

Abstract
Myeloid-derived suppressor cells (MDSCs) are a well-defined population of cells that accumulate in the tissue of tumor-bearing animals and are known to inhibit immune responses. Within 4 days, bone marrow cells cultured in granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor resulted in the generation of CD11b+Ly6GloLy6C+ MDSCs, the majority of which are interleukin-4Rα (IL-4Rα+) and F4/80+. Such MDSCs potently inhibited in vitro allogeneic T-cell responses. Suppression was dependent on L-arginine depletion by arginase-1 activity. Exogenous IL-13 produced an MDSC subset (MDSC-IL-13) that was more potently suppressive and resulted in arginase-1 up-regulation. Suppression was reversed with an arginase inhibitor or on the addition of excess L-arginine to the culture. Although both MDSCs and MDSC-IL-13 inhibited graft-versus-host disease (GVHD) lethality, MDSC-IL-13 were more effective. MDSC-IL-13 migrated to sites of allopriming. GVHD inhibition was associated with limited donor T-cell proliferation, activation, and proinflammatory cytokine production. GVHD inhibition was reduced when arginase-1-deficient MDSC-IL-13 were used. MDSC-IL-13 did not reduce the graft-versus-leukemia effect of donor T cells. In vivo administration of a pegylated form of human arginase-1 (PEG-arg1) resulted in L-arginine depletion and significant GVHD reduction. MDSC-IL-13 and pegylated form of human arginase-1 represent novel strategies to prevent GVHD that can be clinically translated.
Introduction
The use of allogeneic hematopoietic stem cell transplantation is limited by graft-versus-host disease (GVHD). Although GVHD can sometimes be constrained with pharmacologic agents or rigorously T cell–depleted donor grafts, such treatments predispose patients to opportunistic infections and relapse malignancy.
Myeloid-derived suppressor cells (MDSCs) are a well-recognized population of cells known to accumulate in the lymph nodes (LNs), spleen, and liver of tumor-bearing mice and humans where they contribute to tumor evasion of cell-mediated immunity. In mice, MDSCs are CD11b+ (Mac-1) and Gr1+ (Ly6G/Ly6C)1 and are subcategorized based on the differential expression of Ly6G and/or Ly6C. Granulocytic MDSCs are defined as CD11b+Ly6G+Ly6Clow, whereas monocytic MDSCs are defined as CD11b+Ly6Glow/−Ly6Chi.2,3 Although these subsets can have various function and distribution depending on their environment, their capacity to induce T-cell hyporesponsiveness is generally considered equal.2,3 The suppressive capacity of MDSCs has been linked to the expression of specific surface molecules, including interleukin-4Rα (IL-4Rα,4 CD124) and macrophage colony-stimulating factor receptor (CD115).5
Among the mechanisms proposed for the immune-suppressive properties of MDSCs, L-arginine catabolism appears to be important in MDSC-induced T-cell dysfunction. MDSCs can express both arginase-1 and inducible nitric oxide synthase (iNOS), both of which metabolize L-arginine, leading to the production of the byproducts urea, L-ornithine, and citrulline and nitric oxide, respectively.6 Rodriguez et al have shown that L-arginine deprivation results in repressed expression of the T cell–signaling molecule, CD3ζ, as well as T-cell cycle arrest.7,8 Kerr et al have linked higher numbers of MDSCs in SHIP−/− with reduced GVHD lethality compared with wild-type (WT) recipients.9 The GVHD inhibitory effect of embryonic stem cell-derived MDSCs recently has been reported.10
In this study, we evaluated the efficacy of MDSCs generated from a clinically applicable source, the bone marrow (BM) of non–tumor-bearing donors, to inhibit GVHD lethality in a fully major histocompatibility complex (MHC)-mismatched model of bone marrow transplantation (BMT). MDSCs inhibited T-cell alloresponses and GVHD lethality through the depletion of L-arginine. The effects of limiting concentrations of L-arginine on donor T cells included a decrease in proliferation, a decrease in the expression of CD3ζ, and a decrease in interferon-γ (IFN-γ) production. Despite these alterations in T-cell function, allogeneic T cells retained their ability to eliminate lymphoma cells. Moreover, reduced GVHD lethality may be achieved by administration of a pegylated form of arginase-1 (PEG-arg1) instead of MDSC cellular therapy.
Methods
Mice
BALB/c (H2d) and C57BL/6 (H2b)(termed B6) mice were purchased from Charles River or the National Institutes of Health. GCN2 knockout (KO) mice were generated as described.11 B6 GFP transgenic12 mice were bred at the University of Minnesota. All mice were bred and housed in a specific pathogen-free facility in microisolator cages and used at 8 to 12 weeks of age in protocols approved by the Institutional Animal Care and Use Committee.
MDSC generation and culture
BM was harvested from B6 mice and plated at 2 × 105 cells/mL in Dulbecco modified Eagle medium plus 10% fetal calf serum, 50mM 2-mercaptoethanol, 10mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid buffer, 1mM sodium pyruvate, 100 U/mL penicillin, 100 mg/mL streptomycin, and amino acid supplements (1.5mM l-glutamine, L-arginine, and L-asparagine). Granulocyte colony-stimulating factor (G-CSF) was added at 100 ng/mL, and mouse granulocyte-macrophage colony-stimulating factor (GM-CSF) was added at 250 U/mL. Cultures were incubated at 37°C 10% CO2 for 4 days. On day 3, recombinant murine IL-13 (R&D Systems) was added at 80 ng/mL. On day 4 of culture, MDSCs were positively selected with CD11b by magnetic-activated cell sorting. Purified cells were routinely more than 95% CD11b+ Gr1+. B6 arginase-1 KO BM from Dr Rodriguez was differentiated into MDSC as above.
RT-PCR, quantitative PCR, and Western blots
RNA was isolated using RNAqueous4PCR kit. cDNA was synthesized using Superscript III reverse transcriptase. Reverse-transcribed polymerase chain reaction (RT-PCR) was performed using specific primers for arginase-1 and iNOS and amplified for 35 cycles. Quantitative RT-PCR TaqMan primers/probe specific for arginase-1, iNOS, and glyceraldehyde-3-phosphate dehydrogenase were obtained through Applied Biosystems. For Western blots, 20 μg of cell lysates was fractionated and transferred to polyvinylidene difluoride membranes. Antiarginase-1 monoclonal antibody (BD Biosciences) was incubated overnight at a 1:1000 dilution. Secondary antibody (antimouse horseradish peroxidase) was added for 1 hour at a 1:1000 dilution. α-Actin was used as a loading control.
