally redistributed to distinct cellular domains. Thus, in the absence of functional troponin T, different sarcomeric substructures disintegrate independently before the complete loss of sarcomere integrity rather than disintegrating simultaneously.
We were able to examine the function of another component of the fast-twitch-muscle-specific troponin complex by using tnni2a.4 mutants. These mutants showed myofibril degradation with the same progression and timing as observed for tnnt3a MO-injected embryos. Furthermore, similar to observations for tnnt3a, the myosin inhibitor blebbistatin was able to suppress the tnni2a.4 mutant phenotype. As in the case of tnnt3a and tnnt3b, gene redundancy might explain the lack of early phenotype in tnni2a.4 mutants. Six tnni2 genes are indeed reported in ZFIN, some of which are expressed in myotomes during somitogenesis.
Studies in D. melanogaster have shown that the hypercontraction muscle phenotype caused by mutations in the troponin T or troponin I genes is suppressed by myosin mutations that reduce actin–myosin force production (Nongthomba et al., 2003). Our data are consistent with the D. melanogaster data and validate them in a vertebrate animal. Furthermore, our ability to resolve the sarcomere composition with several markers at different time points and to partially suppress the defect with chemical inhibition of myosin provides further insights into the mechanism. We have shown that troponin is required for normal myofibrillogenesis and for proper thin filament assembly, and that partial loss of troponin activity allows sarcomere assembly but that unregulated actin–myosin activity leads progressively to loss of sarcomeric integrity.
Sarcomere assembly models
It has been shown that sarcomeric components can acquire periodicity in the absence of thick filaments (Hawkins et al., 2008; Wohlgemuth et al., 2007). Similarly, one interpretation of our finding that normal thin filaments are absent in the rudimentary sarcomeres restored upon blocking unregulated actin–myosin activity in troponin-T-depleted muscle could be that sarcomere assembly occurs independently of thin filament formation (Fig. 3D and Fig. 9C).
Recent work has suggested that myofibrillogenesis in zebrafish might proceed through stages that include the initial formation of pre-myofibrils containing Z bodies and non-muscle myosin, nascent myofibrils with non-aligned muscle myosin, and eventually mature myofibrils with aligned thick filaments, consistent with previous observations using cultured avian muscle cells (Sanger et al., 2005; Sanger et al., 2009). Alternative models imply the existence of actin stress-fibre-like structures as the first template for sarcomere assembly (Dlugosz et al., 1984) or the stitching of independently forming thick filaments and I–Z–I bodies (Holtzer et al., 1997).
We found that slow muscle myosin becomes localised to sarcomeres in very young somites (supplementary material Fig. S9) and that sarcomeres appear as early as the 15-somite stage, which is earlier than previously reported (Sanger et al., 2009). At this stage, in wild-type embryos, filamentous actin is mainly localised in unsegmented bundles running along the myocyte length, although some is localised to forming sarcomeres, where it overlaps with slow myosin. Our analysis of tnnt2c MO-injected embryos indicates that filamentous actin does not become periodic if the troponin complex is defective, and that muscle myosin remains in rodlets or aggregates. Despite a difference in the timing of muscle myosin appearance, our results indicate a transition from smaller immature sarcomeres to mature sarcomeres during recovery in tnnt2c MO-injected embryos (Fig. 7). This is consistent with the pre-myofibril model. Unregulated actin–myosin contraction that is due to troponin loss of function is able to interfere with the laying of pre-myofibrils because actin and Z lines are randomly distributed in troponin-T-depleted embryos but align in blebbistatin-treated embryos (Fig. 2). By contrast, thick filaments appear to be able to form as independent units, even in the absence of organised thin filaments. This is shown by the striated distribution of T11, which recognises a titin epitope proximal to the ends of thick filaments, and of myomesin, which marks the M lines (Fig. 2), and by the presence of thin filament-free, Z-line-free sarcomeres in electron microscopy micrographs (Figs 1 and 3). In the absence of thin filaments and α-actinin, titin might hold together the thick filament-rich substructures, possibly maintaining the attachment at the position of the Z lines thanks to the telethonin protein (Bertz et al., 2009). The T12 antibody that targets a portion of the protein at the Z line level appears randomly distributed, although this might be explained by the requirement of an intact Z line for recognition of the epitope. Our data do not disprove the pre-myofibril model but indicate that the role of non-muscle myosin and the dynamics of thick filament assembly might still require further definition.
