

Abstract
Aging is a risk factor for heart failure, which is a leading cause of death world-wide. Elderly patients are more likely than young patients to experience a myocardial infarction (MI) and are more likely to develop heart failure following MI. The poor clinical outcome of aging in cardiovascular disease is recapitulated on the cellular level. Increase in stress exposure and shifts in signaling pathways with age change the biology of cardiomyocytes. The progressive accumulation of metabolic waste and damaged organelles in cardiomyocytes blocks the intracellular recycling process of autophagy and increases the cell’s propensity toward apoptosis. Additionally, the decreased cardiomyocyte renewal capacity in the elderly, due to reduction in cellular division and impaired stem cell function, leads to further cardiac dysfunction and maladaptive responses to disease or stress. We review the cellular and molecular aspects of post-infarction remodeling in the aged heart, and relate them to the clinical problem of post-infarction remodeling in elderly patients.
Keywords: aging, left ventricular remodeling, myocardial infarction
Myocardial Infarction (MI), Left Ventricular (LV) Remodeling, and Heart Failure
In the U.S., chronic heart failure (CHF) affects approximately 5.7 million people (1), and has been implicated as an underlying or contributing cause of death in 282,000 individuals per year at an estimated cost of $39 billion annually (2).
MI is one of the most common causes of CHF (2). The way in which MI leads to heart failure is a complex multifactorial process known as LV remodeling. MI, caused by occlusion of an epicardial coronary artery, leads within hours to irreversible death of the cardiomyocytes in the distribution supplied by that artery. MI also initiates a cascade of neurohormonal changes that attempt to compensate for the lack of contractile function caused by the MI. This initially maintains cardiac output and perfusion to the vital organs, but with time, these compensatory mechanisms fail, and there is progressive deterioration of cardiac function. The end result is a dilated, poorly functioning ventricle, and the clinical syndrome of heart failure ensues (3). At the organ level, important pathological features of post-infarction LV remodeling include infarct expansion, myocardial hypertrophy, cardiac fibrosis, and global ventricular dilation (3). However, at the cellular level, the precise pathophysiological changes associated with LV remodeling remain incompletely characterized. Several molecular and cellular processes appear to be particularly important, including ongoing apoptosis of cardiac myocytes, particularly at the border zone of the MI, dysfunctional cardiomyocyte autophagy, and reduction in the proliferative capacity of new cardiomyocytes. This ongoing cell loss and inability to replace lost cells may contribute to the deterioration in cardiac function (Fig. 1). These cardiac changes are particularly pronounced in the elderly. As our population ages (4), the burden of heart failure is likely to increase. Developing targeted therapies for post-infarction heart failure in the elderly will require investigation into the specific pathophysiological changes of LV remodeling in this age group.
Figure 1. Post-MI Remodeling of the Heart at Cellular and Organ Levels.
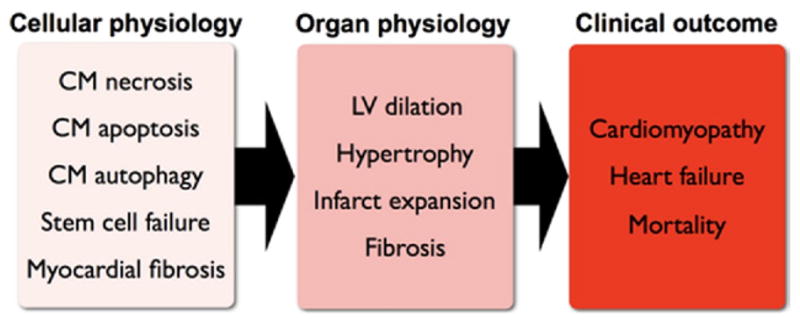
CM = cardiomyocyte; LV = left ventricular.
Epidemiology of MI and Heart Failure With Aging
Aging and MI
The incidence of MI increases dramatically with age. In the Framingham Heart Study, the incidence of MI more than doubles for men and increases more than 5-fold in women from the age group of 55 to 64 years to the age group of 85 to 94 years (5). This increased incidence with age is also seen in the ARIC (Atherosclerosis Risk in Communities) surveillance study (5) (Fig. 2). Elderly patients are not only more likely than young patients to experience an MI, but are more likely to die from their MI. The GISSI-2 (Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico-2) study of thrombolysis for reperfusion of first MI demonstrated an increase in the rate of cardiac rupture with age, as well as an increase in both in-hospital and post-discharge mortality rates with increasing age. In this study, each year of age was associated with a 6% increase in mortality (6). Older patients have a higher likelihood of cardiac arrest following MI (7) and are more likely to develop mechanical complications of MI, including papillary muscle rupture, LV free wall rupture, and acquired ventricular septal defect (8).
Figure 2. Increased Rates of MI With Age.

Incidence of myocardial infarction (MI) increases with age, and is higher in men than in women. Data derived from ARIC (Atherosclerosis Risk in Communities) surveillance (5).
During MI, prompt reperfusion of the infarct-related artery is critical to prevent infarct expansion. Several large randomized controlled clinical trials have shown that timely reperfusion with thrombolysis (9,10) or percutaneous coronary intervention (PCI) (11) limits the extent of myocardial necrosis, and decreases acute and long-term mortality. However, elderly patients are under-represented in clinical trials and real-world registries of reperfusion therapies for MI (12,13). This may be for several reasons. First, elderly patients are at higher risk for procedural complications from PCI (14), and operators may be less inclined to perform PCI on older patients, particularly in the era of public reporting of figures. Second, there is ongoing debate about whether these therapies are effective in older persons (15,16). Third, older patients often present with atypical or no symptoms, or with left bundle branch block rather than ST-segment elevation on electrocardiogram (ECG) (12), thus delaying time to diagnosis and treatment. Finally, the decision not to treat older patients may be based on physicians’ perception of bleeding risk (13), or just therapeutic nihilism about the long-term prognosis of elderly patients. However, a randomized controlled trial of PCI versus fibrinolysis in elderly patients appeared to show a benefit of PCI (17), and in elderly patients with cardiogenic shock, there appears to be benefit in PCI (18,19).