MLR
LNs were harvested from B6 mice and T cells were purified by negative selection using phycoerythrin-conjugated anti-CD19/CD11c/NK1.1 and anti-phycoerythrin magnetic beads. Purity was routinely more than 95%. Spleens were harvested from BALB/c mice, T cell–depleted, and irradiated (1000 cGy). B6 T cells were mixed at a 1:1 with BALB/c splenic stimulators and plated in a 96-well plate (105 T cells/well). MDSCs were plated at 104/well (1:10). Cells were incubated in custom RPMI 1640 containing physiologic levels of L-arginine (150μM) supplemented with 10% fetal calf serum, 50mM 2-mercaptoethanol, 10mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid buffer, 1mM sodium pyruvate, and 100 U/mL penicillin-streptomycin. Cells were pulsed with 3H-thymidine (1 μCi/well) 16 to 18 hours and counted in the absence of scintillant on a β-plate reader. To inhibit arginase activity, Nw-hydroxy-nor-arginine (nor-NOHA) was resuspended at 5 mg/mL in dimethyl sulfoxide and diluted to reach a final concentration of 30 to 300μM. For NOS inhibition, L-NG-monomethyl-arginine-citrate (L-NMMA) was used at a concentration of 300μM. Percentage suppression was calculated by 100 − [(CPM of T cells + stimulators + MDSC)/(CPM of T cells + stimulators) × 100].
Flow cytometry
T cells were stained with 1μM carboxyfluorescein succinimidyl ester (CFSE). For CD3ζ, cells were permeabilized using digitonin (500 ng/mL) before adding anti-CD3ζ-phycoerythrin. Intracellular cytokine staining was performed after a 5-hour incubation of whole splenocytes with anti-CD3/CD28 beads and monensin at 37°C 5% CO2. Samples were analyzed on a FACSLSRII.
Arginase activity and L-arginine quantification
Arginase activity was determined using Quantichrom Arginase Assay Kit. L-Arg concentration in culture medium was measured by high-performance liquid chromatography using an ESA-CoulArray Model 540. Supernatants were deproteinized in methanol and derivatized with 0.2M O-phtaldialdehyde containing 7mM β-mercaptoethanol.
GVHD
BALB/c recipients were lethally irradiated using 600 cGy total body irradiation by x-ray on day −1 followed by infusion of 107 T cell–depleted B6 donor BM, 2 × 106 purified B6 CD25-depleted T cells (CD4/CD8), and 2 to 6 × 106 CD11b positively selected B6 MDSCs on day 0. Controls were given BM alone or BM + T cells. Mice were monitored daily for survival and weighed twice weekly. Migration was determined using MDSC from GFP-transgenic B6 mice. LNs and spleens were explanted and imaged using whole-body fluorescent imager. Images were taken with a Retiga Exi color camera and QCapture software (QImaging) mounted onto a Leica MZFLIII stereomicroscope using a GFP2 bandpass filter and a 1.0× transfer lens (Leica Microsystems). Zoom factors from 1.25× to 6.3× were used for imaging. Exposure times were optimized for each organ, and identical times and settings were used for all mice imaged on any given day.
Assessment of graft-versus-leukemia effect of donor T cells
A20-lymphoma cells were nucleoporated with a Sleeping Beauty transposon construct to permit the coexpression of dsred2 and Renilla luciferase (termed A20luc). On day 0, BMT cohorts were given 3 × 105 A20luc. A Xenogen IVIS imaging system was used for live animal imaging of anesthetized transplanted mice. Renilla luciferin substrate was injected intraperitoneally at 60mM.
Pegylation of human recombinant arginase-1
O-[2-(N-Succinimidyloxycarbonyl)-ethyl]-O′-methylpolyethylene-glycol (PEG) 5000mw (Sigma-Aldrich) was covalently attached to the human recombinant arginase-1, as described by Cheng et al.13 Pegylation was confirmed using glutaraldehyde-based stain solution, finding PEG-Arg-1 at 150 to 225 kDa and the PEG at 5 kDa. The specific activity of the native arginase and the PEG-arg-1 was 400 IU/mg protein. In experiments using PEG-arg-1, reagent was administered intraperitoneally to transplanted mice at 1 mg/mL twice weekly for 4 weeks.
Statistical analysis
Differences between groups in survival studies were determined using log-rank statistics. For all other data, a Student t test was used to analyze differences between groups. Results were considered significant if P less than or equal to .05.
Results
In vitro generated MDSC inhibit T-cell alloresponses via arginase-1 expression, which is up-regulated by IL-13
To generate MDSCs, BM from WT B6 mice was incubated with G-CSF, GM-CSF, or both. After 4 days, cells were harvested and the surface phenotype determined (Figure 1). Cells incubated with GM-CSF had a more granulocytic morphology, as indicated by the increased side scatter profile. The percentage of CD11b+Gr1+ cells increased from 29% in media to 35% with G-CSF, 34% with GM-CSF treated, 44% in the G-CSF + GM-CSF group, and 50% in G-CSF + GM-CSF + IL-13 group. IL-13 was chosen for further studies because the most consistent and largest increase in arginase activity was seen compared with other cytokines reported to induce arginase, such as IL-4 and prostaglandin E2 (Figure 2D; and data not shown). A preferential expansion of the CD11b+ Ly6G−Ly6C+ population was seen in all groups compared with media alone (24% vs 39% vs 59% vs 69% vs 73%). Compared with media or GM-CSF alone, CD11b+Gr1+ cells cultured with G-CSF, G-CSF + GM-CSF, or G-CSF + GM-CSF + IL-13 had a marked increase in IL4Rα expression (28% vs 78% vs 49% vs 62% vs 64%, respectively). GM-CSF had a greater influence on the expression of CD115 (CSF-1R), and GM-CSF, GM-CSF + G-CSF, GM-CSF + G-CSF + IL-13 groups had increased expression of CD115 compared with BM cells in culture media and cells treated with G-CSF alone (12% vs 20% vs 40% vs 40% vs 43%). GM-CSF also up-regulated F4/80 expression from 31% (media) or 36% (G-CSF alone) to 65% (GM-CSF) or 66% (G-CSF + GM-CSF) and 63% (G-CSF + GM-CSF + IL-13). Cells were negative for CD11c, CD14, and MHC class II molecules in all cultures, and there was no change in MHC class I observed between the groups (Figure 1A; and data not shown). As shown in Figure 1B-C, hematoxylin and eosin-stained cytospin preparations demonstrated that MDSCs were highly homogeneous and had mononuclear morphology with a large nuclear-to-cytoplasm ratio. Thus, cells with a phenotype consistent with MDSCs can be generated from whole BM through simple culture techniques in 4 days.
Figure 1.