Relevance to the human diseases
We found an excess number of zebrafish troponin genes as compared with mammalian orthologues and have proposed a new nomenclature for this group of genes, which is summarised in Table 1. Notably, in human and rodent skeletal slow muscle, the foetal troponin T function is provided by the cardiac gene TNNT2, which is then replaced by the slow-twitch-muscle-specific TNNT1 gene after birth (Jin, 1996; Jin et al., 2003). Consistently, we found that embryonic genes expressed in zebrafish skeletal slow-twitch muscle are more similar to the human TNNT2 than to the slow-twitch-muscle-specific TNNT1gene.
Mutations in the human skeletal troponin T subunit are linked to two different pathologies, recessive Nemaline myopathy and dominant distal arthrogryposis. In the case of Nemaline myopathy, a stop mutation in the slow-twitch-specific troponin T described in an Amish family generates a truncated protein that is rapidly degraded (Wang et al., 2005), suggesting that slow muscles in these patients are completely deficient in the troponin function. A hallmark of Nemaline myopathy is the accumulation of Z line proteins in aggregates called nemaline rods, which resemble the accumulations of Z line proteins that we observed in micrographs from tnnt2c MO- or triple tnnt MO-injected embryos, indicating that the troponin T loss of function leads to a similar myopathy in human and zebrafish.
Missense or stop mutations in the fast-twitch-specific troponin T and troponin I genes cause distal arthrogryposis, a syndrome characterised by limb contractures with muscular weakness but no muscle atrophy (Ochala, 2008). We showed that there are no intrinsic differences in the mechanisms of sarcomere assembly and loss between slow and fast fibres in the absence of normal troponin and, therefore, we believe that the differences in the human TNNT1 versus TNNT3 or TNNI2 phenotypes can be explained by the nature of the mutations. In the case of distal arthrogryposis, these are often dominant missense mutations that lead to a reduction in the contractile force rather than causing major changes in the distribution of myofibrillar components.
Materials and Methods
Orthology analysis
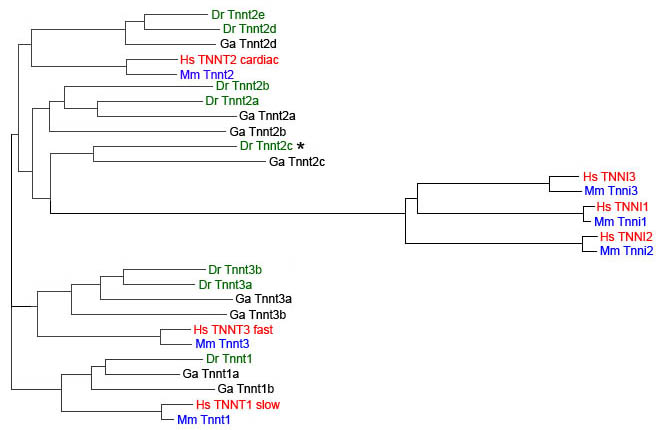
We have used TreeFam, a database of phylogenetic trees of animal genes (www.treefam.org) (Li et al., 2006; Ruan et al., 2008), to verify the number of troponin genes present in the zebrafish genome. We found that there are four genes encoding troponin C (TreeFam TF318191), 13 genes encoding troponin I (TreeFam TF313374) and eight genes encoding troponin T (TreeFam TF313321).