Aging and CHF
The prevalence of CHF increases with age. It rises from 1.9% of men and 1.4% of women at ages 40 to 59 years, to 14.7% of men and 12.8% of women over the age of 80 years (2). Precise causes for the increased incidence of heart failure in the elderly remain unknown. Many risk factors for developing CHF, such as diabetes and hypertension, are more prevalent in elderly populations and have been reviewed elsewhere (20,21). Despite this, MI remains one of the leading causes of CHF in the U.S. (2). Elderly patients are not only more likely to experience MI, they are also more likely to develop heart failure as a consequence of their MI than younger patients (22). As survival from MI increases, the prevalence of heart failure in those patients who survive MI is also increasing (23). In our aging population, the higher incidence of MI will translate into an increasing burden of post-infarction heart failure on our health care system. Therapies specifically designed to limit ventricular remodeling in elderly populations and stem the tide of increasing heart failure prevalence are needed.
Cellular and Molecular Aspects of Ventricular Remodeling
Most animal experiments of MI created by left anterior descending coronary artery (LAD) ligation are performed in young adult or adolescent animals, rather than in middle-aged or older animals. This is in direct contradiction to the human equivalent, where MI is a disease of older adults. In a mouse model of MI, older animals had dramatically worse outcomes compared with young animals. Gould et al. (24) performed LAD ligation on young adult mice (2 months of age), middle-aged mice (10 to 12 months of age), and older mice (14 months of age). They demonstrated an age-dependent reduction in survival in the month following MI. Additionally, the older mice demonstrated greater infarct expansion and septal hypertrophy following MI. Importantly, the changes seen in the older mice could be improved with therapy, in this case, angiotensin-converting enzyme inhibitor (ACE inhibitor) captopril. This highlights the fact that although older animals fare worse following MI than their younger counterparts, this poor prognosis is able to be manipulated with pharmacologic therapies. Therefore, more research into the cellular and molecular events following MI in the elderly may lead to more effective therapies for older patients who suffer an MI.
Cardiomyocyte apoptosis
Apoptosis is an evolutionarily conserved and highly regulated mechanism that results in the death and organized removal of unnecessary or malfunctioning cells. Morphologically, the process is characterized by regulated cellular shrinkage, chromatin condensation, DNA fragmentation, endocytosis of the dead cell by neighboring cells with no spillage of intracellular content nor inflammatory response (25). Cells undergo physiological self-induced death during the developmental process, and as a part of the renewal mechanism to maintain the balance in the number of cells. Additionally, apoptosis is considered to be an antitumor mechanism that keeps cells with DNA damage from proliferating and thus preventing tumorgenesis (26). Aside from its physiological roles in development and homeostasis, apoptosis can also be activated in pathological states (Fig. 3).
Figure 3. Aging of Cardiomyocytes.

Aging is characterized with decreased cardiomyocyte renewal, possibly through the impairment of both the stem cell differentiation into cardiomyoctyes and cardiomyocyte division. Additionally, changes in cardiomyocyte microenvironment and signaling pathways increase the cell’s propensity toward apoptosis. Progressive accumulation of damaged protein and organelles in cardiomyoctyes blocks autophagy mechinary. Failure to clear out the intracellular waste can induce apoptosis of the cell. Compensatory neurohormonal signals induce the remaining cardiomyocytes to undergo hypertrophy and increase metabolic demand, thereby exposing the cellular contents to oxidative damage. Regular arrows = promotion/induction; blocked arrows = inhibition; lightning = detrimental effects of aging. Figure illustration by Craig Skaggs.
For a long time, cardiomyocytes were considered long-lasting cells that do not self-renew. Analysis of cardiomyocyte apoptosis and the observation of cardiomyocyte hypertrophy following events of massive cell death support such theory. At a basal rate of only 10 ± 9 per 100,000 cells (0.01%) committed to programmed cell death at any time (27), it was presumed that the majority of cardiomyocytes share the same lifespan as the organism. In conditions such as ischemic cardiomyopathy or idiopathic dilated cardiomyopathy, the reported extent of cardiomyocyte apoptosis increased to as much as 2% of cardiomyocyte nuclei being apoptotic at the time of the sample collection (27). Even during disease states, the level of cardiomyocyte apoptosis is still low compared with other organs. The liver, for instance, can sustain above 20% apoptosis in hepatocytes without lethal consequence, and the difference is not due to the difference in time taken to complete apoptosis (28). The precise cause for the basal rate of cardiomyocyte apoptosis in the healthy heart is not understood; however, one can speculate on various reasons. Normal cellular stress from day-to-day work may result in some specifically stressed cells, whose gene expression shifts towards up-regulation of pro-apoptotic profile and towards down-regulation of anti-apoptotic genes. Upon reaching a tipping point for apoptosis, the cell falls into self-termination. Alternatively, stochastic accumulation of mutation and errors in protein synthesis and accumulation of misfolded proteins can elicit endoplasmic reticulum stress response that results in activation of caspase cascade (29,30).
The effects of aging predispose cardiomyocytes to apoptosis (31). Cardiomyocytes are exposed to oxidative damage from a high level of metabolism and mechanical stress from constant beating, and yet the low turnover rate requires the majority of them to survive through a human’s lifetime. At the cellular level, the footprint of oxidative damaging in aging leaves cardiomyocytes with a high level of disrupted mitochondria, lipofuscin, and other damaged organelles (32). In addition to the accumulation of somatic wear-and-tear waste, aging cells and long-lived cells such as cardiomyocytes are associated with increased nuclear and mitochondrial DNA damage (33). Together, these molecular characteristics can lead to an increase in pro-apoptotic gene expression profile across the heart. In fact, without the presence of heart disease, an aging human heart on average already loses more than 30% of cardiomyocytes by chronic loss of cells (34). Compounding an aging heart with the extra burden of cardiovascular disease leads to a greater rate of cardiomyocyte loss (35) and worsens the outcome of LV remodeling. Figure 4 demonstrates several possible pathways affected by aging, whereby age may induce cardiomyocyte apoptosis (36 – 43).
Figure 4. Cardiomyocyte Apoptosis.

