

Abstract
A substantial genetic contribution underlies variation in baseline peripheral blood counts. We performed quantitative trait locus/loci analyses to identify chromosome regions harboring genes influencing red cell hemoglobin concentration using the cell hemoglobin concentration mean (CHCM), a directly measured parameter analogous to the mean cell hemoglobin concentration. Fourteen significant loci (gene symbols Chcmq1-Chcmq14) were detected. Seven of these influenced CHCM in a sex-specific fashion, and 2 showed significant interactive effects (epistasis). For quantitative trait locus/loci detected in multiple crosses, confidence intervals were narrowed using statistical and bioinformatic approaches. Two strong candidate genes emerged and were further analyzed: adult β-globin (Hbb) for Chcmq3 on Chr 7, and transferrin (Trf) for Chcmq2 on Chr 9. High and low allele parental strains in crosses detecting Chcmq3 segregate 100% with the known ancestral haplotype blocks, hemoglobin (Hb) diffuse (Hbbd) and Hb single (Hbbs), respectively. Hbbd consists of nonidentical major and minor polypeptides and exhibits an increased positive charge relative to Hbbs due to the net loss of 2 negative residues in the Hbbdminor polypeptide, resulting in a pI of 7.85 versus 7.13. Thus, as shown in human erythrocytes, positively charged Hbs are associated with cell dehydration and increased CHCM in mouse erythrocytes.
Introduction
A substantial genetic component underlies common peripheral blood traits including hemoglobin (Hb), hematocrit, and red cell indices.1 Quantitative trait locus/loci (QTL) studies in mice and genome-wide association studies (GWAS) in human populations have identified an ever-growing list of genomic loci and candidate genes influencing variation in these and other blood traits.2–4 Baseline peripheral blood traits are significant risk factors for complex human diseases with considerable morbidity and mortality. Hb levels are associated with increased all-cause mortality, especially in the elderly,5 which is particularly significant in light of the high rate of anemia in older adults.6 Total Hb is also a risk factor for sickle cell disease severity; high levels increase the risk of pain crises and acute chest syndrome, and low levels increase stroke risk.7,8
Despite the success in identifying polymorphisms associated with human disease phenotypes by GWAS, the fact remains that these polymorphisms account for a small fraction of the total variance in disease phenotypes.9,10 Studies in animal models can be invaluable tools in identifying genetic factors contributing to phenotypic variance. We are using inbred mouse strains as an unbiased approach to identify QTL underlying baseline blood traits. Here, we report our findings for the cell hemoglobin concentration mean, or CHCM. We used CHCM as the quantitative trait in our analyses because it was a much more robust measure than total Hb concentration, mean cell hemoglobin (MCH), or mean cell hemoglobin concentration (MCHC) in terms of the number of QTL identified, although some pleiotropy was observed. The cell hemoglobin concentration is directly measured in picograms per cell using the Advia 120 blood analyzer (Bayer Diagnostics); the CHCM is the mean of the distribution curve.
Our analyses of 12 mouse crosses reveal considerable complexity underlying variability in red cell Hb concentration. Fourteen distinct loci significantly associate with CHCM; 7 influence CHCM in a sex-specific fashion, and 2 show a significant interaction (epistasis). By convention, significant loci are named; we designated the loci Chcmq1-Chcmq14 (CHCM quantitative locus 1 through locus 14). Using statistical and bioinformatic methods, we significantly narrowed the 95% confidence intervals (CIs) for our most significant loci and identified candidate genes. The 2 most compelling candidates identified were the transferrin gene (Trf) for Chcmq2 on mouse chromosome (Chr) 9 and the gene encoding adult β-globin, Hbb, for Chcmq3 on Chr 7. Subsequent analyses provide strong evidence that the adult β-globin gene underlies Chcmq3. However, studies performed to date do not provide clear evidence that Trf is the quantitative trait gene for Chcmq2; hence, further analysis is required.
Methods
Animals
Mice from The Jackson Laboratory were housed in humidity- and temperature-controlled rooms (12-hour light cycle) with free access to acidified water and food (NIH 5K52). The Jackson Laboratory Animal Care and Use Committee approved all protocols.
Crosses
As part of an ongoing project to identify QTL underlying baseline hematopoietic traits, multiple crosses were established (Table 1). Four have been described in part previously: NZxSM and KSxSM,3 Bx129S1,11 and BxCB.12
Table 1.
Crosses used to identify CHCM QTL
| Cross name | Parental strains | No. progeny phenotyped | % Female | No. markers genotyped |
|---|---|---|---|---|
| 129S1xA | 129S1/SvImJ, A/J | 293 | 45 | 188 |
| AxB | A/J, C57BL/6J | 436 | 49 | 179 |
| BxD | C57BL/6J, DBA/2J | 438 | 56 | 172 |
| BxCB | C57BL/6J, CBA/J | 105 | 100† | 91 |
| Bx129S1 | C57BL/6J, 129S1/SvImJ | 483 | 49 | 506 |
| CByx129S1 | BALB/cByJ, 129S1/SvImJ | 437 | 50 | 171 |
| CByxC3 | BALB/cByJ, C3H/HeJ | 437 | 53 | 144 |
| C3xD | C3H/HeJ, DBA/2J | 439 | 53 | 134 |
| MRxSM | MRL/MpJ, SM/J | 363 | 38 | 258 |
| KSxSM | C57BLKS/J, SM/J | 186 | 49 | 86 |
| NZxSM | NZW/LacJ, SM/J | 183 | 57 | 83 |
| BxWSB* | C57BL/6J, WSB/EiJ | 207 | 49 | 208 |
Backcross (to strain C57BL/6J); all others F2 intercrosses.
Cross included only females by design.
Phenotyping
EDTA (ethylenediaminetetraacetic acid)–anticoagulated whole blood was obtained from cross progeny (8 weeks of age) via the retro-orbital sinus as described,13 with the exception of BxWSB progeny, where 20 μL of EDTA-anticoagulated whole blood were obtained via the retro-orbital sinus and mixed with 180 μL of 5% bovine serum albumin in phosphate-buffered saline before analysis using an Advia120 Multispecies Hematology Analyzer (Bayer Diagnostics).
DNA isolation and genotyping
Tail DNA was isolated by phenol-chloroform extraction using standard techniques. For Bx129S1 progeny, genotyping was performed using a single-nucleotide polymorphism (SNP) array, described previously.11 MRxSM progeny were genotyped by the High Throughput Sequenom and Illumina Genotyping facility (http://www.hpcgg.org/) using a 760-SNP array. Progeny from the NZxSM, KSxSM, and BxWSB crosses were genotyped using anonymous DNA simple sequence length polymorphic markers and/or SNPs, as described.3,14 For all other crosses, SNP genotyping was performed by KBiosciences (http://www.kbioscience.co.uk/). Markers genotyped in each cross are available upon request. The total number of markers genotyped per cross is given in Table 1.
QTL analysis
QTL mapping for CHCM was performed using R/qtl v1.07-12 (http://www.rqtl.org).15 Genetic map positions of all markers were updated to the new mouse genetic map using online mouse map converter tool at http://cgd.jax.org.16 The new map resolves inconsistencies between the physical and genetic maps. Phenotypic data were transformed to approximate the normal distribution.17,18 Results for each cross were analyzed using a 3-step approach, as described.11,19 First, genome-wide scans with sex as an additive covariate were performed to detect single loci associated with CHCM (main effect QTL). Single-locus scans that included sex as an interactive covariate were also performed to identify sex-specific QTL.11 The difference in the logarithm of the odds ratio (LOD) scores (ΔLOD) between these 2 scans is a test for sex-specific effects; a ΔLOD > 2.0 indicates significant QTL-by-sex interactions.20 Significance thresholds were determined by permutation testing (1000 permutations).21 LOD scores ≥ the 95th percentile (P < .05) of the permutation distribution were considered significant; scores meeting or exceeding the 37th percentile (P < .63) were considered suggestive.22 QTL CIs were determined by the posterior probability, as described.19 In part 2 of the analysis, simultaneous pair-wise scans to detect additive and epistatic effects were performed. Third, to determine the combined effects of all QTL detected, multiple regression analysis was performed. The analysis included all significant and suggestive QTL and interactions. Terms that failed to meet significance levels (P < .01) were eliminated one by one until all remaining QTL were significant, resulting in the final model for each cross.