Generation and phenotype of cultured MDSCs. (A) Fluorescence-activated cell sorter phenotype of B6 whole BM at day 4 of culture that has been left untreated, treated with G-CSF alone (100 ng/mL), treated with GM-CSF alone (250 U/mL), or treated with both G-CSF and GM-CSF. All gates based on isotype controls. Ly6G/Ly6C graphs were first gated on CD11b+ cells. Representative images of hematoxylin and eosin-stained cytospins of MDSCs with combined treatment (B) and combined treatment plus IL-13 addition on day 3 (C) Photographs were taken using 400× magnification with a RT-Spot camera mounted on an Olympus BX51 microscope (Olympus).
Figure 2.

MDSCs inhibit T-cell alloresponses through the expression of arginase-1. (A) RT-PCR showing that MDSCs express arginase-1 and iNOS and up-regulate arginase-1 on stimulation with IL-13. Hypoxanthine phosphoribosyl transferase shown as endogenous control. M indicates marker. (B) Real-time RT-PCR showing quantitatively the extent of arginase-1 and iNOS up-regulation by IL-13-stimulated MDSCs. Standardized to glyceraldehyde-3-phosphate dehydrogenase endogenous control and relative expression compared with untreated BM. (C) Western blot showing arginase-1 and actin control at the protein level in untreated BM, MDSCs, and MDSC IL-13. (D) Arginase activity was determined by measuring the production of urea over time. MLR was performed by mixing B6 purified T cells with irradiated BALB/c stimulators (1:1 ratio) and MDSCs (1:10 ratio). Cultures were pulsed with 3H-thymidine on the indicated days and harvested after a 16-hour incubation. (E) MLR/MDSC cocultures were left untreated or were treated with arginase inhibitor, nor-NOHA (300μM), nitric oxide inhibitor, L-NMMA (300μM), or both. (F) Arginase inhibitor (nor-NOHA) was added to MLR/MDSC cocultures at increasing concentrations (30, 100, and 300μM). (G) B6 WT T cells or GCN2 KO T cells were used as responder cells, and MDSCs were added at 1:10 ratio. (H) Addition of excess L-arginine (5mM) or tryptophan (5mM) was added back to MLR/MDSC cocultures and assessed for T-cell proliferation.
To enrich MDSCs, CD11b+ selection was performed on day 4. More than 95% of CD11b selected cells expressed both CD11b and Gr1 (data not shown). RT-PCR analysis of CD11b selected cells showed both arginase-1 and iNOS expression (Figure 2A). Adding IL-13 (80 ng/mL) on day 3 resulted in the up-regulation of arginase-1 expression in MDSCs (Figure 2A). To quantify the degree of increased expression, real-time PCR was performed (Figure 2B). Compared with the BM control, the relative expression of arginase-1 was increased 20-fold in MDSCs and increased to a significantly greater extent to 350-fold in MDSC-IL-13 cultures. The expression of iNOS was increased in MDSCs but was not significantly increased in MDSC-IL-13 (5-fold vs 7-fold vs BM controls, respectively; Figure 2B). Western blot analysis of arginase-1 (Figure 2C) and arginase-1 activity determination (Figure 2D) using a urea-based detection method indicated that the increase in the expression levels of arginase-1 in MDSCs and MDSC-IL-13 correlated with the downstream increase in protein levels and activity.
Next, studies were performed to determine the in vitro suppressive capacity of MDSCs in an allogeneic mixed leukocyte reaction (MLR) culture. B6 purified CD4+ and CD8+ T cells (2 × 105) were cocultured with irradiated BALB/c T cell–depleted stimulators (2 × 105) and B6 MDSCs (2 × 104). Cells were plated in complete media containing physiologic concentrations of L-arginine (150μM) and incubated for 3 to 7 days. The percentage suppression by MDSCs ranged from 50% to 60% at a 1:10 ratio of T cells to MDSCs (supplemental Figure 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). MDSC-IL-13 were significantly more suppressive at each time point of the assay, ranging from 65% to 75%. Because the in vitro suppression and in vivo GVHD inhibition (see “Donor MDSC-IL-13 cells migrate to sites of allopriming shortly after BMT and reduce GVHD lethality”) was superior with MDSC-IL-13 versus MDSC, subsequent studies were performed with MDSC-IL-13.
The suppressive capabilities of MDSCs were dominantly cell-contact independent (supplemental Figure 2). To determine the extent to which MLR suppression was dependent on the arginase pathway, the arginase inhibitor (nor-NOHA) was added to MDSC-IL-13 cultures. Nor-NOHA significantly reduced MDSC suppression of an in vitro MLR response to levels of 18% to 35% at a nor-NOHA concentration of 300μM, an effect that was titratable at lower concentrations (Figure 2E-F). NOS inhibition by L-NMMA alone resulted in a slight reduction in suppression at the peak of the response (72% in control vs 64% in L-NMMA at day 5), and the addition of L-NMMA to nor-NOHA was not more effective in reducing suppression than nor-NOHA alone.
The stress response pathway, GCN2, can sense essential amino acid starvation. Therefore, GCN2 KO T cells were used as responders in an MLR culture to which MDSC-IL-13 were added. Consistent with the possibility that L-arginine and perhaps other essential amino acids were depleted by MDSC-IL-13, the percentage suppression by MDSC-IL-13 significantly decreased using WT versus GCN2 KO responders from 60% to 25% at the peak of the response (day 5, P = .01; Figure 2G). Adding excess L-arginine (5mM) to the MLR/MDSC-IL-13 coculture resulted in a 60% reduction in suppression (day 4, P = .001; Figure 2H). This effect was specific for L-arginine because adding excess tryptophan had no such effect. Together, these results indicate that MDSC-IL-13 inhibit T-cell alloresponses by starving T cells of L-arginine.