Apoptosis can be mediated by the intrinsic or extrinsic pathways. Many factors associated with these pathways are increased with aging, resulting in a predisposition toward apoptosis in the aging cardiomyocyte. Intrinsically, apoptosis is activated in the event of hypoxia, oxidative stress, DNA damage, acidosis, or growth factor deprivation (36,37). These stressors activate cell death signaling chiefly through Bcl-2 family–regulated increase of mitochondrial outer membrane permeabilization (MOMP). Cytochrome C from mitochondria diffuses into the cytoplasm following the increase in mitochondrial membrane permeability. In the cytosol, cytochrome C activates a downstream cascade to initiate caspase activities (38,39). Active caspases target nuclear lamins, nuclear inhibitor of caspase-activated deoxyribonuclease/DNA fragmentation factor-45 (ICAD/DEF45), and various other cytoplasmic proteins. Cleavage of nuclear lamins breaks down the nuclear structure (40). Cleavage of ICAD/DEF45, a DNase inhibitor, disinhibits DNase, thereby allowing it to break down DNA (41). Extrinsic apoptosis can be activated by Fas ligand or tumor necrosis factor (TNF)-alpha. Binding of TNF-alpha or Fas ligand to their respective receptors activates TNF receptor–associated death domain (TRADD) and Fas-associated death domain (FADD) proteins (42,43). These proteins act on regulators of mitochondria to change the permeability of the mitochondrial outer membrane, and this change converges with intrinsic apoptosis activation on the caspase cascade. Figure illustration by Craig Skaggs.
Autophagy
Autophagy, or self-eating, is a process in which the cell degrades its own components. Autophagy is a mechanism for the degradation of damaged long-lived proteins and organelles, and is a survival response to starvation. Autophagy is essential for cardiomyocytes. The longevity of cardiomyocytes and their high energy requirements unavoidably expose the cell to a high level of cumulative oxidative damage and stress. Meanwhile, since most cardiomyocytes do not divide, halving cellular waste through mitosis is not an option. Effective autophagy in cardiomyocytes is therefore necessary to continue normal metabolism and to maintain cellular survival. Autophagic failure is known to cause cardiomyopathy in experimental and clinical models. For instance, experimental disruption of the autophagy gene atg5 in the heart causes an accumulation of damaged proteins and leads to hypertrophy, subsequent dilated cardiomyopathy, and heart failure (44).
Although functional autophagy is crucial for normal functioning of cardiomyocytes, the effects of MI on autophagy, and conversely the effects of autophagy on the heart following MI, remain incompletely described. In hearts subjected to reperfused MI, autophagy is activated first by the AMP activated protein kinase (AMPK)-dependent mechanism during the ischemic period, followed by Beclin-1–dependent autophagy after reflow is established (45). In this model, autophagy promotes the survival of cardiomyocytes during the AMPK-dependent (ischemic) phase, but becomes detrimental during the Beclin-1–dependent (reflow) phase.
Cardiomyocytes’ requirement for autophagy increases with age. Damage from reactive oxygen species impairs functional organelles, which then need to be cleared out by autophagy in order to prevent cytotoxic effects. Such damage is especially pronounced with aging. For instance, long-term accumulation of oxidative damage results in mitochondria that are structurally disorganized, enlarged, disabled for energy production, and prone to reactive oxygen species leakage, posing oxidative damage for other cytosolic proteins (46). Moreover, senescence-related damages, such as mutation accumulation and protein aggregation, impair mitochondrial fission (47), which also serves as a waste-clearing mechanism by halving the amount of cytosolic waste. Aging is also associated with reduced autophagy efficiency (Fig. 5) (48,49). For instance, lipofusin, or pigmented waste from incomplete lysosomal digestion that accumulate in aging hearts, can disrupt lysosomal turnover. Lysosomes committed to degrading lipofusin are unable to complete the digestion, and therefore are also unavailable for other uses (50), resulting in a net loss of intracellular lysosomal availability and a bottleneck in autophagy flux. With autophagy impaired, biological waste such as reactive oxygen species can cause additional protein and organelle damage. Impaired autophagy and the accumulation of toxic biological waste together result in a vicious cycle leading to defective cells.
Figure 5. Cardiomyocyte Autophagy.

Autophagy may be triggered by the accumulation of intracellular protein aggregation (48), nutrient deprivation (49), or various intracellular signals. The process initiates with a highly regulated activation step that forms a double membrane autophagosome, which engulfs damaged proteins and organelles. Autophagosomes then fuse with the lysosome, which contain proteases to digest the engulfed content. Aging increases the cardiomyocyte’s need for autopahgy to maintain intracellular homeostasis, but simultaneously reduces the activity of lysosomes and thereby inhibits autophagic flux. Figure illustration by Craig Skaggs.
Cardiomyocyte hypertrophy
Classical paradigm holds that human cardiomyocytes proliferate only through the developmental stages shortly after birth. The growth of the heart in the later part of life is mediated chiefly through cardiomyocyte hypertrophy, or increase in cell size (51). Under the influence of hemodynamic load, neurohormonal factors, or prohypertrophic signals, cardiomyocytes are capable of significant growth in both transverse and longitudinal directions (52). Cardiomyocyte hypertrophy is pronounced in an aging heart and contributes to the development of a dysfunctional heart (53). Clinically, hypertrophy has been a strong predictor of morbidity and mortality for cardiovascular disease (54). Hypertrophy has been considered maladaptive, and can be targeted by interventions to protect against further adverse remodeling which would set the stage for eventual heart failure (55).
Cardiomyocyte hypertrophy can be considered a consequence of a shift in the balance between atrophy and hypertrophy signaling (56). The currently known prohypertrophic signaling mechanisms have been reviewed with possible targets for interventions. Briefly, hypertrophy can be activated by biomechanical stress in response to hemodynamic load increase, G protein pathways in response to extracellular signaling molecules, peroxisome proliferator activation in response to fatty acid oxidation, and phosphoinositide 3-kinase in response to Akt signaling (57).