Narrowing QTL CIs: combining crosses and interval-specific haplotyping
To narrow the CIs for overlapping QTL identified on Chr 2, 6, 7, and 9 in 2 or more crosses, data were combined and analyzed as described.23 QTL CIs were further narrowed using interval-specific haplotype analysis as described,24 which assumes that the sequence change underlying QTL results from ancestral polymorphisms. Regions of the genome that are identical by descent between the parental strains are removed, as they are unlikely to contain ancestral polymorphisms. This is usually a safe assumption, as 97% of the variation between inbred mouse strains is ancestral.25 The assumption is further strengthened when the QTL is found in multiple crosses involving different parental strains. The region(s) of the CIs most likely containing the causal mutation is further honed by identifying regions where the strains contributing the high allele increasing the trait value share a haplotype that differs from the shared haplotype of low allele-contributing strains. This process was performed using the Center for Genome Dynamics “Mouse Strain Comparison” tool (http://cgd.jax.org/) with the imputed diversity array 166 strain SNP panel option. Gene lists were obtained from Mouse Genome Informatics (MGI; http://www.informatics.jax.org/).
HAM
Haplotype association mapping (HAM) was performed as described.26 Briefly, we first applied a Hidden Markov Model (HMM) to restore missing genotypes and to infer haplotype simultaneously using a panel of 63 222 SNP loci for 57 inbred strains. Our HMM algorithm has been described in detail previously.27 Inferred haplotype block sizes range from a single SNP to 10 or more SNPs. Second, we systematically scanned the genome to calculate analysis of variance F statistics for each potential haplotype-phenotype association. A family-wise error rate model is used to set significance level at α of 0.05 level adjusted for multiple testing.
Real-time qPCR
Details meeting minimal information for publication of real-time qPCR experiments (MIQE)28 are provided to facilitate interpretation, accuracy, and reproducibility of results. RNA extraction: adult (8-12 weeks) bone marrow, spleen, and liver RNA was extracted using the TRIzol reagent lysis method of the PureLink RNA Mini kit (Invitrogen). Samples were further purified with PureLink DNAse. Samples were assessed for integrity and concentration using the Agilent Bioanalyzer (Agilent Technologies) and Nanodrop (NanoDrop Technologies), respectively. RNA integrity numbers (RIN) were between 9.0 and 10.0. Samples were stored at −80°C. Reverse-transcription: Total RNA (1 μg) was reverse-transcribed in a 20-μL reaction using RETROscript random decamers (Ambion) and SuperScript II (Invitrogen) according to the manufacturer's directions. Reactions for each sample were performed in triplicate on an MJ Research PTC 200 Peltier thermal cycler. The 3 reactions were pooled and tested for inhibitors using both dilution curve and SPUD assay methods.29 qPCR: reactions were performed on 3 biologic samples per mouse strain using POWER SYBR Green (Applied Biosystems) master mix with 5 μL of 1:1000 dilution of cDNA obtained from RT reactions, and 1-μL primer pairs in 20-μL reactions. Primer pairs (supplemental Table 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article) were designed and provided by PrimerDesign Ltd (http://www.primerdesign.co.uk). A single peak was observed in the melt curve for each primer set indicating desired primer specificity. Each biologic sample was run in triplicate for each primer set on an AB Prism 7900HT Gene Detection System (Applied Biosystems). Cycle conditions consisted of 50°C for 2 minutes and 95°C for 10 minutes followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Each sample and primer pair was tested with a no template control (NTC) and a no reverse transcriptase control (NRT). We are using the Cq (quantification cycle) value in place of CT, according to MIQE guidelines.28 In spleen RNA, the NTC and NRT Cqs ranged from 8.5 to 23.6 cycles, and 7.7 to 23.6 cycles, respectively, beyond that of the samples (supplemental Table 2). Similar results were obtained for all other analyses (not shown). Data analysis: fluorescence data were collected by AB 7900HT v2.3 Sequence Detection System software (Applied Biosystems). Rn (normalized reporter signal) values were exported to LinRegPCR software (v11.3 March 2009) for baseline and Cq determinations and to obtain amplicon group reaction efficiencies.30 Cqs and reaction efficiencies were exported to qBasePlus (v 1.4) to obtain relative normalized quantity (RQN) values.31 Three reference genes, Actb, β2m, and Gapdh were run initially. Gapdh proved unstable and was eliminated from the study.
Sequencing
Amplification and resequencing of rs52263982 were performed using primers complimentary to flanking sequences provided by dbSNP (http://www.ncbi.nlm.nih.gov/SNP): forward 5′-TCTCTGTAACACAGTGGGGTTTTCT-3′, reverse 5′-TCCCATTCCTGGTCCTGGCATTCCC-3′. Amplification products were purified (AMPure; Agencourt Biosciences) and sequenced using the automated dye termination technique (ABI Prism Model 3700 genetic analyzer; Applied Biosystems) as described.13 Sequences were analyzed using Sequencher 4.1 licensed software.
Statistical analysis for allele effects
Between-group comparisons were analyzed by Tukey HSD using JMP Version 7.0 software (SAS Institute).
Results
Identification of QTL for CHCM
In genome-wide scans to detect main effect QTL, multiple significant and suggestive QTL were identified (Figure 1). Details of QTL identified including peak location, CI, LOD score, nearest marker, the strain contributing the allele that increases the trait value (high allele), and homologous regions in the human genome are given in Table 2.
Figure 1.

Genome-wide scans for CHCM. Main effect QTL are shown for single-locus genome-wide scans with sex as an additive covariate in the 12 crosses indicated (top left of each panel). Cross abbreviations are defined in Table 1. Horizontal dashed lines on each panel represent significant (P = .05, top line) and suggestive (P = .63, bottom line) LOD scores as determined by 1000 permutations. For C3xD, the single dashed line represents the suggestive level, as no significant QTL were identified in this cross. x-axis, chromosomes 1 through X with the relative spacing representative of the relative length of each chromosome and marker locations shown by tick marks. y-axis, LOD score.
Table 2.
QTL indentified in single-locus and pairwise scans
| QTL name* | Chr | Cross | Peak (cM) | Peak (Mb)† | 95% CI (Mb)† | LOD | Nearest marker | High allele | Human homologous region(s) surrounding peak§ |
|---|---|---|---|---|---|---|---|---|---|
| Chcmq1 | 2 | NZxSM | 60.2 | 120.0 | 24.03-135.14 | 2.2 | D2MIT100 | SM | 11p13, 2q31.2, 11p14, 15q15 |
| 2 | Bx129SI | 56.7 | 109.6 | 90.44-158.19 | 4.2‡ | rs6318808 | B | 11p12, 11p13, 15q26 | |
| 2 | C3xD | 57.4 | 113.5 | 23.29-134.53 | 2.3 | rs3681744 | C3 | 11p12, 11p13, 15q26 | |
| 2 | KSxSM | 46.2 | 77.5 | 57.52-127.96 | 4.2‡ | D2MIT300 | KS | 2q31-q32, 20q13, 2q21-q23, 11p12-p13 | |
| 2 | MRxSM | 22.3 | 33.0 | 10.95-52.49 | 2.9 | rs13476460 | MR | 9q33-q34 | |