We aligned the sequences of the human, mouse, zebrafish and stickleback troponin T proteins using ClustalW2 (http://www.ebi.ac.uk/Tools/clustalw2/index.html). We found five zebrafish genes in the human TNNT2 branch, one is the cardiac-specific gene known as tnnt2, the silent heart locus, which we suggest should be renamed tnnt2a; therefore the gene most closely related to tnnt2a, LOC562296, should be called tnnt2b. Genes tnnt2a and tnnt2b both show obvious synteny to the human TNNT2 gene. There are three other genes encoding zebrafish troponin T2 isoforms as defined by orthology. These were known as tnnt1, zgc:114184 and si:ch211-136g2.1. The tnnt1 gene was described in literature as the slow-twitch-specific gene (Hsiao et al., 2003); however, it does not branch together with the human TNNT1 gene and it appears to be a teleost-specific gene. It shows conserved synteny with cryptochrome 1a, between zebrafish and stickleback, and we suggest it should be renamed tnnt2c. The other two are duplicated genes in zebrafish, with apparently only one copy in stickleback and we suggest that these should be renamed tnnt2d (zgc:114184) and tnnt2e (si:ch211-136g2.1), respectively. The zebrafish gene that clusters together with the human TNNT1 gene is presently called zgc:193865, which we suggest should be called tnnt1. We have obtained approval for the proposed new nomenclature and summarise the changes in Table 1.
Maintenance of zebrafish
Breeding zebrafish (Danio rerio) lines were maintained at 28°C on a cycle of 14 hours light and 10 hours dark. Fertilised eggs were obtained from natural spawning and grown in incubators at a temperature between 24°C and 28°C, depending on the stage required. Embryos were staged according to standard references (Kimmel et al., 1995). For imaging purposes, live embryos were manually dechorionated with #5 watchmaker's forceps, when necessary anaesthetised with 0.02% tricaine (ethyl 3-aminobenzoate methanesulphonic acid), and mounted for viewing in 3% methylcellulose in embryo medium (Westerfield, 2000). Pictures were taken using a Zeiss Axio Imager M1 microscope and a Zeiss AxioCam HRc digital camera.
Morpholino injections
The following morpholino-modified antisense oligonucleotides were ordered from Genetools: tnnt2c MO, 5′-CCACAAACTCTTCGGTGTCGCACAT-3′ (ATG MO); tnnt2d MO, 5′-GAAGTAAACACTCACGATTCTGTTG-3′ (ATG MO); tnnt3a MO, 5′-CTCAATGTCCTCTGTGTCTGACATG-3′ (ATG MO); tnnt3b MO, 5′-AACATCCTCAGTATCAGACATGATG-3′ (ATG MO); and standard control oligo, 5′-CCTCTTACCTCAGTTACAATTTATA-3′.
Oligonucleotides were diluted in morpholino buffer (5 mg/ml phenol red, 4 mM HEPES pH 7.2, 160 mM KCl) and injected in 1–2 cell stage embryos. We used 6 ng for tnnt2c MO, 6 ng for tnnt2d MO, 3 ng for tnnt3a MO, 6 ng for tnnt3b MO and equal amounts of the control MO. Needles were pulled from glass capillary tubes using a needle puller (Kopf Instruments) and injections were performed using a PV 820 Pneumatic Picoump micro-injector (World Precision Instruments).
Plasmids, mRNA and DNA injections
The coding sequence for the mCherry fluorescent protein (Shaner et al., 2004) was amplified with the following primers: FmCheClav2, 5′-ggcggccatcgatATGGTCTCTAAGGGCGAG-3′ and RmCheEcoRInostop, 5′-ggcggccGAATTCaTATTTGTACAGTTCATC-3′.