An exception where hypertrophy has no association with poor clinical prognosis is in athletes’ hearts. Differences between pathological hypertrophy (in diseased or aging hearts) and physiological hypertrophy (in athletes’ hearts) have been recently reviewed in great depth (58). Moreover, it is certainly possible that enlargement of cardiomyocytes due to pathological responses is not the sole cause of dysfunctional hearts. For instance, the increase in protein synthesis in response to hypertrophic signaling in the context of inefficient autophagy and cytosol awash with oxidative agents leaked from damaged mitochondria can lead to the runaway accumulation of damaged protein that strains the already inefficient autophagy machinery. Moreover, an increase in the energy demands of hypertrophied cardiomyocytes require more energy output from a pool of poorly performing and highly damaged mitochondria, which can further wear down the already stressed mitochondria, causing cytotoxic effects such as leakage of reactive oxygen species or cytochrome C release, resulting in cardiomyocyte apoptosis (Fig. 3).
Renewal failure
The number of cardiomyocytes in both the rodent and human heart declines with age (59). The very low rate of cell death in a normal heart, occurring in approximately 0.001% to 0.01% of cells at any given time point, as well as the availability of cardiomyocyte hypertrophy and fibrosis as compensatory mechanisms for cell loss in aging hearts, were thought to reduce the requirement for cardiomyocyte regeneration (59). Additionally, after events such as coronary ischemia that lead to massive loss of cardiomyocytes, hearts are left with permanent scarring and an irreversible decrease in function. Until recently, there was no firm evidence to support the existence of cardiomyocyte renewal. Now it appears there are 2 sources for renewal of the cardiomyocyte pool: division of existing adult cardiomyocytes, and differentiation of precursor cells (or stem cells) into new cardiomyocytes (60,61). Both of these processes may be subject to the ravages of aging.
Adult cardiomyocytes possess a limited capacity for proliferation (62). The rarity of cardiomyocyte mitosis at baseline, estimated at around 14 cells per million (63), makes the role of cardiomyocyte renewal in aging difficult to study or speculate upon using current laboratory techniques. Interestingly, this number is within the same order of magnitude to the apoptotic index found in a normal adult heart (27). Assuming the time course of mitosis is similar to apoptosis (both process can be completed in the order of hours), the mitotic and apoptotic indexes suggest a significant contribution to cardiomyocyte renewal through cell division. It is then tempting to speculate whether the reduction of cardiomyocyte renewal with aging is a result of reduction in cardiomyocyte mitosis. Many factors may lead to cell cycle arrest in aging, including telomere shortening, mutation accumulation, oxidative stress, altered gene expression, and paracrine signaling, as reviewed by Campisi et al. (64). The overlap of these factors with senescent cardiomyocyte phenotype may provide possible targets for antiaging therapy.
Stem cells or precursor cells can give rise to all the cell types within the adult heart (65). Furthermore, many types of stem or precursor cells have been used to regenerate cardiac tissue in experimental models. These include bone marrow–derived cells (66), mesenchymal stem cells (67), umbilical cord blood cells (68), adipose-derived stem cells (69), endothelial progenitor cells (70), and resident cardiac progenitor cells (CPC) (71). It is known that stem cells, which are popularly considered to be the fountain of youth, can be drained with aging. Old stem cells are associated with replicative senescence or stress-induced senescence, rendering the cells incapable of further division and self-renewal (72). Replicative senescence involves the shortening of the telomere, a repeated sequence capping the end of chromosomes. Telomeres protect the integrity of coding sequences on chromosomes from the unavoidable loss of DNA content due to the inability to synthesize DNA on the lagging strand at its very end (73). Increasing metabolic assaults due to aging can also induce cellular senescence by arresting cells from entering cycle (64). It has been found that CPC senesce as well. Phenotypes of senescence such as telomere shortening and accumulation of p16INK4a are observed in CPC from aged and diseased hearts (74), which may lead to a decrease in regenerative capacity.
A recent groundbreaking work to pinpoint the extent of cardiomyocyte renewal in aging was carried out by a global scale pulse-chase experiment. By taking advantage of the decade-long surge in nuclear bomb testing, which leads a pulse that incorporates 14C in DNA, and examining the post-mortem 14C content in cardiomyocyte nuclei, Bergmann et al. were able to establish a mathematical model to describe cardiomyocyte renewal in the human heart (75). Their model suggests that in young hearts, 1% of cardiomyocytes are renewed annually. The rate decreases to 0.45% in aging hearts. Aging cardiomyocytes, however, do not exhibit a lower rate of cell death compared with the young ones (31); therefore, the low cardiomyocyte turnover phenomenon more likely reflects the decrease in the formation of new cells. Reduced cardiomyocyte renewal in aging may represent a therapeutic target in post-infarction LV remodeling in elderly patients.
Aging of other cell types
Aging of the heart as an organ is certainly not confined to the aging of cardiomyocytes alone. Although cardiomyocytes make up most of the mass of the heart, numerically they constitute only one-half of the cell population. Cardiac fibroblasts, endothelial cells lining capillaries, and smooth muscle cells make up most of the remaining one-half of the cell population (76). Cardiac fibroblasts are responsible for laying out the cardiac tissue scaffold through their production, maintainance, and remodeling of extracellular matrix (ECM). Fibroblasts from aged hearts have impaired proliferative capacity (77) and a blunted response to profibrotic stimuli in vitro (78). Although aged hearts have more fibrosis than young hearts at baseline, a pathological finding, when rapid elaboration of fibrous tissue is necessary, such as following MI, the aged fibroblast is unable to respond adequately. Aging hearts display reduced collagen deposition in the scar, delayed stablization of scarring, and reduced but prolonged inflammation (78), suggesting that cardiac fibroblasts become dysfunctional with age. This may contribute to increased risk of cardiac rupture following MI in the elderly. Little is known about the effects of age on endothelial cells and smooth muscle cells in the post-infarction setting. Last, aging of cardiac stem cells, which have shown to posess differentiation capacity into various cardiac cell lineages (71), may contribute to the deterioration and reduction in regeneration of nonmuscle cells in myocardium.
In addition to cellular changes with age, the ECM also changes. Age is associated with increased expression of several ECM proteins, including collagen, fibronectin, and alpha 1 and alpha 5 integrin (79). Furthermore, there is increased collagen cross-linkng with advanced age, which can contribute to ventricular stiffness, and this can be modulated with exercise (80). Just as the function of a heart is achieved by the interactions of its various cell types, studying the aging heart requires consideration of the age-related change in ECM signaling, as well as cell–cell interactions.