| Chcmq2 | 9 | KSxSM | 58.7 | 107.6 | 73.28-114.49 | 7‡ | D9MIT198 | SM | 3p21, 3q23, 3p22 |
| 9 | AxB | 58.5 | 107.5 | 56.57-114.93 | 3.9‡ | rs3700340 | A | 3p21, 3q23, 3p22 | |
| 9 | Cbyx129SI | 56.4 | 104.0 | 92.59-114.38 | 6.1‡ | rs3706728 | CBy | 3q22, 3p21, 3q23, | |
| 9 | 129SIxA | 54.4 | 102.3 | 96.27-107.52 | 15.5‡ | rs3696932 | A | 3q22-q23, 3p21 | |
| Chcmq3 | 7 | Bx129SI | 54.9 | 110.0 | 103.25-114.37 | 80.8‡ | rs3717027 | 129SI | 11p15, 16p12, 16p13, |
| 7 | BxWSB | 55.7 | 112.1 | 96.64-117.94 | 15.7‡ | D7MIT220 | WSB | 16p13, 16p12, 11p15 | |
| 7 | AxB | 56.8 | 114.5 | 97.82-122.27 | 6.7‡ | rs3681072 | A | 11p15, 16p12, 16p13 | |
| 7 | BxCB | 60 | 121.8 | 101.62-127.28 | 4.6‡ | rs3684285 | CB | 16p11, 16p12, 10q25-q26 | |
| 7 | BxD | 48.8 | 94.4 | 85.93-127.52 | 7.3‡ | rs3654319 | D | 11q14, 11q13, 11q15, 4p16 | |
| 7 | MRxSM | 54.9 | 110.6 | 104.10-124.71 | 20.2‡ | rs3717027 | MR | 16p12, 16p13, 11p15 | |
| 7 | NZxSM | 53.8 | 105.6 | 96.63-117.87 | 18.9‡ | D7MIT30 | NZ | 11p15, 16p13-p12 | |
| Chcmq4 | 11 | NZxSM | 47.8 | 80.2 | 46.17-90.59 | 4‡ | D11MIT30 | SM | 17q21, 17q11, 17q12, 17p13, 17q22-q23, 17p13 |
| Chcmq5 | 6 | Bx129SI | 43.8 | 94.9 | 88.49-125.75 | 3.7‡ | rs30129127 | B | 3p26 |
| 6 | CByxC3 | 28.6 | 59.4 | 42.22-116.98 | 3.9‡ | rs3680652 | CBy | 4q22, 4q21, 7q14, 7p11 | |
| 6 | KSxSM | 53.8 | 116.5 | 63.71-137.88 | 2.3 | D6MIT149 | KS | 22q11, 12p13, 12p11, 10q11, 7q36 | |
| 6 | NZxSM | 59.7 | 125.7 | 94.82-137.84 | 2.3 | D6MIT52 | SM | 12p13, 12p12-p11, 12q13 | |
| 6 | MRxSM | 4.9 | 11.0 | 6.56-136.45 | 2.5 | rs29926568 | MR | 7q21-q22, 7q31-q32 | |
| Chcmq6‖ | 12 | C3xD | 33.7 | 77.7 | 68.04-89.05 | — | rs3700554 | C3/D (M), D (F) | 14q23-q24 |
| Chcmq7‖ | 18 | KSxSM | 40.5 | 68.3 | 49.49-73.54 | — | D18MIT208 | SM (F) | 18q21, 18p11 |
| Chcmq8‖ | 11 | BxD | 70.9 | 107.8 | 94.65-113.41 | — | rs13481179 | D (F) | 17q23-q24, 17q25 |
| Chcmq9‖ | 9 | KSxSM | 38.7 | 69.6 | 56.85-110.67 | — | D9MIT198 | SM (M) | 15q21, 15q21-q23 |
| Chcmq10‖ | 8 | MRxSM | 8.7 | 16.9 | 16.86-131.24 | — | rs13479624 | MR (M), SM (F) | 8p23-p22, 8p12, 8p11, 13q14 |
| Chcmq11‖ | 9 | CBy129SI | 32.4 | 59.3 | — | — | rs3685575 | CBy (M) | 11q23, 15q22, 15q23-q25 |
| Chcmq12‖ | 8 | 129SIxA | 31.9 | 64.9 | 34.25-128.56 | — | rs3678134 | 129S1 (M) | 4q31, 8p23-p21, 4q34, 4q31-q32, 4q33, 19p13-p12 |
| Chcmq13¶ | 8 | 129S1xA | 65.8 | 120.5 | — | — | rs13479965 | — | 16q24, 16q22-qter, 1q42, 10p11 |
| Chcmq14¶ | 14 | 129S1xA | 34.6 | 67.6 | — | — | rs13482140 | — | 8p21, 13q14 |
| 1 | C3xD | 41.8 | 81.7 | 66.58-195.10 | 2.4 | rs3022821 | C3 | 2q35, 2q33-q35, 2q35-q36 | |
| 1 | BxWSB | 75.1 | 169.6 | 12.8-194.3 | 1.5 | D1MIT16 | WSB | 1q31, 1q32, 1q25, | |
| 5 | BxWSB | 60.4 | 120.3 | 111.9-139.6 | 2.4 | D5Mit367 | WSB | 22q12, 12q24, 22q11, 1p22, 12q24 | |
| 9 | BxWSB | 14.4 | 28.4 | 28.4-75.7 | 1.6 | D9MIT64 | 11q24, | 11q24, 11q23 | |
| 17 | BxWSB | 17.9 | 32.9 | 39.2-72.4 | 1.5 | D17MIT175 | WSB | 19p13, 6p21, 21q22 | |
| 15 | CByx129SI | 33.8 | 72.6 | 41.12-84.31 | 2.2 | rs13482655 | CBy | 8q24, 8q23-q24 | |
| 19 | KSxSM | 29.4 | 34.3 | 14.90-37.32 | 2.6 | D1MIT88 | KS | 10q23, 10q25, 10q26 |
LOD scores are not given for sex-specific QTL, which are identified by the difference in LOD scores between sex-additive and sex-interactive scans (see “Methods”). LOD scores and 95% CIs are not given for QTL that interact significantly but by themselves are not significant.
QTL names are given for the cross in which each was first identified. Loci on the same chromosome identified in subsequent crosses may or may not be the same QTL (see text).
Megabase (Mb) positions (Build 37) were determined using the Center for Genome Dynamics (http://cgd.jax.org) Mouse Map Converter Tool.
Significant LOD score.
Homologous positions identified using Mouse Genome Informatics Mouse-Human Orthology Maps (http://www.informatics.jax.org).
QTL identified from pairwise search for gene interactions.
Sex-specific QTL: M, male; F, female.
Two significant QTL, Chcmq1 (CHCM quantitative locus 1) on Chr 2 and Chcmq2 on Chr 9, were identified previously in the KSxSM cross3 and confirmed in new crosses included in this study. In addition, 3 new significant QTL were identified: Chcmq3 on Chr 7, Chcmq4 on Chr 11, and Chcmq5 on Chr 6. The shapes of the LOD curves (Figure 1) on Chr 9 (most evident in crosses AxB and KSxSM) and Chr 11 (NZxSM) suggest that 2 closely linked QTL may be present on these chromosomes. To test this possibility, we determined the ΔLOD score between 1- and 2-QTL models; evidence for a 2-QTL model (ΔLOD ≥ 2) was not found in any case. Similar model fitting in cross CByxC3, where the double peak on Chr 6 probably results from the known paracentric inversion on Chr 6 in strain C3,32 also failed to provide evidence for more than one QTL.
We also identified 7 significant QTL showing sex-specific effects in sex-interactive genome-wide scans (ΔLOD > 2 in the interactive versus additive scans): Chcmq6 (Chr 12), Chcmq7 (Chr 18), Chcmq8 (Chr 11), Chcmq9 (Chr 9 @ 69.6 Mb), Chcmq10 (Chr 8 @ 16.8 Mb), Chcmq11 (Chr 9 @ 59.3 Mb), and Chcmq12 (Chr 8 @ 64.9 Mb; Table 2). Two of these QTL show significant effects in females only—Chcmq7 on Chr 18 and Chcmq8 on Chr 11 (Figure 2A-B). Chcmq11 and Chcmq12 show significant effects in males only (Figure 2C-D). Chcmq6, Chcmq9, and Chcmq10 show effects in both sexes but they differ substantially. At Chcmq6, opposing allele effects are seen in males versus females homozygous for both C3 and D alleles (Figure 2E). At Chcmq9, homozygosity for the KS allele in males significantly decreases the CHCM compared with both heterozygous males and males homozygous for the SM allele (Figure 2F). In females, heterozygotes show the highest CHCM (Figure 2F). At Chcmq10, homozygosity for MR alleles reduces CHCM in females and increases it in males (Figure 2G).
Figure 2.

Sex-specific and interacting QTL. (A-G) Allele effects determined at the peak marker for 7 sex-specific QTL showing a ΔLOD > 2 between sex-interactive and sex-additive LOD scores. Female effects are shown in red, males in blue. The 3 possible genotypes for each QTL are given on the ordinate, with the QTL name and Chr position below. CHCM values are shown on the y-axis. Alleles: K, C57BLKS/J; S, SM/J; B, C57BL/6J; D, DBA/2J; Z, 129S1/SvImJ; A, A/J; M, MRL/MpJ; C, C3H/HeJ; *P < .05. (H) Chcmq11 was detected in cross CByx129SI as a significant “shoulder peak” closely linked to Chcmq2 on Chr 9 with a ΔLOD (dotted purple line) > 2 between the sex-interactive (solid red line) and sex-additive (solid black line). x-axis, Chr position on Chr9 in cM. y-axis, LOD score. (I) Pairwise scans detected 2 additional QTL, Chcmq13 and Chcmq14, showing significant gene interaction (epistasis). The effects of gene interaction predicted by multiple regression are shown for the 9 possible genotypes for Chcmq14 and Chcmq13, indicated at the bottom. Complex allele-specific interactions are apparent. Alleles: A, A/J; Z, 129S1/SvImJ. y-axis, CHCM value. *P < .001 compared with AZ and ZZ Chcmq13 genotypes with heterozygosity at the Chcmq14 locus. **P < .001 vs heterozygosity for Chcmq13 with homozygosity for 129S1 alleles at Chcmq14. No other significant differences.