The second primer was a modified reverse primer that abolished the stop codon. This insert was cloned in pCS2 using ClaI and EcoRI to generate the pCS2mCherry vector. The tropomyosin 3 (tpma) and troponin T2c (tnnt2c) coding sequences were amplified by RT-PCR from 48 hpf embryo RNA with the following primers: FTpmaEcoRI, 5′-ggcggccGAATTCATGGATGCCATTAAGAAG-3′; RTpmaXbaI, 5′-ggcggccTCTAGATTAAATGGAGGTCATGTC-3′; FTnnt2cEcoRInew, 5′-ggcggccGAATTCATGTGCGACACCGAAGAG-3′ and RTnnt2cXhoInew, 5′-ggcggccCTCGAGTTACTTCCAGGACTTCCT-3′.
PCR fragments were digested with EcoRI and XbaI or EcoRI and XhoI and cloned downstream of the mCherry coding sequence in the pCS2mCherry vector, and plasmid were sequence verified. The pCS2mCherry–Tpma plasmid was linearised with NotI and used as a template to obtain mRNA in an in vitro transcription reaction with the RNA polymerase SP6 (Biolabs). Typically, 150 pg of mRNA were injected in one-cell embryos. For DNA injections, the coding sequence for the fusion protein mCherry–Tnnt2c was amplified from the pCS2mCherry–Tnnt2c vector with the following primers: FNheI–mCherry, 5′-ggcggccGCTAGCatgGTCTCTAAGGGCGAG-3′ and RBglII–Tnnt2c, 5′-ggcggccAGATCTTTACTTCCAGGACTTCCT-3′.
The PCR fragment was digested with NheI and BglII and cloned in the backbone of the pECFP–ER vector (Clontech) digested with the same restriction enzymes to remove the 846 bp CFP–ER sequence. The pEmCherry–Tnnt2c vector was linearised with StuI and purified with phenol/chloroform; 60 pg was injected into one-cell embryos. Both in the mRNA and DNA injections experiments, fluorescence was detected in live 24 and 48 hpf embryos using a 40× water immersion lens on a Leica SP5 confocal microscope. Fluorescent embryos were fixed in 4% paraformaldehyde and processed as described in the following subsection.
Immunolabelling and imaging
For phalloidin staining, embryos were fixed at the appropriate stage in 4% paraformaldehyde in PBS overnight at 4°C, washed with PBS containing 2% Triton X-100, incubated with Alexa Fluor 488 or Alexa Fluor 568 conjugated to phalloidin (Invitrogen), resuspended according to the manufacturer's instructions and diluted 1:100 in PBS, washed in PBS containing 0.1% Tween and mounted with Vectashield Mounting Media with DAPI (Vector Laboratories). For fluorescence immunohistochemistry, embryos were fixed at the appropriate stage overnight in methanol at −20°C or in 4% paraformaldehyde in PBS at 4°C. B4 (myomesin), F59 (slow myosin), EB165 (fast myosin), CH1 (tropomyosin) and Ct3 (troponin T) antibodies were obtained from the Developmental Studies Hybridoma Bank and used 1:5 (if obtained as supernatant) or 1:100 (if obtained as concentrate). Anti-titin T12, a gift from Yaniv Hinits (King's College, London, UK) was used 1:10. Anti α-actinin, anti-titin T11 and anti-troponin JLT-12 were purchased from Sigma (A7811, T9030 and T6277) and used 1:100. The secondary antibody used was Alexa-Fluor-488-labelled goat anti-mouse IgG (A-11001; Invitrogen). Embryos were washed several times in PBS containing 0.1% Triton X-100 and 0.2% BSA, incubated overnight in the same buffer with 5% of goat serum and primary antibodies, washed again, incubated with secondary antibody overnight, then washed and mounted with Vectashield Mounting Media with DAPI (Vector Laboratories). For double-staining, phalloidin was added together with the secondary antibody. Samples were imaged with the Leica SP5 confocal microscope using a 40× or a 63× oil immersion objective.