Clinical Sequelae of Cellular Changes in LV Remodeling With Age
The molecular and cellular changes described in the previous text have direct effects on cardiac function, and thereby on patient outcomes. Loss of cardiomyocytes with age appears to be due to 2 separate mechanisms: loss of cells due to wear-and-tear that are not replaced (impaired cardiomyocyte division and cardiac stem cell senescence) and active loss of cells due to activation of apoptosis. In addition, cardiomyocyte function is impaired with age. The result is a predisposition toward LV impairment and heart failure at baseline. Furthermore, with the stress of MI, there is resultant inability of the aged heart to cope, and post-infarction LV remodeling is more pronounced. This translates into the higher clinical incidence of post-infarction heart failure in elderly patients.
Cardiac fibroblast dysfunction with age contributes to higher baseline levels of fibrosis, which likely contributes to diastolic heart failure in the elderly. Following MI, aged fibroblasts are unable to elaborate a strong, mature infarct scar, and there is resultant infarct expansion, heart failure, and excess mortality, as seen in animal models and clinical studies described in the previous text. Table 1 summarizes these changes.
Table 1.
Effects of Aging on the Heart
| Cellular and Molecular Changes | Effect of Aging* | Clinical Effects at Baseline | Clinical Effects After MI |
|---|---|---|---|
| Cardiomyocyte survival | ↓ | Impaired systolic function | LV dilation, eccentric remodeling, and systolic dysfunction |
| Cardiomyocyte proliferation | ↓ | Impaired systolic function | LV dilation, eccentric remodeling, and systolic dysfunction |
| Cardiac stem cell number | ↓ | Impaired systolic function | LV dilation, eccentric remodeling, and systolic dysfunction |
| Cardiomyocyte diameter | ↑ | LV hypertrophy | Increased wall stress, concentric remodeling |
| Myocardial fibrosis | ↑ | Diastolic dysfunction | Fibrosis of noninfarct zone, increased arrhythmias |
| Dysfunctional fibroblasts | ↑ | Diastolic dysfunction | Infarct expansion |
Downward-pointing arrow (↓) indicates a decrease in effects; upward-pointing arrow (↑) indicates an increase.
LV = left ventricular; MI = myocardial infarction.
Management of Elderly Patients With Post-Infarction LV Remodeling
Guidelines from the American College of Cardiology (ACC) and American Heart Association (AHA) for management of patients with ST-segment elevation MI (81), and the 2009 update (82) make no distinction based on age for the assessment and initial management of MI. All patients with ST-segment elevation MI should undergo rapid evaluation for reperfusion therapy and have a reperfusion strategy implemented promptly after contact with the medical system (Level of Evidence: A). Elderly patients appear to have a modest benefit from PCI over fibrinolysis, although the benefit is mainly in recurrent ischemia (17,83). This suggests that suitable elderly patients can be offered primary PCI and may benefit from this approach. The class of recommendation for both primary PCI and rescue PCI in patients with cardiogenic shock is Class II in patients over 75 years of age and Class I in younger patients. This is mainly because younger patients demonstrated clear benefit in the SHOCK (Should We Emergently Revascularize Occluded Coronaries for Cardiogenic Shock) trial (84), whereas older patient data are based on registries (18,19). When PCI is not available and fibrinolysis is the treatment of choice for reperfusion, elderly patients have improved outcomes when treated with fibrinolytic agents compared with placebo (10), but this benefit may not extend beyond 85 years of age.
The guidelines for standard adjunctive therapies during the index admission, such as aspirin, beta-blockers, and antiplatelet agents, make no differential recommendations based on age. However, the addition of prasugrel in the 2009 update comes with a Federal Drug Administration warning about use in those 75 years of age or older, because of lack of benefit and increased risk of intracranial hemorrhage in this age group (85). In addition, low molecular weight heparin should not be used as an alternative to unfractionated heparin in combination with fibrinolytic therapy in patients over 75 years of age.
After reperfusion of MI, therapies to prevent and treat LV remodeling include beta-blockers, ACE inhibitors, aldosterone antagonists, and cardiac rehabilitation. In the ACC/AHA guidelines, these are recommended for all, with no stipulation on age. Aspirin, ACE inhibitors, beta-blockers, and statins appear to be at least as effective in elderly patients as in younger patients following MI (12). In summary, with the exception of prasugrel and low molecular weight heparin after fibrinolysis, elderly patients should be considered for the same therapies as young patients in the setting of acute MI and for prevention of long-term adverse LV remodeling.
Acknowledgments
This work has been supported by grants from the NIH K08HL090915-01 and the Ellison Medical Foundation AG-NS-0507-08.
Abbreviations and Acronyms
- ACE
angiotensin-converting enzyme
- CHF
chronic heart failure
- CPC
cardiac progenitor cell
- ECM
extracellular matrix
- LV
left ventricle/ventricular
- MI
myocardial infarction
- PCI
percutaneous coronary intervention
Footnotes
The authors have reported that they have no relationships to disclose.
References
- 1.National Institutes of Health. [Accessed June 1, 2010];National Heart Lung and Blood Institute Factbook Fiscal Year 2008. Available at: http://www.nhlbi.nih.gov/about/factbook/FactBookFinal.pdf.