In single-locus scans with sex as an additive covariate, a single significant QTL with a peak at 104.0 Mb was identified on Chr 9 in cross CByx129S1 (Figure 1 and Table 2). However, a second QTL, Chcmq11, was also detected on Chr 9 in CByx129S1 in the sex-interactive scan as a “shoulder” peak distinct from Chcmq2 that met the criteria for significance as a sex-specific QTL (Figure 2H).
Pairwise genome scans detected 2 additional QTL in cross 129S1xA that showed significant epistatic interactions, Chcmq13 (Chr 8) and Chcmq14 (Chr 14; Table 2). When Chcmq14 is homozygous for the A allele, no significant differences are seen between the 3 possible Chcmq13 genotypes (Figure 2I first panel). However, when Chcmq14 is heterozygous for A and 129S1 alleles, homozygosity for the A allele at Chcmq13 significantly reduces the CHCM versus either Chcmq13 heterozygotes (P = .0005) or 129S1 homozygotes (P = .0013), which do not differ from each other (P = .8425) (Figure 2I second panel). Most notably, the highest CHCM is found in 129S1 double homozygotes. When Chcmq14 is homozygous 129S1, Chcmq13 heterozygous and AA genotypes are not significantly different from each other (P = .0581), but 129S1 homozygosity significantly increases CHCM over 129S1 heterozygotes (P = .0007; Figure 2I third panel).
For each cross, we performed regression analysis to establish the relative contribution of all significant, suggestive, and interacting loci together. Not all QTL identified in single-locus scans meet these criteria and remain in the final model (Table 3). Clearly, multiple loci influence baseline CHCM levels. In addition to the QTL listed in Table 3, sex contributes significantly to the total variance in all but one cross (BxWSB). The total CHCM variance explained ranges from 11.7% (cross BxD) to as high as 61.3% (Bx129S1).
Table 3.
Multiple regression models for each cross
| Chr@Mb | df | LOD | % Variance* | F | P |
|---|---|---|---|---|---|
| Cross 129S1xA | |||||
| Sex | 1 | 12.03 | 12.7 | 58.51 | 3.24 × 10−13 |
| 8@120.5 | 6 | 6.88 | 7.0 | 5.34 | 3.08 × 10−5 |
| 14@67.6 | 6 | 5.71 | 5.7 | 4.39 | 2.91 × 10−4 |
| 9@102.3 | 2 | 14.53 | 15.7 | 36.06 | 1.15 × 10−14 |
| 8@120.5:14@67.6 | 4 | 5.53 | 5.5 | 6.37 | 6.37 × 10−5 |
| Total | 38.9 | ||||
| Cross AxB | |||||
| Sex | 1 | 4.48 | 4.2 | 20.88 | 6.42 × 10−6 |
| 7@114.5 | 2 | 6.75 | 6.3 | 15.90 | 2.19 × 10−7 |
| 9@107.5 | 2 | 4.03 | 3.7 | 9.34 | 1.07 × 10−4 |
| Total | 14.4 | ||||
| Cross BxD | |||||
| Sex | 1 | 5.36 | 5.1 | 25.16 | 7.71 × 10−7 |
| 7@94.4 | 2 | 7.37 | 7.1 | 17.48 | 4.99 × 10−8 |
| Total | 11.7 | ||||
| Cross BxCB | |||||
| 7@121.8 | 2 | 4.45 | 17.7 | 11.00 | 4.73 × 10−5 |
| Cross Bx129S1 | |||||
| Sex | 1 | 16.37 | 6.5 | 80.22 | < 2.0 × 10−16 |
| 2@109.6 | 2 | 5.38 | 2.0 | 12.49 | 5.16 × 10−6 |
| 6@94.9 | 2 | 3.45 | 1.3 | 7.95 | 4.1 × 10−4 |
| 7@110.0 | 2 | 83.57 | 47.2 | 289.39 | < 2.0 × 10−16 |
| Total | 61.3 | ||||
| Cross CByx129S1 | |||||
| Sex | 3 | 11.11 | 10.3 | 17.76 | 6.97 × 10−11 |
| 9@59.3 | 4 | 4.86 | 4.4 | 5.63 | 2.0 × 10−4 |
| 9@104.0 | 2 | 6.31 | 5.7 | 14.75 | 6.40 × 10−7 |
| 9@59.3:sex | 2 | 4.46 | 4.0 | 10.32 | 4.19 × 10−5 |
| Total | 16.9 | ||||
| Cross CByxC3 | |||||
| Sex | 1 | 11.00 | 10.7 | 53.23 | 1.43 × 10−12 |
| 6@59.4 | 2 | 3.61 | 3.36 | 8.40 | 2.64 × 10−4 |
| Total | 13.3 | ||||
| Cross C3xD | |||||
| Sex | 3 | 6.58 | 6.3 | 10.21 | 1.65 × 10−6 |
| 1@81.7 | 2 | 2.58 | 2.4 | 5.89 | .01 |
| 2@113.5 | 2 | 2.32 | 2.2 | 5.29 | .01 |
| 12@77.7 | 4 | 3.33 | 3.1 | 3.82 | .01 |
| 12@77.7:sex | 2 | 2.53 | 2.4 | 5.76 | .01 |
| Total | 12.1 | ||||
| Cross MRxSM | |||||
| Sex | 3 | 2.80 | 2.6 | 4.26 | .01 |
| 2@33.0 | 2 | 3.07 | 2.8 | 7.00 | .01 |
| 7@110.6 | 2 | 21.11 | 21.6 | 54.19 | < 2.0 × 10−16 |
| 8@16.9 | 4 | 4.23 | 3.9 | 4.86 | .001 |
| 8@16.9:sex | 2 | 2.67 | 2.4 | 6.08 | .01 |
| Total | 29.5 | ||||
| Cross KSxSM | |||||
| Sex | 3 | 8.75 | 12.8 | 13.86 | 3.88 × 10−8 |
| 2@77.5 | 2 | 3.97 | 5.5 | 8.87 | 2.15 × 10−4 |
| 6@116.5 | 2 | 2.72 | 3.7 | 5.99 | .01 |
| 9@69.6 | 4 | 4.99 | 7.0 | 5.65 | .001 |
| 9@107.6 | 2 | 3.77 | 5.2 | 8.42 | .001 |
| 18@68.3 | 2 | 2.81 | 3.8 | 6.19 | .01 |
| 9@69.6:sex | 2 | 2.67 | 3.6 | 5.88 | .01 |
| Total | 47.1 | ||||
| Cross NZxSM | |||||
| Sex | 1 | 7.97 | 11.8 | 39.33 | 2.68 × 10−9 |
| 7@105.6 | 2 | 19.37 | 33.4 | 55.64 | < 2.0 × 10−16 |
| 11@80.2 | 2 | 4.15 | 5.9 | 9.74 | 9.72 × 10−5 |
| Total | 47.2 | ||||
| Cross BxWSB | |||||
| 7@112.1 | 1 | 18.8 | 33.7 | 106.06 | < 2.0 × 10−16 |
df indicates degrees of freedom (includes main effect QTL and interactions).
Variance is the percentage of the total phenotypic variance in the F2 population associated with each location.
Narrowing QTL intervals using combined cross analysis
QTL on Chr 2, 6, 7, and 9 were detected in multiple crosses (Table 2). In the statistical method of combined cross-analysis (CCA), data from multiple crosses are combined into a single, large dataset by recoding all genotypic data to a phenotype-based code, as determined by allele effects, and reanalyzing in R/qtl, as described.23 CCA assumes that loci detected on the same Chr represent the same QTL. If so, CCA increases the statistical power by increasing the numbers of F2 progeny and markers analyzed. If the assumption that overlapping QTL identified in 2 or more crosses is invalid, CCA often resolves linked peaks into distinct QTL.
Four crosses were combined for Chcmq1 on Chr 2—Bx129S1, C3xD, KSxSM, and MRxSM (Table 4). NZxSM was excluded due to conflicting SM allele effects with cross KSxSM; strain SM contributes the high allele in NZxSM, but the low allele in KSxSM, suggesting that the 2 crosses detect distinct, closely linked Chr 2 QTL. We also suspected that a distinct QTL was detected in cross MRxSM based on its peak position, which is significantly proximal to the QTL peak positions on Chr 2 for the other crosses (Table 2). Indeed, a distinct shoulder with a peak at 28.4 cM was resolved in the CCA (Figure 3A). The peak location of Chcmq1 on Chr 2 was refined to 58.2 cM with a CI of 52.4-62.2 cM in the CCA (Table 4). Thus, the Chcmq1 CI was reduced to 31 Mb, a reduction of more than 50% compared with the smallest interval obtained in the individual crosses (68 Mb, Bx129S1).
Table 4.