Donor MDSC-IL-13 cells migrate to sites of allopriming shortly after BMT and reduce GVHD lethality
We then determined whether MDSC-IL-13 migratory patterns were such that it allowed them to traffic to secondary lymphoid organs, sites of allorecognition and priming, and thus have the potential to suppress GVHD. MDSC-IL-13 cells expressed the secondary lymphoid organ selectin/adhesion molecules, CD62L and LFA-1, as well as CCR9 (small intestine/colon) and CCR7 (LN/afferent lymphatics) but not α4β7 (Peyer patches/lamina propria; Figure 3A). Lethally irradiated BALB/c recipients were given 107 B6 BM cells plus 2 × 106 purified B6 T cells. A cohort also received MDSC-IL-13 (6 × 106 cells/mouse) expanded from eGFP transgenic B6 donor BM. On days 7 and 14 after BMT, LN and spleen were explanted and imaged using a whole-body fluorescent imager. A bright fluorescent signal emanated from the spleen and from the mesenteric/axillary/inguinal LNs (Figure 3B). As indicated by flow cytometric analysis, eGFP expressing cells in the LN and spleen increased from days 7 to 14 (LN, 3.3 vs 11 × 104; spleen, 6.5 vs 15 × 104; Figure 3C). On day 7, 75% of donor CD11b+ cells coexpressed Gr-1 (Figure 3D). On day 21 after transplantation, more than 80% of the donor MDSCs retained their coexpression of CD11b+Gr1+. The cell surface phenotype of this population of cells was CD11c+ (60%), MHC class II+ (65%), Ly6C+ (80%), Ly6Glo (5%), F4/80+ (75%), CD115+ (55%), and IL4Ra+ (55%) (supplemental Figure 3). At this time point, CD45.1 cells were reisolated from the spleen via positive selection and applied to in vitro MLR cultures to determine whether they had preserved their suppressive capacity. Reisolated donor MDSC-IL-13 potently suppressed a primary allogeneic (B6 > BALB/c) T-cell reaction and were comparable with fresh MDSC-IL-13 (supplemental Figure 4). These results demonstrate that MDSC-IL-13 migrate to sites of alloactivation in a GVHD setting and maintain their suppressive potential on day 21 after transplantation. Additional studies were performed to determine whether the donor CD45.1 MDSC-IL-13 were expanded further beyond day 14 in allogeneic recipients undergoing a GVHD reaction. In examining the spleen and the LNs on day 14 vs 21, there were comparable numbers of MDSC-IL-13 in the spleen and LNs (data not shown), suggesting that MDSC-IL-13 persisted during the later phases of the GVHD reaction.
Figure 3.

MDSC-IL-13 migrate to sites of allopriming in a GVHD setting. (A) Fluorescence-activated cell sorter analysis of pretransplantation MDSC-IL-13 was gated on Gr1+CD11b+ and analyzed for the surface expression of molecules associated with homing and recruitment. Gates are based on isotype controls. (B) Lethally irradiated BALB/c recipients were given 107 BM cells plus 2 × 106 T cells with 6 × 106 eGFP transgenic MDSC-IL-13. Control mice were given BM and T cells only. On days 7 and 14, LN and spleen were harvested and imaged using macroscopic fluorescent imaging. (C) The absolute number of eGFP+ cells that had migrated to the LNs and spleen was determined using flow cytometry. (D) Spleen was harvested from day 7 transplanted mice, and flow cytometry was performed to determine whether MDSC-IL-13 retained Gr1+CD11b+ phenotype. Cells were gated on donor (GFP+) CD11b+ cells.
Next, we wanted to determine whether MDSC-IL-13 had a beneficial impact on GVHD survival. Lethally irradiated BALB/c recipients were given 107 B6 BM cells plus 2 × 106 purified B6 T cells. Cohorts received 2 × 106 or 6 × 106 B6-derived MDSC-IL-13 at the time of transplantation. As shown in Figure 4A, mice given MDSC-IL-13 had significantly improved survival compared with controls (P < .001). Recipients of higher versus lower MDSC-IL-13 had a trend toward increased survival (P = .06). Mice given MDSC-IL-13 at either dose lost significantly less weight than mice without MDSC-IL-13; albeit weight loss was greater than the BM only group indicating that MDSC-IL-13 mice were not GVHD-free (Figure 4B). In other studies and consistent with the MLR data, recipients of MDSC-IL-13 had increased survival compared with those receiving MDSCs (P = .001; supplemental Figure 5), validating the choice of MDSC-IL-13 for further analysis.
Figure 4.

Cultured MDSC-IL-13 enhance GVHD survival in an arginase-1-dependent fashion. Lethally irradiated BALB/c recipients were given 107 B6 BM cells plus 2 × 106 purified CD25-depleted T cells plus either 2 × 106 MDSC-IL-13 or 6 × 106 MDSC-IL-13, both IL-13-treated. Kaplan-Meier survival curve of transplanted mice (A). Data represent 2 pooled experiments (BM only, N = 18; BM + T, N = 18; BM + T + 2 × 106 MDSC IL-13-tx, N = 22; BM + T + 6 × 106 MDSC- IL-13, N = 22; BM + T vs 2 × 106 MDSC IL-13, P < .001; BM + T vs 6 × 106 MDSC IL-13, P < .001; 2 × 106 MDSC IL-13 vs 6 × 106 MDSC- IL-13, P = .06). (B) Corresponding weights. (C) Kaplan-Meier survival curve of BM transplantation using 6 × 106 MDSC IL-13 generated from either WT mice or arginase-1 KO mice. Data represent one experiment (BM only, N = 8; BM + T, N = 12; BM + T + 6 × 106 WT MDSC-IL-13, N = 12; BM + T + 6 × 106 arginase-I KO MDSC-IL-13, N = 12; BM + T vs 6 × 106 WT MDSCs, P = .05; BM + T vs 6 × 106 arginase-I KO MDSC-IL-13, P = .08; arginase-1 KO MDSC-IL-13 vs WT MDSC-IL-13, P = .1). (D) Corresponding weights.
MDSC-IL-13 suppress donor T-cell activation, proliferation, and IFN-γ production and GVHD lethality via an arginase-1 dependent mechanism
Because the MDSC-IL-13 expression of arginase-1 was essential to achieve maximum suppression of a T-cell alloresponse in vitro, we sought to determine whether the same mechanism of action was involved in vivo. MDSC-IL-13 were generated from WT or arginase-1 KO mice. Survival analysis indicated that WT MDSC-IL-13 significantly enhanced survival compared with mice receiving BM + T alone (BM + T vs BM + T + 6 × 106 WT MDSC-IL-13, P = .05). Arginase-1 contributed to the GVHD inhibitory effect of MDSC-IL-13 because arginase-1 KO MDSC-IL-13 were not sufficiently potent in reducing GVHD to reach statistical significance with 12 mice/group (BM + T vs BM + T + 6 × 106 arginase-1 KO MDSC, P = .08; Figure 4C). Weight loss substantiated survival data (Figure 4D). These data confirm arginase-1 expression as a contributing mechanism for MDSC-IL-13 inhibition of GVHD lethality.