Electron microscopy
Embryos were immersed in freshly prepared primary fixative containing 2% paraformaldehyde with 2% glutaraldehyde in 0.1M sodium cacodylate buffer (pH 7.42) containing 0.1% magnesium chloride and 0.05% and calcium chloride at 20°C for 10 minutes before transferring to an ice bath for the remainder of 2 hours. Samples were rinsed three times for 10 minutes each in sodium cacodylate buffer with added chlorides on ice. Secondary fixation with 1% osmium tetroxide in sodium cacodylate buffer only was carried out at room temperature for 1 hour. All following steps were performed at room temperature. Embryos were rinsed three times in cacodylate buffer over 30 minutes and mordanted with 1% tannic acid for 30 minutes followed by a rinse with 1% sodium sulphate for 10 minutes. Samples were dehydrated through an ethanol series of 20, 30 (staining en bloc with 2% uranyl acetate at this stage), 50, 70, 90 and 95% for 20 minutes each then 100% for 3×20 minutes. Ethanol was exchanged for propylene oxide (PO) for 2×15 minutes followed by 1:1 PO to Epon resin mixture for at least 1 hour, and neat Epon (with a few drops of PO) overnight. Embryos were embedded in a flat moulded tray with fresh resin and cured in an oven at 65°C for 24 hours. We cut 40-nm sections on a Leica UCT ultramicrotome, stained the sections with uranyl acetate and lead citrate for contrast, and imaged on an FEI 120kV Spirit Biotwin using an F415 Tietz CCD camera.
Genotyping
We genotyped tnni2sa0058 embryos using the following nested primers: tnni2-4-1, 5′-GAGAACAAACATCTGACAATGG-3′; tnni2-4-2, 5′-GGTCAATTAAATGATTTGAAGC-3′; tnni2-4-3, 5′-TTCTTGAACTTGCCCTTCAG-3′; and tnni2-4-4, 5′-CAGCAGAGCCTGAAGCATAG-3′. Whole 5 dpf embryos were fixed in methanol and then dried on a heat block. Genomic DNA was extracted by incubating with proteinase K at 55°C overnight. Mutant slou45 embryos are easily distinguished from wild-type siblings because they are completely immotile.
Blebbistatin treatment
Blebbistatin was purchased from Sigma (B0560-1MG) and resuspended in DMSO. A final concentration of 6 μM was added to embryo medium at 28 hpf, and fish were collected at 48 hpf. DMSO only was added to untreated controls.
Supplementary Material
Acknowledgments
We are grateful to Elisabeth Busch-Nentwich, Gavin Wright, Steven Harvey, Bill Saxton and Simon Hughes for helpful discussion and critical reading of this manuscript, and to Laurel Lam Hung for her assistance. We also thank the Sanger Zebrafish Mutation Resource. This work was supported by the Wellcome Trust (grant numbers WT 077037/Z/05/Z, WT 077047/Z/05/Z). Deposited in PMC for release after 6 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/124/4/565/DC1
References
- Ahmad F., Banerjee S. K., Lage M. L., Huang X. N., Smith S. H., Saba S., Rager J., Conner D. A., Janczewski A. M., Tobita K., et al. (2008). The role of cardiac troponin T quantity and function in cardiac development and dilated cardiomyopathy. PLoS ONE 3, e2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts B., Johnson A., Lewis J., Raff M., Roberts K., Walter P. (eds) (2007). Molecular Biology of the Cell, 5th edn. New York: Garland Science; [Google Scholar]
- Bassett D. I., Bryson-Richardson R. J., Daggett D. F., Gautier P., Keenan D. G., Currie P. D. (2003). Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development 130, 5851-5860 [DOI] [PubMed] [Google Scholar]