Combined cross analysis
| QTL | Chr | Crosses combined | CCA Peak, cM (Mb) | 95% CI, cM (Mb) | Other QTL detected cM (Mb) | HAM peak (Mb)* |
|---|---|---|---|---|---|---|
| Chcmq1 | 2 | B6x129S1, C3xD, KSxSM, MRxSM | 58.2 (115.5) | 52.4-62.2 (96.7-127.9) | 28.4 (48.7) | None |
| Chcmq2 | 9 | AxB, CByx129S1, KSxSM, 129S1xA | 56.4 (104.0) | 52.4-60.4 (99.9-110.6) | None | 102.16-103.38 |
| Chcmq3 | 7 | AxB, BxCB, BxD, MRxSM, NZxSM | 54.9 (110.0) | 53.5-56.8 (104.5-114.5) | None | 108.50-111.58 |
| Chcmq5 | 6 | Bx129S1, KSxSM | 43.8 (94.9) | 39.5-53.8 (88.5-116.5) | 1.8 (4.8) | None |
Only peaks within the interval common to CCA and interval-specific haplotyping are listed. The peak Mb intervals given include more than one suggestive peak, but addition of 500 kb on either side results in a single overlapping interval.
Figure 3.

Combined cross analysis. Ninety-five percent confidence intervals for (A) Chcmq1, (B) Chcmq2, (C) Chcmq3, and (D) Chcmq5 following combined cross analysis are shown. x-axis, map position in cM. y-axis, LOD score. Dashed lines denote significant (P = .05, top lines) and suggestive (P = .63, bottom lines) LOD scores as determined by permutation testing.
CCA for Chr 9 revealed a single peak at 56.4 cM with a CI spanning from 52.4 to 60.4 cM (Figure 3B and Table 4). For Chcmq3, we included all intercrosses in the CCA with the exception of cross Bx129S1 due to its inordinately large effect (LOD > 80; Table 2), which would skew all other cross data. Analysis of the remaining 5 intercrosses resolved a single peak at 54.9 cM with a CI spanning from 53.5 to 56.8 cM (Figure 3C and Table 4). These coordinates are almost exactly the same as seen in the Bx129S1 cross alone, providing strong evidence for its position.
In the CCA of Chcmq5, we excluded crosses MRxSM, whose peak position is located at a significant distance from Chr 6 QTL detected in other crosses, and CByxC3 due to the inversion present on Chr 6 in strain C3H. We also excluded cross NZxSM due to inconsistent allele effects vs. cross KSxSM. As above, these observations suggest that more than one QTL is present in this region of Chr 6. Indeed, even when just the 2 remaining crosses were analyzed, the shape of the CCA QTL scan, showing several shoulders, indicated the presence of multiple, linked QTL peaks (Figure 3D). A suggestive peak at 1.8 cM, likely corresponding to the suggestive peak detected in cross MRxSM, was also resolved in the analysis (Figure 3D and Table 4).
Narrowing QTL intervals by interval-specific haplotype analysis
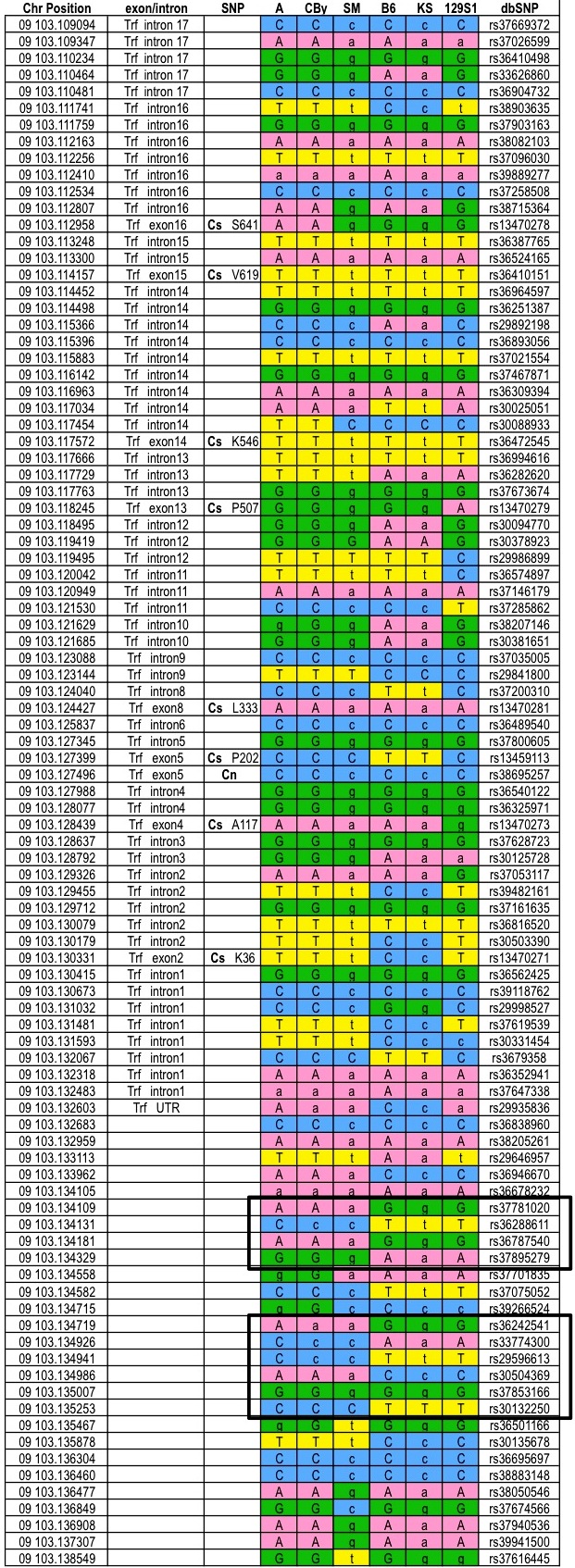
We performed interval-specific haplotype analysis of Chcmq1 on Chr 2 using parental strains from the same crosses used in the CCA with the exception of MR and SM, which were excluded because cross MRxSM clearly detects a QTL distinct from Chcmq1 on Chr 2 (Tables 2 and 4). Using windows of 3 contiguous SNPs to define haplotypes, we identified regions of the genome within the Chcmq1 CCA interval (Table 4) that were shared by the high allele strains (B6, C3, and KS) and differed from the shared haplotype of the low allele strains (129S1, D, and SM). This analysis narrowed the chromosomal interval most likely to contain the Chcmq1 QTL gene to 0.34 Mb of noncontiguous sequence, which includes 23 known or predicted genes (Figure 4 and gene list in supplemental Table 3). It is noteworthy that no microRNAs are contained within the Chcmq1 or any other CHCM QTL interval, according to MGI. By a similar analysis, the Chcmq2 interval on Chr 9 was reduced to 1.2 Mb (33 genes; Figure 4 and supplemental Table 4). Notably, the transferrin gene (Trf), a strong candidate gene, is located at 103.11 Mb on Chr 9, very near the CCA QTL peak for Chcmq2 (Table 4).
Figure 4.

Interval-specific haplotype analysis and HAM. (A) Each QTL was reduced by interval-specific haplotype analysis of the strains involved to the intervals indicated by the blue bars, prioritizing regions most likely to contain the QTL gene. QTL names and Chr locations are given to the left, with Chr positions (Mb) below. (B) Example of results obtained by HAM. Positions of the most significant peaks within the common combined cross/interval-specific haplotyping interval are given in Table 4.
In addition to the strains from the CCA (Table 4), we included parental strains from crosses Bx129S1 and BxWSB (Table 2) in the interval-specific haplotype analysis for Chcmq3. The analysis reduced the genomic region containing Chcmq3 to 0.42 Mb (Figure 4) containing 35 genes (supplemental Table 5). Of these, 16 were olfactory receptor genes. Notably, the interval contains the hemoglobin cluster including duplicated adult β-globin genes.
For Chcmq5 on Chr 6, interval specific haplotyping with just 4 parental strains (including 2 closely related strains, B6 and KS) from 2 crosses substantially reduced the QTL interval, to 1.6 Mb versus 28 Mb that remained after CCA (Figure 4 and Table 4). The reduced interval contains just 11 genes (supplemental Table 6).
HAM
HAM in inbred mouse strains is analogous to GWAS in human populations. HAM is fraught with false positives but when used in combination with experimental crosses is a powerful tool to narrow QTL intervals.33,34 We used CHCM data from male (28 strains) and female (30 strains) mice at age 12 months that were available through the Jackson Aging Center (http://agingmice.jax.org/)35 to perform HAM analysis.