- 2.Lloyd-Jones D, Adams RJ, Brown TM, et al. Heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation. 2010;121:e46–215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 3.Yousef ZR, Redwood SR, Marber MS. Postinfarction left ventricular remodelling: where are the theories and trials leading us? Heart. 2000;83:76–80. doi: 10.1136/heart.83.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Administration on Aging. A Profile of Older Americans. Department of Health and Human Services; 2008. [Accessed June 1, 2010]. Available at: http://www.csrees.usda.gov/nea/economics/pdfs/profile_older_american_2008.pdf. [Google Scholar]
- 5.National Heart, Lung, and Blood Institute. Incidence and Prevalence: 2006 Chart Book on Cardiovascular and Lung Diseases. Bethesda, MD: National Institutes of Health; 2006. [Google Scholar]
- 6.Maggioni AA, Maseri A, Fresco C, et al. Age-related increase in mortality among patients with first myocardial infarctions treated with thrombolysis. N Engl J Med. 1993;329:1442–8. doi: 10.1056/NEJM199311113292002. [DOI] [PubMed] [Google Scholar]
- 7.Ornato JP, Peberdy MA, Tadler SC, Strobos NC. Factors associated with the occurrence of cardiac arrest during hospitalization for acute myocardial infarction in the second national registry of myocardial infarction in the US. Resuscitation. 2001;48:117–23. doi: 10.1016/s0300-9572(00)00255-0. [DOI] [PubMed] [Google Scholar]
- 8.French JK, Hellkamp AS, Armstrong PW, et al. Mechanical complications after percutaneous coronary intervention in ST-elevation myocardial infarction (from APEX-AMI) Am J Cardiol. 2010;105:59–63. doi: 10.1016/j.amjcard.2009.08.653. [DOI] [PubMed] [Google Scholar]
- 9.The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med. 1993;329:673–82. doi: 10.1056/NEJM199309023291001. [DOI] [PubMed] [Google Scholar]
- 10.Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Lancet. 1994;343:311–22. [PubMed] [Google Scholar]
- 11.Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet. 2003;361:13–20. doi: 10.1016/S0140-6736(03)12113-7. [DOI] [PubMed] [Google Scholar]
- 12.Alexander KP, Newby LK, Armstrong PW, et al. Acute coronary care in the elderly, Part II: ST-segment-elevation myocardial infarction: a scientific statement for healthcare professionals from the American Heart Association Council on Clinical Cardiology: in collaboration with the Society of Geriatric Cardiology. Circulation. 2007;115:2570–89. doi: 10.1161/CIRCULATIONAHA.107.182616. [DOI] [PubMed] [Google Scholar]
- 13.Krumholz HM, Murillo JE, Chen J, et al. Thrombolytic therapy for eligible elderly patients with acute myocardial infarction. JAMA. 1997;277:1683–8. [PubMed] [Google Scholar]
- 14.Singh M, Peterson ED, Roe MT, et al. Trends in the association between age and in-hospital mortality after percutaneous coronary intervention: National Cardiovascular Data Registry experience. Circ Cardiovasc Interv. 2009;2:20–6. doi: 10.1161/CIRCINTERVENTIONS.108.826172. [DOI] [PubMed] [Google Scholar]
- 15.de Labriolle A, Giraudeau B, Pacouret G, et al. Revascularization algorithm in acute STEMI should take into account age. Cardiovasc Revasc Med. 2007;8:90–3. doi: 10.1016/j.carrev.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 16.Wenaweser P, Ramser M, Windecker S, et al. Outcome of elderly patients undergoing primary percutaneous coronary intervention for acute ST-elevation myocardial infarction. Catheter Cardiovasc Interv. 2007;70:485–90. doi: 10.1002/ccd.21128. [DOI] [PubMed] [Google Scholar]
- 17.de Boer M-J, Ottervanger J-P, van’t Hof AWJ, Hoorntje JCA, Suryapranata H, Zijlstra F. Reperfusion therapy in elderly patients with acute myocardial infarction: a randomized comparison of primary angioplasty and thrombolytic therapy. J Am Coll Cardiol. 2002;39:1723–8. doi: 10.1016/s0735-1097(02)01878-8. [DOI] [PubMed] [Google Scholar]
- 18.Dauerman HL, Ryan TJ, Jr, Piper WD, et al. Outcomes of percutaneous coronary intervention among elderly patients in cardiogenic shock: a multicenter, decade-long experience. J Invasive Cardiol. 2003;15:380–4. [PubMed] [Google Scholar]
- 19.Dzavik V, Sleeper LA, Cocke TP, et al. Early revascularization is associated with improved survival in elderly patients with acute myocardial infarction complicated by cardiogenic shock: a report from the SHOCK Trial Registry. Eur Heart J. 2003;24:828–37. doi: 10.1016/s0195-668x(02)00844-8. [DOI] [PubMed] [Google Scholar]
- 20.Forman DE, Rich MW. Heart failure in the elderly. Congest Heart Fail. 2003;9:311–23. doi: 10.1111/j.1527-5299.2003.00798.x. [DOI] [PubMed] [Google Scholar]
- 21.Spiecker M. Heart failure in elderly patients. Exp Gerontol. 2006;41:549–51. doi: 10.1016/j.exger.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 22.Ezekowitz JA, Kaul P, Bakal JA, Armstrong PW, Welsh RC, McAlister FA. Declining in-hospital mortality and increasing heart failure incidence in elderly patients with first myocardial infarction. J Am Coll Cardiol. 2009;53:13–20. doi: 10.1016/j.jacc.2008.08.067. [DOI] [PubMed] [Google Scholar]
- 23.Velagaleti RS, Pencina MJ, Murabito JM, et al. Long-term trends in the incidence of heart failure after myocardial infarction. Circulation. 2008;118:2057–62. doi: 10.1161/CIRCULATIONAHA.108.784215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gould KE, Taffet GE, Michael LH, et al. Heart failure and greater infarct expansion in middle-aged mice: a relevant model for postinfarction failure. Am J Physiol Heart Circ Physiol. 2002;282:H615–21. doi: 10.1152/ajpheart.00206.2001. [DOI] [PubMed] [Google Scholar]
- 25.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 26.Sjostrom J, Bergh J. How apoptosis is regulated, and what goes wrong in cancer. BMJ. 2001;322:1538–9. doi: 10.1136/bmj.322.7301.1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olivetti G, Abbi R, Quaini F, et al. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–41. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 28.Hayakawa K, Takemura G, Koda M, et al. Sensitivity to apoptosis signal, clearance rate, and ultrastructure of fas ligand-induced apoptosis in in vivo adult cardiac cells. Circulation. 2002;105:3039–45. doi: 10.1161/01.cir.0000018651.89208.69. [DOI] [PubMed] [Google Scholar]