- Bertz M., Wilmanns M., Rief M. (2009). The titin-telethonin complex is a directed, superstable molecular bond in the muscle Z-disk. Proc. Natl. Acad. Sci. USA 106, 13307-13310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkeen A. K., Maday S. L., Rybicka K. K., Sulcove J. A., Ward J., Huang M. M., Barstead R., Franzini-Armstrong C., Allen T. S. (2004). Disruption of Caenorhabditis elegans muscle structure and function caused by mutation of troponin I. Biophys. J. 86, 991-1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devoto S. H., Melancon E., Eisen J. S., Westerfield M. (1996). Identification of separate slow and fast muscle precursor cells in vivo, prior to somite formation. Development 122, 3371-3380 [DOI] [PubMed] [Google Scholar]
- Dlugosz A. A., Antin P. B., Nachmias V. T., Holtzer H. (1984). The relationship between stress fiber-like structures and nascent myofibrils in cultured cardiac myocytes. J. Cell Biol. 99, 2268-2278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu C. Y., Lee H. C., Tsai H. J. (2009). The molecular structures and expression patterns of zebrafish troponin I genes. Gene Expr. Patterns 9, 348-356 [DOI] [PubMed] [Google Scholar]
- Fyrberg E., Fyrberg C. C., Beall C., Saville D. L. (1990). Drosophila melanogaster troponin-T mutations engender three distinct syndromes of myofibrillar abnormalities. J. Mol. Biol. 216, 657-675 [DOI] [PubMed] [Google Scholar]
- Galinska-Rakoczy A., Engel P., Xu C., Jung H., Craig R., Tobacman L. S., Lehman W. (2008). Structural basis for the regulation of muscle contraction by troponin and tropomyosin. J. Mol. Biol. 379, 929-935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granato M., van Eeden F. J., Schach U., Trowe T., Brand M., Furutani-Seiki M., Haffter P., Hammerschmidt M., Heisenberg C. P., Jiang Y. J., et al. (1996). Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development 123, 399-413 [DOI] [PubMed] [Google Scholar]
- Hawkins T. A., Haramis A. P., Etard C., Prodromou C., Vaughan C. K., Ashworth R., Ray S., Behra M., Holder N., Talbot W. S., et al. (2008). The ATPase-dependent chaperoning activity of Hsp90a regulates thick filament formation and integration during skeletal muscle myofibrillogenesis. Development 135, 1147-1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchcock-DeGregori S. E., Heald R. W. (1987). Altered actin and troponin binding of amino-terminal variants of chicken striated muscle alpha-tropomyosin expressed in Escherichia coli. J. Biol. Chem. 262, 9730-9735 [PubMed] [Google Scholar]
- Holtzer H., Hijikata T., Lin Z. X., Zhang Z. Q., Holtzer S., Protasi F., Franzini-Armstrong C., Sweeney H. L. (1997). Independent assembly of 1.6 microns long bipolar MHC filaments and I-Z-I bodies. Cell Struct. Funct. 22, 83-93 [DOI] [PubMed] [Google Scholar]
- Hsiao C. D., Tsai W. Y., Horng L. S., Tsai H. J. (2003). Molecular structure and developmental expression of three muscle-type troponin T genes in zebrafish. Dev. Dyn. 227, 266-279 [DOI] [PubMed] [Google Scholar]
- Huang W., Zhang R., Xu X. (2009). Myofibrillogenesis in the developing zebrafish heart: A functional study of tnnt2. Dev. Biol. 331, 237-249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J. P. (1996). Alternative RNA splicing-generated cardiac troponin T isoform switching: a non-heart-restricted genetic programming synchronized in developing cardiac and skeletal muscles. Biochem. Biophys. Res. Comm. 225, 883-889 [DOI] [PubMed] [Google Scholar]
- Jin J. P., Brotto M. A., Hossain M. M., Huang Q. Q., Brotto L. S., Nosek T. M., Morton D. H., Crawford T. O. (2003). Truncation by Glu180 nonsense mutation results in complete loss of slow skeletal muscle troponin T in a lethal nemaline myopathy. J. Biol. Chem. 278, 26159-26165 [DOI] [PubMed] [Google Scholar]