Multiple suggestive HAM peaks were detected (Figure 4B). Table 4 includes HAM peaks common to haplotype blocks identified by interval-specific haplotyping. The peak interval size includes an additional 500 kb on each side, as has been recommended to account for lack of precision inherent to both HAM and GWAS.33 Although suggestive HAM peaks were obtained for Chcmq1 and Chcmq5, they do not coincide with regions identified by interval-specific haplotyping. Whether these HAM peaks are false positives or represent real loci cannot be ascertained without additional data. For Chcmq2, 17 genes are contained within the HAM peak interval (MGI) but only 5 are common with those within interval-specific haplotype blocks (supplemental Table 4). Of these, transferrin is the strongest candidate gene.
For Chcmq3, 143 genes, including many olfactory receptor genes, are contained within the HAM peak, but only 26, of which 12 are olfactory receptor genes, overlap with haplotype blocks (supplemental Table 5). The interval includes the 4 genes of the β-globin cluster: adult β-globins Hbb-b1 and Hbb-b2, and embryonic globins Hbb-bh1 and Hbb-γ (ϵY). As our QTL analysis was done using adult mice, adult β-globin is the most likely candidate gene.
Further analysis of Chcmq3
In mice, polymorphic differences in duplicated β-globin genes produce the classical “single” and “diffuse” hemoglobin electrophoretic variants, Hbbs and Hbbd, respectively. In single strains, the 2 loci, designated βS and βT, are identical at the amino acid level; in diffuse strains, the duplicated loci, βdmajor and βdminor, produce 2 polypeptides differing by 9 amino acids.36 As the reference sequence strain is B6, a single Hb strain, the gene symbols Hbb-b1 and Hbb-b2 are the duplicated BS and βT loci.
All 7 high allele strains from the crosses that detect Chcmq3 are diffuse variants, while the 2 strains contributing the low alleles are single variants. The high allele strains carry an extended haplotype at the β-globin locus that differs from the extended haplotype shared by the 2 low strains (Figure 5A). The only exception is one SNP difference in strain WSB, not unexpected given that WSB is a recently inbred wild-derived strain. One SNP, rs52263982, was typed as a guanine in strains 129S1 and D (MPD: source Perlegen 2). Resequencing, however, confirmed that rs52263982 is really a cytosine in both strains (Figure 5B).
Figure 5.

Haplotype blocks at Hbb-b1. (A) The high allele contributing strains from crosses in which Chcmq3 was detected are all hemoglobin diffuse and the low allele strains hemoglobin single. Lowercase indicates imputed loci. Asterisks indicate resequenced SNPs. (B) Resequencing of SNP rs52263982 in strains 129S1/SvImJ and DBA2 confirms they carry the C allele.
In the congenic strain C.B6-Tyr+Hbbs/J, the Hbbs and tyrosinase loci from C57BL/6J mice were transferred to the BALBc/By genetic background; BALBc/By is a hemoglobin diffuse (Hbbd) strain. Notably, a significant reduction in CHCM (P < .001) is seen in strain C.B6-Tyr+Hbbs/J versus BALB/cByJ (Table 5). Thus, replacing the diffuse hemoglobin locus with the single locus in an otherwise genetically identical background reduces CHCM. Furthermore, in crosses where we failed to detect Chcmq3, the parental strains were of the same β-globin haplotype, meaning they were either both diffuse or both single. This is added evidence that β-globin is the Chcmq3 gene, because in the absence of allelic variation one could not expect to detect QTL.
Table 5.
Congenic analysis
| Strain | CHCM |
|
|---|---|---|
| Females | Males | |
| BALB/cBy | 33.2 ± 0.3 (10) | 32.5 ± 0.2 (10) |
| C.B6-Tyr+Hbbs/J | 31.8 ± 0.4 (10)* | 31.5 ± 0.4 (9)* |
All values ± SD.
P < .001 vs BALBc/By.
Finally, qPCR using a primer that exactly matched sequences common to βS, βT, βdmajor, and βdminor reveals higher relative expression in most high allele Hbbd strains versus low allele Hbbs strains (Figure 6A-B), suggesting that expression differences account for some of the Chcmq3 variance.
Figure 6.

qPCR. β-globin (Hbb) expression of high allele strains (color-coded) relative to the corresponding low allele strain in (A) spleen and (B) bone marrow (BM) in crosses detecting Chcmq3 (Table 2). qPCR primers (supplemental Table 1) exactly matched sequences in βS, βT, βdmajor, and βdminor. Bone marrow (C) and liver (D) Trf expression in crosses in which Chcmq2 was detected. Because strain A was the high allele in 2 different crosses, the specific cross is indicated directly on the bar graph. *Expression in high and low allele strains differ significantly (P ≤ .028).
Further analysis of Chcmq2
No such extended haplotype structure defining high versus low allele strains is evident at the Trf locus, although 2 small SNP blocks are seen upstream of Trf (supplemental Figure 1). High allele strains express Trf at lower rather than higher levels than do low allele strains in the bone marrow (Figure 6C), while no significant Trf expression differences are seen in the spleen (not shown). In the liver, the major site of transferrin synthesis, high allele strains from 3 of the crosses showed increased expression (Figure 6D).
Moreover, the Trf gene shows a cis-eQTL in the livers from a BxA cross, which is one of the crosses that contributes to this QTL (unpublished microarray data publically available at http://cgd.jax.org/datasets/expression.shtml). In cross CByx129S1, however, the high allele strain (CBy) showed decreased Trf expression. Thus, the data are conflicting; 3 of the 4 crosses show that Trf is the likely candidate gene, but the fourth cross does not support that hypothesis. Further work will be necessary to resolve this issue.
Discussion
Using a QTL analysis approach, we have identified multiple sex-independent and sex-dependent loci influencing baseline CHCM in mice, underscoring enormous complexity in the genetic regulation of baseline cell Hb concentration. We chose CHCM as our trait because it is a direct, per cell measure of Hb. Perhaps not unexpectedly, other erythroid traits used in our studies (Hb, MCH, MCHC, hematocrit, red cell count) demonstrate overlap of loci detected (data not shown), as recently noted for human erythroid traits as well.37 The identification of genetic loci influencing cell Hb is a critical first step in elucidating novel or previously unsuspected pathways that may modify clinical disease severity in the hemoglobinopathies and anemia of aging.
Our strongest QTL in terms of LOD score was Chcmq3 on Chr 7. All statistical and bioinformatics methods used reduced the Chcmq3 interval to one that includes the β-globin locus. In mice, duplicated β-globin genes are maintained in natural populations by balancing selection.38,39 Several studies suggest possible functional mechanisms that prevent evolutionary divergence of β-globin polypeptides and thus allow their maintenance, including an overall fitness advantage of Hbbd homozygotes as a result of higher Hb concentration40 leading to increased cold-adaptation responses39 and increased availability of reduced glutathione for detoxification.41 The identification of the adult β-globin locus as the quantitative trait gene for Chcmq3 is supported by several criteria established by the Complex Trait Consortium.42 High and low allele parental strains in multiple crosses detecting Chcmq3 segregate 100% with the known ancestral haplotype blocks, Hbbd and Hbbs, respectively.36 In crosses in which both parents were Hbbd or Hbbs, Chcmq3 was not detected, and a significant reduction in CHCM is seen in the congenic strain, C.B6-Tyr+Hbbs/J, compared with BALB/cBy.
Examination of the Mouse Phenome Database (www.jax.org/phenome) reveals that, in general, MCHC/CHCM is high in Hbbd versus Hbbs strains (not shown), in line with earlier observations on overall Hb levels.40 By qPCR, we show that total β-globin, regardless of the locus from which it derives, is highest in spleen and bone marrow of Hb diffuse parental strains in the majority of the crosses, in accordance with calculated allele effects. That this relationship was not absolute, however, suggests that expression differences alone do not account for the entire phenotypic effect. Coding differences between the 2 haplotypes likely influence CHCM as well, possibly via effects on red blood cell hydration status. Positively charged Hb S and Hb C are associated with dehydration via up-regulation of the KCl cotransporter.43–45 Notably, diffuse Hb strains would be predicted to have increased positive charge relative to single strains because βdminor has a net loss of 2 negative residues, resulting in a pI of 7.85 versus 7.13 in βS/T and βdmajor. Whether this translates into a corresponding increase in KCl cotransport activity awaits further study.
The haplotype blocks associated with high and low CHCM strains include the embryonic βh1 and ϵY genes and beyond. Indeed, the haplotype structure is maintained from bp 110956036-110053860 (Ensembl gene coordinates). Within this region, only minor variations (4 SNPs in total) in strain WSB are seen. Beyond this range, however, WSB shows significant SNP variation compared with the other 8 strains involved in crosses detecting Chcmq3, which eventually become essentially identical by descent (supplemental Table 7). Of the other genes in the Chcmq3 interval common to all analyses, Olfr66 is of interest in light of a recent study identifying a significant association of HbF with 2 olfactory receptor genes (OR51B5, OR51B6) just upstream of the β-globin cluster in humans.46 Olfr66 lies upstream of the mouse β-globin cluster and contains a nonsynonymous coding SNP in single versus diffuse strains. In both humans and mice, the role of olfactory receptor genes in globin expression awaits further analysis.