To further investigate the direct effects MDSC-IL-13 had on donor T-cell alloresponses in vivo, CFSE-labeled B6 T cells (15 × 106) and T cells plus MDSC-IL-13 (10 × 106; 1:0.67 ratio) were given to lethally irradiated mice. Compared with the GVHD group, there was a significant reduction in the percentage of both CD4+ and CD8+ donor T cells that had divided on day 4 after BMT when MDSC-IL-13 were coadministered (40.2 ± 7 vs 17.3 ± 1 for CD4, P = .01; 51.5 ± 13 vs 16.1 ± 1 for CD8, P = .01; Figure 5A). L-arginine deprivation has been shown to be associated with a decrease in the expression of CD3ζ on CD4+ and CD8+ T cells. Compared with controls, a decrease in CD3ζ expression was observed in donor CD4+ and CD8+ T cells when MDSC-IL-13 was coadministered (86% ± 2% vs 56% ± 3% for CD4+ T cells, P < .001; 85% ± 1% vs 68% ± 1% for CD8+ T cells, P < .001; Figure 5B). The activation status of donor T cells was assessed by examining the expression of CD25 and L-selectin (CD62L; Figure 5C). Compared with controls, CD8+ T cells coadministered with MDSC-IL-13 had a less activated phenotype, indicated by a lower frequency of CD25+ cells (32% ± 2% vs 19% ± 1%, P < .001). There was no statistical difference in the percentage of CD4+ T cells coexpressing CD25 between the 2 groups (37% ± 3% vs 33% ± 1%, P = .3). However, both CD4+ and CD8+ T cells have fewer CD62L− cells when MDSC-IL-13 are coadministered (74% ± 2% vs 50% ± 3% for CD4+ T cells, P < .001; 77% ± 4% vs 41% ± 3% for CD8+ T cells, P < .001). Comparing control with MDSC-IL-13 coadministration, there was a significant reduction in the percentage of cells expressing IFN-γ in both CD4+ and CD8+ T-cell populations (37% ± 5% vs 24% ± 2% for CD4+IFN-γ+, P = .04; 30% ± 4% vs 15% ± 1% for CD8+IFN-γ+, P = .007; Figure 5D). Cumulatively, these results show that MDSC-IL-13 diminished donor T-cell activation, proliferation, and proinflammatory cytokine production, which is associated with reduced GVHD lethality.
Figure 5.

MDSC-IL-13 negatively impact proliferation, activation, and effector function of donor T cells. Lethally irradiated BALB/c mice were transplanted with 15 × 106 CFSE-labeled CD25-depleted B6 Ly5.1-expressing T cells alone or with 10 × 106 CD11b purified MDSC-IL-13. (A) Day 4 splenocytes were analyzed via flow cytometry for CFSE dilution on CD4 and CD8 T cells. (B) Flow cytometry was used to detect the amount of CD3ζ present on CD4 and CD8 splenic T cells. (C) Graph of common activation markers (CD25 and CD62L) on CD4 and CD8 splenic T cells. (D) Intracellular cytokine staining was performed on LN CD4 and CD8 T cells to determine the percentage of cells producing IFN-γ.
MDSC-IL-13 preserve the GVL effect of allogeneic T cells
To determine whether the reduction of GVHD lethality on coadministration of MDSCs would impair a graft-versus-leukemia (GVL) effect, we used an A20-lymphoma model known to be sensitive toward T cell–mediated elimination. Irradiated BALB/c recipients were given BM with or without purified B6-T cells (2 × 106). Other cohorts received MDSC-IL-13 (6 × 106) and/or A20luc cells (3 × 105) on day 0 (Figure 6). Coadministration of MDSCs on day 0 resulted in a significant reduction of GVHD-induced mortality (BM + T vs BM + T + MDSC-IL-13, P < .001; Figure 6A) and weight loss compared with mice receiving T cells alone (Figure 6B). Recipients of T cells plus A20luc cells all eventually died of GVHD. However, recipients of T cells/A20luc/MDSC-IL-13 had a significantly improved survival, and 37% of mice survived long-term (BM + T + A20 vs BM + T + A20 + MDSC-IL-13, P < .001; Figure 6C-D), in marked contrast to mice receiving BM + A20luc cells, which all died of lymphoma by day 25 (Figure 6C). Despite a reduction in GVHD, recipients of MDSC-IL-13/T cells eliminated A20luc cells to the same extent as recipients of T cells only (Figure 6E). These data show that the dampening of GVHD by MDSC-IL-13 does not hinder the ability of donor T cells to mediate a GVL effect when given at the time of allo-BMT.
Figure 6.

MDSCs preserve GVL effect of allogeneic donor T cells. Lethally irradiated BALB/c mice were given 10 × 106 B6 T cell–depleted BM cells alone or with 2 × 106 CD25-depleted B6 T cells. Cohorts of mice also received 3 × 105 A20 cells and/or 6 × 106 B6 MDSC-IL-13. All cells were given intravenously on day 0 of transplantation. (A) Kaplan-Meier survival curve of mice receiving BM only (N = 10), BM + T (N = 10), or BM + T + MDSC-IL-13 (N = 10). (B) Corresponding weights from these mice. (C) Kaplan-Meier survival curve of mice receiving BM + A20 (N = 10), BM + T + A20 (N = 10), or BM + T + A20 + MDSC-IL-13 (N = 10). (D) Corresponding weights. (E) Mice were monitored on days 14, 18, and 50 using bioluminescent imaging to detect Renilla luciferase-expressing A20 cells. BM + T versus BM + T + MDSCs, P < .001; BM + A20 versus BM + T + A20, P < .001; BM + T + A20 versus BM + T + A20 + MDSCs, P < .001. Data represent one experiment.
PEG-arg1 administration has similar GVHD inhibitory properties as MDSC-IL-13
Arginase-1 contributed to the MDSC-IL-13 suppression of GVHD lethality. To determine whether arginase-1 itself was sufficient to inhibit GVHD lethality, arginase-1 was conjugated to PEG-arg1 to prolong its half-life in vivo. Lethally irradiated BALB/c mice were given 106 BM cells plus 2 × 106 CD25− B6 T cells. Cohorts were given PEG-arg1 intraperitoneally at 1 mg/mouse 2 times weekly for a total of 4 weeks. Other cohorts received 6 × 106 MDSC-IL-13. Mice receiving PEG-arg1 had a significant improvement in survival compared with mice receiving BM + T alone (P = .003; Figure 7A), comparable with mice receiving 6 × 106 MDSC-IL-13 (P = .4; Figure 7A). Notably, the most impressive survival differences between PEG-arg1 and T-cell only control was evident when PEG-arg1 continued to be administered with 75% of treated mice versus 0% of controls surviving. The precipitous loss of mice from GVHD in the PEG-arg1-treated group occurred on discontinuation of PEG-arg1 treatment (ie, between days 30 and 62). Weights of these mice demonstrated that mice receiving PEG-arg1 were comparable and different from the T-cell only controls (Figure 7B). When we quantify the concentration of L-arginine in peripheral blood from these animals at day 14, we observed a significant decrease of this amino acid in mice given 6 × 106 MDSC-IL-13 and PEG-arg1 (Figure 7C). These data indicate that PEG-arg1 administration was able to reduce GVHD lethality, especially during the time period of continued injections.
Figure 7.