- Johnston J. J., Kelley R. I., Crawford T. O., Morton D. H., Agarwala R., Koch T., Schaffer A. A., Francomano C. A., Biesecker L. G. (2000). A novel nemaline myopathy in the Amish caused by a mutation in troponin T1. Am. J. Hum. Genet. 67, 814-821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., Schilling T. F. (1995). Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253-310 [DOI] [PubMed] [Google Scholar]
- Kimura A., Harada H., Park J. E., Nishi H., Satoh M., Takahashi M., Hiroi S., Sasaoka T., Ohbuchi N., Nakamura T., et al. (1997). Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat. Genet. 16, 379-382 [DOI] [PubMed] [Google Scholar]
- Landstrom A. P., Parvatiyar M. S., Pinto J. R., Marquardt M. L., Bos J. M., Tester D. J., Ommen S. R., Potter J. D., Ackerman M. J. (2008). Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J. Mol. Cell. Cardiol. 45, 281-288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Coghlan A., Ruan J., Coin L. J., Heriche J. K., Osmotherly L., Li R., Liu T., Zhang Z., Bolund L., et al. (2006). TreeFam: a curated database of phylogenetic trees of animal gene families. Nucleic Acids Res. 34, D572-D580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen J., Kubo T., Duque M., Uribe W., Shaw A., Murphy R., Gimeno J. R., Elliott P., McKenna W. J. (2003). Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J. Clin. Invest. 111, 209-216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen J., Murphy R. T., Shaw T., Bahl A., Redwood C., Watkins H., Burke M., Elliott P. M., McKenna W. J. (2004). Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 44, 2033-2040 [DOI] [PubMed] [Google Scholar]
- Murphy R. T., Mogensen J., Shaw A., Kubo T., Hughes S., McKenna W. J. (2004). Novel mutation in cardiac troponin I in recessive idiopathic dilated cardiomyopathy. Lancet 363, 371-372 [DOI] [PubMed] [Google Scholar]
- Myers C. D., Goh P. Y., Allen T. S., Bucher E. A., Bogaert T. (1996). Developmental genetic analysis of troponin T mutations in striated and nonstriated muscle cells of Caenorhabditis elegans. J. Cell Biol. 132, 1061-1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasevicius A., Ekker S. C. (2000). Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 26, 216-220 [DOI] [PubMed] [Google Scholar]
- Nishii K., Morimoto S., Minakami R., Miyano Y., Hashizume K., Ohta M., Zhan D. Y., Lu Q. W., Shibata Y. (2008). Targeted disruption of the cardiac troponin T gene causes sarcomere disassembly and defects in heartbeat within the early mouse embryo. Dev. Biol. 322, 65-73 [DOI] [PubMed] [Google Scholar]
- Nongthomba U., Cummins M., Clark S., Vigoreaux J. O., Sparrow J. C. (2003). Suppression of muscle hypercontraction by mutations in the myosin heavy chain gene of Drosophila melanogaster. Genetics 164, 209-222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nongthomba U., Ansari M., Thimmaiya D., Stark M., Sparrow J. (2007). Aberrant splicing of an alternative exon in the Drosophila troponin-T gene affects flight muscle development. Genetics 177, 295-306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochala J. (2008). Thin filament proteins mutations associated with skeletal myopathies: defective regulation of muscle contraction. J. Mol. Med. 86, 1197-1204 [DOI] [PubMed] [Google Scholar]