The gene encoding transferrin, Trf, stands out as a strong candidate for Chcmq2 on Chr 9, falling within the final interval defined by CCA, interval-specific haplotyping, and HAM. However, supporting experimental evidence is unconvincing so far. Expression levels in adult tissues are conflicting (Figure 6), and no coding changes are seen in the parental strains according to the SNP databases. Still, Trf cannot be formally excluded as the QTL gene without further study, including studies in congenic strains and resequencing.
Candidate gene lists for the major loci identified are provided in supplemental Tables 3-6. From these lists, the most compelling candidates for functional analysis can be gleaned by examining expression databases such as BioGPS (http://biogps.gnf.org) and identifying the phenotype of available mutant strains. All available classes of mutant strains (eg, spontaneous, targeted) for any gene are identified in MGI. For example, of the genes in the final Chcmq2 list (supplemental Table 4), some are restricted in expression to testis (4930519F24Rik, 4921517D21Rik), and others show no erythroid phenotype in knockout mouse strains (Nck1, Rab6b, Dag1, Arih2, Ip6k2, Slc26a6). Although a knockout cannot always be assumed to produce the same phenotype as a polymorphic variant, these genes would be considered less likely candidates for initial analysis. On the other hand, Stag1 (stromal antigen 1) is expressed in bone marrow and spleen (BioGPS) and is a high priority candidate.
A great deal of concordance exists for rodent and human QTL for many traits including high-density lipoprotein cholesterol,47 kidney function,26,48 and bone density.49 Emerging data suggest that such is also the case for erythroid traits. Recent, large-scale GWAS4,37,50 reveal candidate genes for Hb, MCH, and MCHC, of which several fall into conserved regions of the mouse genome corresponding to QTL CIs identified in our study (Table 6). Interestingly, however, we did not identify CHCM QTL corresponding to HBS1L-MYB or BCL11A, loci associated with several hematologic parameters in humans including MCHC and Hb F levels.4,37,50 However, we detected several QTL potentially concordant with BCL11A using other red blood cell traits, as summarized in Table 7. The laboratory mouse has significant power to narrow candidate intervals and positionally clone underlying modifier loci. For many mapped human traits, experimental analysis in mouse models will be necessary to confirm the identity of the underlying causative genes and/or SNPs. Doing so will further our understanding of complex human disorders, opening up possibilities for new targeted therapies.
Table 6.
Concordance of human GWAS for Hb, MCH, and MCHC, and Mouse CHCM QTL
| Candidate gene(s) | Human chr | QTL cross | Mouse Chr | QTL peak position (Mb) | CI (Mb) | MGI gene location (Mb) |
|---|---|---|---|---|---|---|
| Abo | 9 | MRxSM | 2 | 33.0 | 10.95-52.49 | 26.7 |
| Tmprss6 | 22 | CBy129S1 | 15 | 72.6 | 41.12-84.31 | 78.3 |
| Cdt1 | 16 | 129S1xA (Chcmq13)* | 8 | 120.5 | — | 125.1 |
| March8-Anubl1-Fam21c | 10 | KSxSM | 6 | 116.5 | 63.71-137.88 | 116.3 |
| Ncaph2-Sco2-Tymp-Klhdc7b | 22 | CByx129S1 | 15 | 72.6 | 41.12-84.31 | 89.2 |
| Rcl1 | 9 | KSxSM | 19 | 34.3 | 14.90-37.32 | 29.2 |
| Gcdh | 19 | 129S1xA (Chcmq12) | 8 | 64.9 | 34.25-128.56 | 87.4 |
| Spna1 | 1 | C3xD | 1 | 81.7 | 66.58-195.10 | 176.1 |
| Aldh2 | 12 | BxWSB | 5 | 120.3 | 111.9-139.6 | 122.0 |
| Trafd1 | 12 | BxWSB | 5 | 120.3 | 111.9-139.6 | 121.8 |
| Bysl | 6 | BxWSB | 17 | 32.9 | 39.2-72.4 | 47.7 |
QTL identified in pairwise search for gene interactions; Hb, hemoglobin; MCH, mean cell hemoglobin; MCHC, mean cell hemoglobin concentration; and CHCM, cell hemoglobin concentration mean.
Table 7.
Suggestive QTL peaks near Bcl11a (Chr 11, 24 Mb)
| Cross | Trait | Peak (Mb) | 95% CI (Mb) | LOD | Nearest marker | High allele |
|---|---|---|---|---|---|---|
| MRxSM | RBC | 11.2 | 7.3-32.0 | 3.1 | rs13480871 | SM |
| CByxC3 | RBC | 5.7 | 5.6-114.5 | 2.8 | rs4222040 | C3 |
| MRxSM | Hct | 19.8 | 7.3-36.9 | 2.6 | rs13480889 | SM |
| MRxSM* | Hct | 29.6 | — | — | rs6398304 | — |
| NZxSM | MCHC | 19.4 | 17.1-80.2 | 2.5 | D11Mit2 | SM |
| Bx129S1 | MCHC | 8.9 | 3.5-98.6 | 2.6 | rs29414290 | 129S1 |
RBC indicates red blood cell count; Hct, hematocrit; and MCHC, mean cell hemoglobin concentration.
QTL identified in pairwise search for gene interactions.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grant nos. HL085480 (to L.L.P.), HL081497 (to C.B.), GM070638 (to G.A.C.), HL39693 (to D.G.), P01-HL057346 (to D.G.), HD028820 (to J.A.S); American Heart Association 0675025N (to J.A.S.); National Hemophilia Foundation Clinical Fellowship Program (to J.A.S.); and The National Cancer Institute CA34196 (The Jackson Laboratory).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: L.L.P. and C.B. designed the study, analyzed data, and wrote the manuscript; A.J.L. established and maintained experimental crosses, phenotyped and genotyped mice, performed experiments, analyzed data, and edited the manuscript; S.-W.T., Q.L., and G.A.C. performed statistical analyses and edited the manuscript; Z.S. and B.J.P. designed, generated, and genotyped crosses Bx129S1 and BxCB, and edited the manuscript; M.S.L. and B.J.P. designed, generated, and genotyped cross MRxSM, and edited the manuscript; and J.A.S. and D.G. designed, generated, phenotyped, and genotyped cross BxWSB, analyzed data, and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Luanne L. Peters, The Jackson Laboratory, 600 Main St, Bar Harbor, ME 04609; e-mail: luanne.peters@jax.org.