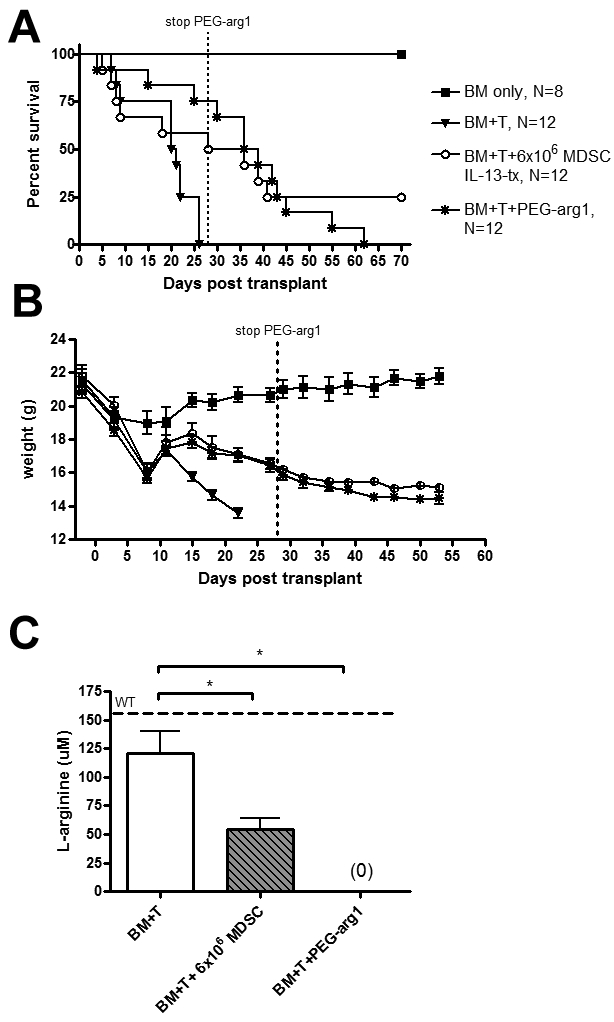
PEG-arg1 has similar protective effects as MDSC IL-13-tx. Lethally irradiated BALB/c mice were given 10 × 106 B6 BM cells alone or with 2 × 106 CD25-depleted T cells. In addition to this, cohorts were also given 6 × 106 MDSC IL-13 on day 0 or PEG-arg1 at 1 mg/mouse, 2 times weekly. (A) Kaplan-Meier survival curve. (B) Corresponding weights. P values for survival are BM + T versus 6 × 106 MDSCs, P = .03; BM + T versus PEG-arg1, P = .003. (C) L-Arginine was quantified using high performance liquid chromatography on day 14 from the peripheral blood of transplanted mice. WT B6 mice were used as controls. Data are representative of 2 replicate experiments with similar results.
Discussion
We report a novel cellular therapy using MDSC-IL-13 generated within 4 days from the BM of tumor-free animals using G-CSF + GM-CSF followed by the addition of IL-13 to up-regulate arginase-1 expression. MDSC-IL-13 were more potent in inhibiting in vitro alloresponses and in vivo GVHD lethality than MDSCs generated without IL-13. MDSC-IL-13 limited the proliferation, activation, and IFN-γ secretion of donor T cells exposed to allogeneic stimuli in vivo while preserving their GVL alloreactivity. Arginase-1 expression was critical for GVHD reduction. Similarly, the administration of PEG-arg1 inhibited GVHD lethality.
Several studies have proposed a link between the increased production of tumor-derived G-CSF/GM-CSF and increasing numbers of MDSCs.14–16 We have used these cytokines to develop a culture system to produce CD11b+Gr1+ MDSCs from whole BM. Phenotypic analysis revealed these cells express IL4Rα and CD115; both have previously been linked to enhanced suppressive capacity of MDSCs.4,5 MDSCs also expressed both Ly6C and F4/80, placing them within the monocytic subset of MDSCs described earlier.1,3 Adding IL-13 to the culture for the last 24 hours markedly increased arginase-1 expression (Figure 2B), which proved to be vital to the suppressive effects of MDSC-IL-13.
Successful prevention of GVHD using cellular approaches is dependent on migration of suppressor cells to areas, which harbor alloreactive donor T cells. Our laboratory has previously shown that regulatory T cells (Tregs) expressing high levels of CD62L (CD62Lhi) efficiently migrated to secondary lymphoid organs and potently inhibited GVHD and BM graft rejection, whereas CD62L− Tregs had no such effect.17 More recent studies using multipotent adult progenitor cells further elucidated the need for suppressor cells to be located within the vicinity of alloreactive T cells. Only when multipotent adult progenitor cells were administered within the splenic microenvironment was there was a positive effect on GVHD survival that resulted from decreased activation and expansion of donor T cells in vivo.18 In the current study, we have shown that in vitro-generated MDSCs and MDSC-IL-13 express appropriate homing molecules that guide them to secondary lymphoid organs, including CD62L. Furthermore, we show that the absolute number of MDSC-IL-13 in the secondary lymphoid organs increases from day 7 to 14, indicative of in vivo proliferation of MDSC-IL-13. Because most of the injected MDSC-IL-13 retained their cell surface phenotype of CD11b+Gr-1+ at 7 days after transplantation, MDSC-IL-13 adoptively transferred in vivo should retain the capacity to modulate immune responses. When we reisolated donor MDSC-IL-13 from recipients at day 21 after transplantation, they proved to be equally as capable at suppressing alloimmune responses in vitro as freshly generated MDSC-IL-13.