- 29.Nakagawa T, Zhu H, Morishima N, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-[beta] Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 30.Ferri KF, Kroemer G. Organelle-specific initiation of cell death pathways. Nat Cell Biol. 2001;3:E255–63. doi: 10.1038/ncb1101-e255. [DOI] [PubMed] [Google Scholar]
- 31.Centurione L, Antonucci A, Miscia S, et al. Age-related death-survival balance in myocardium: an immunohistochemical and biochemical study. Mech Ageing Dev. 2002;123:341–50. doi: 10.1016/s0047-6374(01)00378-5. [DOI] [PubMed] [Google Scholar]
- 32.Ozawa T. Mitochondrial DNA mutations and age. Ann N Y Acad Sci. 1998;854:128–54. doi: 10.1111/j.1749-6632.1998.tb09898.x. [DOI] [PubMed] [Google Scholar]
- 33.Dubec SJ, Aurora R, Zassenhaus HP. Mitochondrial DNA mutations may contribute to aging via cell death caused by peptides that induce cytochrome c release. Rejuvenation Res. 2008;11:611–9. doi: 10.1089/rej.2007.0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olivetti G, Melissari M, Capasso J, Anversa P. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ Res. 1991;68:1560–8. doi: 10.1161/01.res.68.6.1560. [DOI] [PubMed] [Google Scholar]
- 35.Azhar G, Liu L, Zhang X, Wei JY. Influence of age on hypoxia/reoxygenation-induced DNA fragmentation and Bcl-2, Bcl-xl, Bax and Fas in the rat heart and brain. Mech Ageing Dev. 1999;112:5–25. doi: 10.1016/s0047-6374(99)00048-2. [DOI] [PubMed] [Google Scholar]
- 36.Pollack M, Phaneuf S, Dirks A, Leeuwenburgh C. The role of apoptosis in the normal aging brain, skeletal muscle, and heart. Ann N Y Acad Sci. 2002;959:93–107. doi: 10.1111/j.1749-6632.2002.tb02086.x. [DOI] [PubMed] [Google Scholar]
- 37.Crow MT, Mani K, Nam Y-J, Kitsis RN. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res. 2004;95:957–70. doi: 10.1161/01.RES.0000148632.35500.d9. [DOI] [PubMed] [Google Scholar]
- 38.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 39.Meier P, Vousden KH. Lucifer’s labyrinth—ten years of path finding in cell death. Mol Cell. 2007;28:746–54. doi: 10.1016/j.molcel.2007.11.016. [DOI] [PubMed] [Google Scholar]
- 40.Henkart PA. ICE family proteases: mediators of all apoptotic cell death? Immunity. 1996;4:195–201. doi: 10.1016/s1074-7613(00)80428-8. [DOI] [PubMed] [Google Scholar]
- 41.Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–9. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- 42.Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296:1634–5. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 43.Chao W, Shen Y, Li L, Rosenzweig A. Importance of FADD signaling in serum deprivation- and hypoxia-induced cardiomyocyte apoptosis. J Biol Chem. 2002;277:31639–45. doi: 10.1074/jbc.M204104200. [DOI] [PubMed] [Google Scholar]
- 44.Nakai A, Yamaguchi O, Takeda T, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–24. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 45.Matsui Y, Takagi H, Qu X, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–22. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 46.Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging. Eur J Biochem. 2002;269:1996–2002. doi: 10.1046/j.1432-1033.2002.02869.x. [DOI] [PubMed] [Google Scholar]
- 47.Terman A, Dalen H, Eaton JW, Neuzil J, Brunk UT. Mitochondrial recycling and aging of cardiac myocytes: the role of autophagocytosis. Exp Gerontol. 2003;38:863–76. doi: 10.1016/s0531-5565(03)00114-1. [DOI] [PubMed] [Google Scholar]
- 48.Tannous P, Zhu H, Nemchenko A, et al. Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation. 2008;117:3070–8. doi: 10.1161/CIRCULATIONAHA.107.763870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brady NR, Hamacher-Brady A, Yuan H, Gottlieb RA. The autophagic response to nutrient deprivation in the hl-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic reticulum calcium stores. FEBS J. 2007;274:3184–97. doi: 10.1111/j.1742-4658.2007.05849.x. [DOI] [PubMed] [Google Scholar]
- 50.Rajawat YS, Hilioti Z, Bossis I. Aging: central role for autophagy and the lysosomal degradative system. Ageing Res Rev. 2009;8:199–213. doi: 10.1016/j.arr.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 51.Chien KR, Olson EN. Converging pathways and principles in heart development and disease. Cell. 2002;110:153–62. doi: 10.1016/s0092-8674(02)00834-6. [DOI] [PubMed] [Google Scholar]
- 52.Gosse P. Left ventricular hypertrophy as a predictor of cardiovascular risk. J Hypertens Suppl. 2005;23:S27–33. doi: 10.1097/01.hjh.0000165625.79933.9a. [DOI] [PubMed] [Google Scholar]
- 53.Susic D, Frohlich ED. The aging hypertensive heart: a brief update. Nat Clin Pract Cardiovasc Med. 2008;5:104–10. doi: 10.1038/ncpcardio1091. [DOI] [PubMed] [Google Scholar]
- 54.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–6. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- 55.Verdecchia P, Angeli F, Pittavini L, Gattobigio R, Benemio G, Porcellati C. Regression of left ventricular hypertrophy and cardiovascular risk changes in hypertensive patients. Ital Heart J. 2004;5:505–10. [PubMed] [Google Scholar]
- 56.Razeghi P, Taegtmeyer H. Hypertrophy and atrophy of the heart. Ann Nw Y Acad Sci. 2006;1080:110–9. doi: 10.1196/annals.1380.011. [DOI] [PubMed] [Google Scholar]
- 57.Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–9. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- 58.McMullen JR, Jennings GL. Differences between pathological and physiological cardiac hypertrophy: novel therapeutic strategies to treat heart failure. Clin Exp Pharmacol Physiol. 2007;34:255–62. doi: 10.1111/j.1440-1681.2007.04585.x. [DOI] [PubMed] [Google Scholar]
- 59.Anversa P, Palackal T, Sonnenblick E, Olivetti G, Meggs L, Capasso J. Myocyte cell loss and myocyte cellular hyperplasia in the hypertrophied aging rat heart. Circ Res. 1990;67:871–85. doi: 10.1161/01.res.67.4.871. [DOI] [PubMed] [Google Scholar]