References
- 1.Garner C, Tatu T, Reittie JE, et al. Genetic influences on F cells and other hematologic variables: a twin heritability study. Blood. 2000;95(1):342–346. [PubMed] [Google Scholar]
- 2.Liu P, Vikis H, Lu Y, Wang D, You M. Large-scale in silico mapping of complex quantitative traits in inbred mice. PLoS ONE. 2007;2(7):e651. doi: 10.1371/journal.pone.0000651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peters LL, Lambert AJ, Zhang W, Churchill GA, Brugnara C, Platt OS. Quantitative trait loci for baseline erythroid traits. Mamm Genome. 2006;17(4):298–309. doi: 10.1007/s00335-005-0147-3. [DOI] [PubMed] [Google Scholar]
- 4.Soranzo N, Spector TD, Mangino M, et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet. 2009;41(11):1182–1190. doi: 10.1038/ng.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Penninx BW, Pahor M, Woodman RC, Guralnik JM. Anemia in old age is associated with increased mortality and hospitalization. J Gerontol A Biol Sci Med Sci. 2006;61(5):474–479. doi: 10.1093/gerona/61.5.474. [DOI] [PubMed] [Google Scholar]
- 6.Guralnik JM, Eisenstaedt RS, Ferrucci L, Klein HG, Woodman RC. Prevalence of anemia in persons 65 years and older in the United States: evidence for a high rate of unexplained anemia. Blood. 2004;104(8):2263–2268. doi: 10.1182/blood-2004-05-1812. [DOI] [PubMed] [Google Scholar]
- 7.Castro O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood. 1994;84(2):643–649. [PubMed] [Google Scholar]
- 8.Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91(1):288–294. [PubMed] [Google Scholar]
- 9.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet. 2009;10(4):241–251. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]
- 11.Su Z, Korstanje R, Tsaih SW, Paigen B. Candidate genes for obesity revealed from a C57BL/6J × 129S1/SvImJ intercross. Int J Obes (Lond) 2008;32(7):1180–1189. doi: 10.1038/ijo.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burgess-Herbert SL, Tsaih SW, Stylianou IM, Walsh K, Cox AJ, Paigen B. An experimental assessment of in silico haplotype association mapping in laboratory mice. BMC Genet. 2009;10:81. doi: 10.1186/1471-2156-10-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robledo RF, Ciciotte SL, Gwynn B, et al. Targeted deletion of alpha-adducin results in absent beta- and gamma-adducin, compensated hemolytic anemia, and lethal hydrocephalus in mice. Blood. 2008;112(10):4298–4307. doi: 10.1182/blood-2008-05-156000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shavit JA, Manichaikul A, Lemmerhirt HL, Broman KW, Ginsburg D. Modifiers of von Willebrand factor identified by natural variation in inbred strains of mice. Blood. 2009;114(26):5368–5374. doi: 10.1182/blood-2009-07-233213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broman KW, Wu H, Sen S, Churchill GA. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 2003;19(7):889–890. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- 16.Cox A, Ackert-Bicknell CL, Dumont BL, et al. A new standard genetic map for the laboratory mouse. Genetics. 2009;182(4):1335–1344. doi: 10.1534/genetics.109.105486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basrak B, Klaassen CA, Beekman M, Martin NG, Boomsma DI. Copulas in QTL mapping. Behav Genet. 2004;34(2):161–171. doi: 10.1023/B:BEGE.0000013730.63991.ba. [DOI] [PubMed] [Google Scholar]
- 18.Li M, Boehnke M, Abecasis GR, Song PX. Quantitative trait linkage analysis using Gaussian copulas. Genetics. 2006;173(4):2317–2327. doi: 10.1534/genetics.105.054650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sen S, Churchill GA. A statistical framework for quantitative trait mapping. Genetics. 2001;159(1):371–387. doi: 10.1093/genetics/159.1.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Solberg LC, Baum AE, Ahmadiyeh N, et al. Sex- and lineage-specific inheritance of depression-like behavior in the rat. Mamm Genome. 2004;15(8):648–662. doi: 10.1007/s00335-004-2326-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138(3):963–971. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11(3):241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 23.Li R, Lyons MA, Wittenburg H, Paigen B, Churchill GA. Combining data from multiple inbred line crosses improves the power and resolution of quantitative trait Loci mapping. Genetics. 2005;169(3):1699–1709. doi: 10.1534/genetics.104.033993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peters LL, Robledo RF, Bult CJ, Churchill GA, Paigen BJ, Svenson KL. The mouse as a model for human biology: a resource guide for complex trait analysis. Nat Rev Genet. 2007;8(1):58–69. doi: 10.1038/nrg2025. [DOI] [PubMed] [Google Scholar]
- 25.Frazer KA, Wade CM, Hinds DA, Patil N, Cox DR, Daly MJ. Segmental phylogenetic relationships of inbred mouse strains revealed by fine-scale analysis of sequence variation across 4.6 mb of mouse genome. Genome Res. 2004;14(8):1493–1500. doi: 10.1101/gr.2627804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsaih SW, Pezzolesi MG, Yuan R, Warram JH, Krolewski AS, Korstanje R. Genetic analysis of albuminuria in aging mice and concordance with loci for human diabetic nephropathy found in a genome-wide association scan. Kidney Int. 2010;77(3):201–210. doi: 10.1038/ki.2009.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szatkiewicz JP, Beane GL, Ding Y, Hutchins L, Pardo-Manuel de Villena F, Churchill GA. An imputed genotype resource for the laboratory mouse. Mamm Genome. 2008;19(3):199–208. doi: 10.1007/s00335-008-9098-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bustin SA, Benes V, Garson JA, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55(4):611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 29.Nolan T, Hands RE, Ogunkolade W, Bustin SA. SPUD: a quantitative PCR assay for the detection of inhibitors in nucleic acid preparations. Anal Biochem. 2006;351(2):308–310. doi: 10.1016/j.ab.2006.01.051. [DOI] [PubMed] [Google Scholar]
- 30.Ruijter JM, Ramakers C, Hoogaars WM, et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009;37(6):e45. doi: 10.1093/nar/gkp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8(2):R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akeson EC, Donahue LR, Beamer WG, et al. Chromosomal inversion discovered in C3H/HeJ mice. Genomics. 2006;87(2):311–313. doi: 10.1016/j.ygeno.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 33.Burgess-Herbert SL, Cox A, Tsaih SW, Paigen B. Practical applications of the bioinformatics toolbox for narrowing quantitative trait Loci. Genetics. 2008;180(4):2227–2235. doi: 10.1534/genetics.108.090175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manenti G, Galvan A, Pettinicchio A, et al. Mouse genome-wide association mapping needs linkage analysis to avoid false-positive Loci. PLoS Genet. 2009;5(1):e1000331. doi: 10.1371/journal.pgen.1000331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan R, Tsaih SW, Petkova SB, et al. Aging in inbred strains of mice: study design and interim report on median lifespans and circulating IGF1 levels. Aging Cell. 2009;8(3):277–287. doi: 10.1111/j.1474-9726.2009.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erhart MA, Simons KS, Weaver S. Evolution of the mouse beta-globin genes: a recent gene conversion in the Hbbs haplotype. Mol Biol Evol. 1985;2(4):304–320. doi: 10.1093/oxfordjournals.molbev.a040353. [DOI] [PubMed] [Google Scholar]
- 37.Ganesh SK, Zakai NA, van Rooij FJ, et al. Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat Genet. 2009;41(11):1191–1198. doi: 10.1038/ng.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Storz JF, Baze M, Waite JL, Hoffmann FG, Opazo JC, Hayes JP. Complex signatures of selection and gene conversion in the duplicated globin genes of house mice. Genetics. 2007;177(1):481–500. doi: 10.1534/genetics.107.078550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berry RJ. Genetic variation in wild house mice: where natural selection and history meet. Am Sci. 1978;66(1):52–60. [PubMed] [Google Scholar]
- 40.Newton MF, Peters J. Physiological variation of mouse haemoglobins. Proc R Soc Lond B Biol Sci. 1983;218(1213):443–453. doi: 10.1098/rspb.1983.0050. [DOI] [PubMed] [Google Scholar]
- 41.Hempe JM, Ory-Ascani J, Hsia D. Genetic variation in mouse beta globin cysteine content modifies glutathione metabolism: implications for the use of mouse models. Exp Biol Med (Maywood) 2007;232(3):437–444. [PubMed] [Google Scholar]
- 42.Abiola O, Angel JM, Avner P, et al. The nature and identification of quantitative trait loci: a community's view. Nat Rev Genet. 2003;4(11):911–916. doi: 10.1038/nrg1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olivieri O, De Franceschi L, Capellini MD, Girelli D, Corrocher R, Brugnara C. Oxidative damage and erythrocyte membrane transport abnormalities in thalassemias. Blood. 1994;84(1):315–320. [PubMed] [Google Scholar]
- 44.Olivieri O, Vitoux D, Galacteros F, et al. Hemoglobin variants and activity of the (K+Cl-) cotransport system in human erythrocytes. Blood. 1992;79(3):793–797. [PubMed] [Google Scholar]
- 45.Romero JR, Suzuka SM, Nagel RL, Fabry ME. Expression of HbC and HbS, but not HbA, results in activation of K-Cl cotransport activity in transgenic mouse red cells. Blood. 2004;103(6):2384–2390. doi: 10.1182/blood-2003-01-0237. [DOI] [PubMed] [Google Scholar]
- 46.Solovieff N, Milton JN, Hartley SW, et al. Fetal hemoglobin in sickle cell anemia: genome-wide association studies suggest a regulatory region in the 5′ olfactory receptor gene cluster. Blood. 2010;115(9):1815–1822. doi: 10.1182/blood-2009-08-239517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Paigen B. Genetics of variation in HDL cholesterol in humans and mice. Circ Res. 2005;96(1):27–42. doi: 10.1161/01.RES.0000151332.39871.13. [DOI] [PubMed] [Google Scholar]
- 48.Garrett MR, Pezzolesi MG, Korstanje R. Integrating human and rodent data to identify the genetic factors involved in chronic kidney disease. J Am Soc Nephrol. 2010;21(3):398–405. doi: 10.1681/ASN.2009080881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ackert-Bicknell CL, Karasik D, Li Q, et al. Mouse BMD quantitative trait loci show improved concordance with human genome wide association loci when recalculated on a new, common mouse genetic map. J Bone Miner Res. 2010;25(8):1808–1820. doi: 10.1002/jbmr.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kamatani Y, Matsuda K, Okada Y, et al. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat Genet. 2010;42(3):210–215. doi: 10.1038/ng.531. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}