Recently, Zhou et al have described a culture system, which generates CD115+Ly6C+ MDSCs from embryonic stem cells and purified hematopoietic stem cells.10 The suppressive capacity of their MDSCs is in line with our current report, although there are significant differences worth mentioning. First, the length of time needed to generate MDSCs is longer than ours (17 days for embryonic stem-MDSC, 8 days for hematopoietic stem cell-MDSC) and uses a larger array of reagents/cytokines (stem cell factor, IL-6, IL-3, thrombopoietin, vascular endothelial growth factor, Fms-like tyrosine kinase 3 ligand, and macrophage colony-stimulating factor), complicating the logistics of translation into the clinic.10 Although the MDSCs derived from Zhou et al10 and those described in this paper have a similar cell surface phenotype, there is an apparent discordance in the suppressive mechanism used. For example, these investigators demonstrated that inhibiting arginase-1 had minimal effects on the suppressive capacity of MDSCs in vitro and show data, which indicate iNOS, IL-10, and the induction of Tregs as the mediators of T-cell inhibition.10 In contrast, our data show an important role for arginase-1. In addition, T-cell production of IL-10, the induction of Tregs, or the inhibition of iNOS did not differ in the in vitro MLR suppression assay between conditions that contained or did not contain MDSC-IL-13 (data not shown). We speculate that these differences may be the result of the addition of IL-13 in our culture system, which is known to increase arginase-1 activity at the expense of iNOS.19 Second, the effect of embryonic stem-MDSC on GVHD was tested using 5 × 105 allogeneic T cells as GVHD inducers, whereas we used 2 × 106 CD25-depleted T cells, thereby eliminating the possibility that Tregs within the donor inoculum contributed to the GVHD inhibitory effect of MDSC-IL-13. Lastly, the ratio of MDSCs to T cells administered in vivo differed between the studies. Zhou et al administered 3 total treatments of 2 × 106 MDSCs each, effectively making MDSC/input T-cell ratio 12:1.10 Our model used 2 × 106 T cells with one treatment of 6 × 106 MDSC-IL-13, which is a 3:1 MDSC-IL-13/input T-cell ratio. Taken together, these differences may explain the decreased survival we observed in our system. Consistent with data not shown but mentioned by Zhou et al10 is that the GVL activity of donor T cells remained intact in mice given CD115+Gr-1+F4/80+ MDSCs. We now report that MDSC-IL-13 preserve the GVL response as analyzed by a day 0 challenge of BMT recipients with a luciferase-labeled lymphoma cell line (A20). Allogeneic T cells given with MDSC-IL-13 retained their GVL activity and were equally effective at eradicating tumor as T cells given alone. The net result of MDSC-IL-13 was to reduce GVHD lethality, eliminate A20-lymphoma cells, and permit survival of 37% of recipients versus 0% of controls not given MDSC-IL-13 or allogeneic T cells alone.
Amino acid starvation results in T-cell dysfunction. IDO, the best studied enzyme that induces this effect, degrades the essential amino acid tryptophan and activates the GCN2 stress-kinase pathway.20 This results in the phosphorylation of eIF2α, initiating cell cycle arrest and T-cell anergy.20 Munn et al have shown that GCN2 KO T cells bypass this pathway and can proliferate in the absence of tryptophan.11 L-Arginine deprivation also induces the GCN2 kinase pathway in T cells, and GCN2 KO T cells are also resistant to these inhibitory effects in vitro.7 In line with this, we show that GCN2 KO T cells are more resistant to MDSC-IL-13-mediated suppression compared with WT T cells. Further, the suppressive effects are specific for L-arginine starvation. Only the addition of exogenous L-arginine to MLR cultures, and not tryptophan, had the capacity to restore T-cell proliferation.
It is known that, under conditions of low concentrations of L-arginine, the reductase domain of iNOS generates superoxide ion (O2−),21 which is converted to hydrogen peroxide (H2O2) and oxygen or, in the presence of nitric oxide, to peroxynitrite (ONOO−).22 Peroxynitrite inhibits T-cell activation and proliferation by reducing tyrosine phosphorylation and inducing apoptosis.23 Kusmartsev et al demonstrated that peroxynitrite was involved in T-cell inhibition by Gr-1+ MDSCs derived from tumor-bearing mice and the addition of the peroxynitrite scavenger, uric acid, completely abrogated the antigen-specific inhibitory effect of MDSCs.24 Others have also shown that the stimulation of T cells in an L-arginine poor microenvironment results in an inhibition of T-cell proliferation, decreased CD3ζ expression, and decreased production of IFN-γ.8,25–27 Under these circumstances, inhibiting the production of H202 did not reverse these effects, and only arginase-1 inhibitors or excess L-arginine allowed for CD3ζ reexpression, indicating that different MDSC types may use one or more distinct pathways to achieve suppression.27 Because we have shown that MDSC-IL-13 express both arg-1 and iNOS, it remains possible that these are acting synergistically and are producing toxic metabolites, such as superoxide ion or peroxynitrite, which can affect T-cell function. We attribute much, but not all, of the immunosuppressive capabilities of MDSC-IL-13 to the consumption of L-arginine. Thus, it is not surprising that most of the MDSC-IL-13 suppression occurs independently of cell contact. We hypothesize that the component of MDSC-IL-13-induced suppression that occurs via a contact-dependent suppression may be mediated through negative regulators of the immune response. For instance, PD1-PDL1 interactions, known to regulate tolerance, have been documented to reduce both proliferation and IFN-γ secretion by CD3 activated T cells.28 Indeed, MDSC-IL-13 express high levels of PDL1 (data not shown). Future studies will be needed to search for additional contributing mechanisms that can account for the residual suppression observed by MDSC-IL-13.
Because human MDSCs do not express CAT-2B transporter and therefore cannot uptake L-arginine, MDSCs release arginase-1 into the environment where it depletes L-arginine and induces T-cell dysfunction. Although recombinant human arginase-1 has been successfully cloned, the short circulatory half-life in the human body (t1/2 < 30 minutes) makes it difficult to achieve therapeutically effective blood arginine concentrations in patients.29 The attachment of PEG is an established technique used to extend the circulatory half-life and increase stability of numerous proteins. Studies have shown the effectiveness of PEG-arg1 for treatment of cancers dependent on L-arginine, including hepatocellular carcinoma and acute lymphoblastic T-cell leukemia.13,30 These studies have led us to hypothesize that we may be able to replicate the inhibitory effects of MDSCs by creating an L-arginine-free microenvironment. On administration of PEG-arg1, we show a significant benefit in the survival of mice with lethal GVHD, which is virtually the same as that seen when administering a higher (6 × 106) MDSC-IL-13 dose. It is interesting to note that the survival kinetics were more favorable early after transplantation for mice given PEG-arg1 than mice given MDSC-IL-13. However, in contrast to the single infusion of MDSC-IL-13, PEG-arg1 suppression of GVHD lethality appeared to require continuous PEG-arg1 administration because discontinuation on day 28 after BMT was followed by GVHD-induced deaths between days 30 and 62. We speculate that a longer duration of PEG-arg1 treatment may have an even more significant impact on long-term survival. Whether PEG-arg1 is immune suppressive or can be considered tolerogenic is unknown at present. Although future studies will be required to optimize PEG-arg1 dose and schedule, clearly PEG-arg1 has a beneficial effect on reducing GVHD lethality.
In conclusion, MDSC-IL-13 generated from murine BM have the ability to enhance GVHD survival. Arginase-1 expression contributes to the GVHD inhibitory effect of MDSC-IL-13. A GVL effect was not aborted by MDSC-IL-13. The administration of PEG-arg1 had similar effects as a higher-dose MDSC-IL-13 (6 × 106 cells) and was modestly superior to a lower dose of MDSC-IL-13 (2 × 106 cells; data not shown). Future studies will be needed to optimize the dose and schedule of MDSC-IL-13 and PEG-arg1. Nonetheless, these studies hold promise for GVHD prevention and perhaps GVHD therapy in the clinic.