- 60.Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Myocyte death, growth, and regeneration in cardiac hypertrophy and failure. Circ Res. 2003;92:139–50. doi: 10.1161/01.res.0000053618.86362.df. [DOI] [PubMed] [Google Scholar]
- 61.Beltrami AP, Urbanek K, Kajstura J, et al. Evidence that human cardiac myocytes divide after myocardial infarction. N Engl J Med. 2001;344:1750–7. doi: 10.1056/NEJM200106073442303. [DOI] [PubMed] [Google Scholar]
- 62.Amerongen MJv, Engel FB. Features of cardiomyocyte proliferation and its potential for cardiac regeneration. J Cell Mol Med. 2008;12:2233–44. doi: 10.1111/j.1582-4934.2008.00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kajstura J, Leri A, Finato N, Di Loreto C, Beltrami CA, Anversa P. Myocyte proliferation in end-stage cardiac failure in humans. Proc Natl Acad Sci U S A. 1998;95:8801–5. doi: 10.1073/pnas.95.15.8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 65.Anversa P, Leri A, Kajstura J. Cardiac regeneration. J Am Coll Cardiol. 2006;47:1769–76. doi: 10.1016/j.jacc.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 66.Yeghiazarians Y, Zhang Y, Prasad M, et al. Injection of bone marrow cell extract into infarcted hearts results in functional improvement comparable to intact cell therapy. Mol Ther. 2009;17:1250–6. doi: 10.1038/mt.2009.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Amado LC, Saliaris AP, Schuleri KH, et al. Cardiac repair with intramyocardial injection of allogeneic mesenchymal stem cells after myocardial infarction. Proc Natl Acad Sci U S A. 2005;102:11474–9. doi: 10.1073/pnas.0504388102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moelker AD, Baks T, Wever KMAM, et al. Intracoronary delivery of umbilical cord blood derived unrestricted somatic stem cells is not suitable to improve LV function after myocardial infarction in swine. J Mol Cell Cardiol. 2007;42:735–45. doi: 10.1016/j.yjmcc.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 69.Bai X, Yan Y, Song YH, et al. Both cultured and freshly isolated adipose tissue-derived stem cells enhance cardiac function after acute myocardial infarction. Eur Heart J. 2010;31:394–7. doi: 10.1093/eurheartj/ehp568. [DOI] [PubMed] [Google Scholar]
- 70.Boyle AJ, Schuster M, Witkowski P, et al. Additive effects of endothelial progenitor cells combined with ACE inhibition and beta-blockade on left ventricular function following acute myocardial infarction. J Renin Angiotensin Aldosterone Syst. 2005;6:33–7. doi: 10.3317/jraas.2005.004. [DOI] [PubMed] [Google Scholar]
- 71.Messina E, De Angelis L, Frati G, et al. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ Res. 2004;95:911–21. doi: 10.1161/01.RES.0000147315.71699.51. [DOI] [PubMed] [Google Scholar]
- 72.Torella D, Rota M, Nurzynska D, et al. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ Res. 2004;94:514–24. doi: 10.1161/01.RES.0000117306.10142.50. [DOI] [PubMed] [Google Scholar]
- 73.Blackburn EH. Telomere states and cell fates. Nature. 2000;408:53–6. doi: 10.1038/35040500. [DOI] [PubMed] [Google Scholar]
- 74.Chimenti C, Kajstura J, Torella D, et al. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ Res. 2003;93:604–13. doi: 10.1161/01.RES.0000093985.76901.AF. [DOI] [PubMed] [Google Scholar]
- 75.Bergmann O, Bhardwaj RD, Bernard S, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol. 2007;293:H1883–91. doi: 10.1152/ajpheart.00514.2007. [DOI] [PubMed] [Google Scholar]
- 77.Lindsey ML, Goshorn DK, Squires CE, et al. Age-dependent changes in myocardial matrix metalloproteinase/tissue inhibitor of metalloproteinase profiles and fibroblast function. Cardiovasc Res. 2005;66:410–9. doi: 10.1016/j.cardiores.2004.11.029. [DOI] [PubMed] [Google Scholar]
- 78.Bujak M, Kweon HJ, Chatila K, Li N, Taffet G, Frangogiannis NG. Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction. J Am Coll Cardiol. 2008;51:1384–92. doi: 10.1016/j.jacc.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Burgess ML, McCrea JC, Hedrick HL. Age-associated changes in cardiac matrix and integrins. Mech Ageing Dev. 2001;122:1739–56. doi: 10.1016/s0047-6374(01)00296-2. [DOI] [PubMed] [Google Scholar]
- 80.Thomas P, Cotter T, Li X, McCormick R, Gosselin L. Exercise training attenuates aging-associated increases in collagen and collagen crosslinking of the left but not the right ventricle in the rat. Eur J Appl Physiol. 2001;85:164–9. doi: 10.1007/s004210100447. [DOI] [PubMed] [Google Scholar]
- 81.Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction—executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1999 Guidelines for the Management of Patients With Acute Myocardial Infarction) J Am Coll Cardiol. 2004;44:671–719. doi: 10.1016/j.jacc.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 82.Kushner FG, Hand M, Smith SC, Jr, et al. 2009 focused updates: ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction (updating the 2004 guideline and 2007 focused update) and ACC/AHA/SCAI guidelines on percutaneous coronary intervention (updating the 2005 guideline and 2007 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2009;54:2205–41. doi: 10.1016/j.jacc.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 83.Goldenberg I, Matetzky S, Halkin A, et al. Primary angioplasty with routine stenting compared with thrombolytic therapy in elderly patients with acute myocardial infarction. Am Heart J. 2003;145:862–7. doi: 10.1016/S0002-8703(02)94709-5. [DOI] [PubMed] [Google Scholar]
- 84.Hochman JS, Sleeper LA, Webb JG, et al. Early revascularization in acute myocardial infarction complicated by cardiogenic shock. N Engl J Med. 1999;341:625–34. doi: 10.1056/NEJM199908263410901. [DOI] [PubMed] [Google Scholar]
- 85.Wiviott SD, Braunwald E, McCabe CH, et al. Intensive oral anti-platelet therapy for reduction of ischaemic events including stent thrombosis in patients with acute coronary syndromes treated with percutaneous coronary intervention and stenting in the TRITON-TIMI 38 trial: a subanalysis of a randomised trial. Lancet. 2008;371:1353–63. doi: 10.1016/S0140-6736(08)60422-5. [DOI] [PubMed] [Google Scholar]