- Peddy S. B., Vricella L. A., Crosson J. E., Oswald G. L., Cohn R. D., Cameron D. E., Valle D., Loeys B. L. (2006). Infantile restrictive cardiomyopathy resulting from a mutation in the cardiac troponin T gene. Pediatrics 117, 1830-1833 [DOI] [PubMed] [Google Scholar]
- Phillips G. N., Jr, Fillers J. P., Cohen C. (1986). Tropomyosin crystal structure and muscle regulation. J. Mol. Biol. 192, 111-131 [DOI] [PubMed] [Google Scholar]
- Ruan J., Li H., Chen Z., Coghlan A., Coin L. J., Guo Y., Heriche J. K., Hu Y., Kristiansen K., Li R., et al. (2008). TreeFam: 2008 Update. Nucleic Acids Res. 36, D735-D740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger J. W., Kang S., Siebrands C. C., Freeman N., Du A., Wang J., Stout A. L., Sanger J. M. (2005). How to build a myofibril. J. Muscle Res. Cell Motil. 26, 343-354 [DOI] [PubMed] [Google Scholar]
- Sanger J. W., Wang J., Holloway B., Du A., Sanger J. M. (2009). Myofibrillogenesis in skeletal muscle cells in zebrafish. Cell Motil. Cytoskeleton 66, 556-566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehnert A. J., Huq A., Weinstein B. M., Walker C., Fishman M., Stainier D. Y. (2002). Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat. Genet. 31, 106-110 [DOI] [PubMed] [Google Scholar]
- Shaner N. C., Campbell R. E., Steinbach P. A., Giepmans B. N., Palmer A. E., Tsien R. Y. (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567-1572 [DOI] [PubMed] [Google Scholar]
- Skwarek-Maruszewska A., Hotulainen P., Mattila P. K., Lappalainen P. (2009). Contractility-dependent actin dynamics in cardiomyocyte sarcomeres. J. Cell Sci. 122, 2119-2126 [DOI] [PubMed] [Google Scholar]
- Straight A. F., Cheung A., Limouze J., Chen I., Westwood N. J., Sellers J. R., Mitchison T. J. (2003). Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science 299, 1743-1747 [DOI] [PubMed] [Google Scholar]
- Sung S. S., Brassington A. M., Grannatt K., Rutherford A., Whitby F. G., Krakowiak P. A., Jorde L. B., Carey J. C., Bamshad M. (2003). Mutations in genes encoding fast-twitch contractile proteins cause distal arthrogryposis syndromes. Am. J. Hum. Genet. 72, 681-690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierfelder L., Watkins H., MacRae C., Lamas R., McKenna W., Vosberg H. P., Seidman J. G., Seidman C. E. (1994). Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell 77, 701-712 [DOI] [PubMed] [Google Scholar]
- Thisse C., Thisse B. (2005). High throughput expression analysis of ZF-models consortium clones. ZFIN Direct Data Submission (http://zfin.org).
- Trombitas K., Pollack G. H. (1993). Elastic properties of the titin filament in the Z-line region of vertebrate striated muscle. J. Muscle Res. Cell Motil. 14, 416-422 [DOI] [PubMed] [Google Scholar]
- Wang X., Huang Q. Q., Breckenridge M. T., Chen A., Crawford T. O., Morton D. H., Jin J. P. (2005). Cellular fate of truncated slow skeletal muscle troponin T produced by Glu180 nonsense mutation in Amish nemaline myopathy. J. Biol. Chem. 280, 13241-13249 [DOI] [PubMed] [Google Scholar]
- Westerfield M. (2000) The zebrafish book: A guide for the laboratory use of zebrafish (Danio rerio). Eugene, Orego: University of Oregon Press; [Google Scholar]
- Wohlgemuth S. L., Crawford B. D., Pilgrim D. B. (2007). The myosin co-chaperone UNC-45 is required for skeletal and cardiac muscle function in zebrafish. Dev. Biol. 303, 483-492 [DOI] [PubMed] [Google Scholar]
- Zot A. S., Potter J. D. (1987). Structural aspects of troponin-tropomyosin regulation of skeletal muscle contraction. Annu. Rev. Biophys. Biophys. Chem. 16, 535-559 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}