Supplementary Material
Acknowledgments
The authors thank Michelle Smith, Rachel Sutlif, Klara Noble, and Christopher Lees for technical and editorial assistance and Dr Dorota Wyczechowska (Louisiana State University) for providing arginase-1 KO BM.
This work was supported in part by the National Institutes of Health (grants R01 AI34495, HL56067, and HL49997) (B.R.B.) and the Children's Cancer Research Fund (S.L.H.).
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.L.H. designed, organized, and supervised research, performed experiments, analyzed data, designed the figures, and wrote the paper; P.C.R. designed research, synthesized PEG-recombinant human arginase-1, and edited the paper;Q.Z. designed research and performed experiments; B.H.K. and R.V. performed experiments; C.A.G. performed Western blot experiments; P.A.T. and J.T. designed research; A.P.-M., D.H.M., and A.C.O. designed research and edited the paper; J.S.S. provided eGFP mice and edited the paper; and B.R.B. designed, organized, and supervised research and edited the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Bruce R. Blazar, University of Minnesota Masonic Cancer Center and Department of Pediatrics, Division of Bone Marrow Transplantation, MMC 109, 420 Delaware St SE, Minneapolis, MN 55455; e-mail: blaza001@umn.edu.
References
- 1.Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells. Adv Exp Med Biol. 2007;601:213–223. doi: 10.1007/978-0-387-72005-0_22. [DOI] [PubMed] [Google Scholar]
- 2.Movahedi K, Guilliams M, Van den Bossche J, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111(8):4233–4244. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 3.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181(8):5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gallina G, Dolcetti L, Serafini P, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest. 2006;116(10):2777–2790. doi: 10.1172/JCI28828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang B, Pan PY, Li Q, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66(2):1123–1131. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 6.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5(8):641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood. 2007;109(4):1568–1573. doi: 10.1182/blood-2006-06-031856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC. Regulation of T cell receptor CD3zeta chain expression by L-arginine. J Biol Chem. 2002;277(24):21123–21129. doi: 10.1074/jbc.M110675200. [DOI] [PubMed] [Google Scholar]
- 9.Ghansah T, Paraiso KH, Highfill S, et al. Expansion of myeloid suppressor cells in SHIP-deficient mice represses allogeneic T cell responses. J Immunol. 2004;173(12):7324–7330. doi: 10.4049/jimmunol.173.12.7324. [DOI] [PubMed] [Google Scholar]
- 10.Zhou Z, French DL, Ma G, et al. Development and function of myeloid-derived suppressor cells generated from mouse embryonic and hematopoietic stem cells. Stem Cells. 2010;28(3):620–632. doi: 10.1002/stem.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munn DH, Sharma MD, Baban B, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22(5):633–642. doi: 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 12.Serody JS, Burkett SE, Panoskaltsis-Mortari A, et al. T-lymphocyte production of macrophage inflammatory protein-1alpha is critical to the recruitment of CD8(+) T cells to the liver, lung, and spleen during graft-versus-host disease. Blood. 2000;96(9):2973–2980. [PubMed] [Google Scholar]
- 13.Cheng PN, Lam TL, Lam WM, et al. Pegylated recombinant human arginase (rhArg-peg5,000mw) inhibits the in vitro and in vivo proliferation of human hepatocellular carcinoma through arginine depletion. Cancer Res. 2007;67(1):309–317. doi: 10.1158/0008-5472.CAN-06-1945. [DOI] [PubMed] [Google Scholar]
- 14.Abe F, Dafferner AJ, Donkor M, et al. Myeloid-derived suppressor cells in mammary tumor progression in FVB Neu transgenic mice. Cancer Immunol Immunother. 2010;59(1):47–62. doi: 10.1007/s00262-009-0719-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bronte V, Chappell DB, Apolloni E, et al. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. 1999;162(10):5728–5737. [PMC free article] [PubMed] [Google Scholar]
- 16.Shojaei F, Wu X, Qu X, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A. 2009;106(16):6742–6747. doi: 10.1073/pnas.0902280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor PA, Panoskaltsis-Mortari A, Swedin JM, et al. L-Selectin(hi) but not the L-selectin(lo) CD4+25+ T-regulatory cells are potent inhibitors of GVHD and BM graft rejection. Blood. 2004;104(12):3804–3812. doi: 10.1182/blood-2004-05-1850. [DOI] [PubMed] [Google Scholar]
- 18.Highfill SL, Kelly RM, O'Shaughnessy MJ, et al. Multipotent adult progenitor cells can suppress graft-versus-host disease via prostaglandin E2 synthesis and only if localized to sites of allopriming. Blood. 2009;114(3):693–701. doi: 10.1182/blood-2009-03-213850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munder M, Eichmann K, Modolell M. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J Immunol. 1998;160(11):5347–5354. [PubMed] [Google Scholar]
- 20.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117(5):1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xia Y, Roman LJ, Masters BS, Zweier JL. Inducible nitric-oxide synthase generates superoxide from the reductase domain. J Biol Chem. 1998;273(35):22635–22639. doi: 10.1074/jbc.273.35.22635. [DOI] [PubMed] [Google Scholar]
- 22.Squadrito GL, Pryor WA. The formation of peroxynitrite in vivo from nitric oxide and superoxide. Chem Biol Interact. 1995;96(2):203–206. doi: 10.1016/0009-2797(94)03591-u. [DOI] [PubMed] [Google Scholar]
- 23.Groves JT. Peroxynitrite: reactive, invasive and enigmatic. Curr Opin Chem Biol. 1999;3(2):226–235. doi: 10.1016/S1367-5931(99)80036-2. [DOI] [PubMed] [Google Scholar]
- 24.Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172(2):989–999. doi: 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez PC, Quiceno DG, Zabaleta J, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004;64(16):5839–5849. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- 26.Zea AH, Rodriguez PC, Atkins MB, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005;65(8):3044–3048. doi: 10.1158/0008-5472.CAN-04-4505. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez PC, Zea AH, DeSalvo J, et al. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol. 2003;171(3):1232–1239. doi: 10.4049/jimmunol.171.3.1232. [DOI] [PubMed] [Google Scholar]
- 28.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ash DE. Structure and function of arginases. J Nutr. 2004;134(10) suppl:2760S–2764S. doi: 10.1093/jn/134.10.2760S. discussion 2765S–2767S. [DOI] [PubMed] [Google Scholar]
- 30.Hernandez CP, Morrow K, Lopez-Barcons LA, et al. Pegylated arginase I: a potential therapeutic approach in T-ALL. Blood. 2010;115(25):5214–5221. doi: 10.1182/blood-2009-12-258822. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.