

Abstract
BACKGROUND & AIMS
Krüppel-like factor 5 (KLF5) is a transcription factor that promotes proliferation; is highly expressed in dividing crypt cells of the gastrointestinal epithelium and is induced by various stress stimuli. We sought to determine the role of KLF5 in colonic inflammation and recovery by studying mice with dextran sulfate sodium (DSS)-induced colitis.
METHODS
Wild-type (WT) and Klf5+/− mice were given DSS in the drinking water to induce colitis. For recovery experiments, mice were given normal drinking water for 5 days after DSS administration. The extent of colitis was determined using established clinical and histological scoring systems. Immunohistochemical and immunoblotting analyses were used to examine proliferation, migration, and expression of the epidermal growth factor receptor (EGFR).
RESULTS
Klf5 expression was increased in colonic tissues of WT mice given DSS; induction of Klf5 was downstream of mitogen-activated protein kinase signaling. In DSS-induced colitis, Klf5+/− mice exhibited greater sensitivity to DSS than WT mice, with significantly higher clinical and histological colitis scores. In recovery experiments, Klf5+/− mice showed poor recovery, with continued weight loss and higher mortality than WT mice. Klf5+/− mice from the recovery period had reduced epithelial proliferation and cell migration at sites of ulceration compared to WT mice; these reductions correlated with reduced expression of EGFR.
CONCLUSIONS
Epithelial repair is an important aspect of recovery from DSS-induced colitis.
The transcription factor KLF5 regulates mucosal healing through its effects on epithelial proliferation and migration.
Keywords: inflammatory bowel disease, animal model, inflammatory response, mouse
The mammalian gut epithelium forms a dynamic barrier between the organism and various external factors present within the lumen, including diet, microorganisms, and drugs. In the setting of rapid cell turnover and constant changes in luminal content, the intestinal mucosa maintains its integrity through stringent regulation of pathways involving proliferation, migration, differentiation, and cell death. Maintaining tissue homeostasis becomes critical in instances of inflammatory bowel disease (IBD), where dysregulation of these pathways can result in excessive tissue injury, inadequate regeneration and an increased risk of developing cancer. While normal maintenance of the intestinal epithelium by the Wnt signaling pathway has been well characterized 1, 2, little is known about the molecular events that regulate restoration of tissue homeostasis in response to external stress or injury.
Krüppel-like factor 5 (KLF5) is a zinc finger-containing transcription factor that belongs to a family of mammalian Krüppel-like transcription factors. These factors are often tissue-specific and serve diverse roles in processes such as cell proliferation, differentiation and embryogenesis 3, 4. KLF5 is highly expressed in the gut and is enriched in epithelial cells in the proliferative compartment of the intestinal tract 5, indicating a role as a pro-proliferative factor. Indeed, several in vitro studies show that ectopic expression of KLF5 enhances proliferation of non-transformed cultured epithelial cells 6–8. KLF5 has also been shown to mediate the growth and transforming effects of oncogenic KRAS in intestinal epithelial cells 9. Furthermore, mouse studies reveal that Klf5 promotes proliferation in the settings of bacterial infection and intestinal tumor initiation 10, 11. The pro-proliferative effects of KLF5 are thought to be mediated through its transcriptional targets, which include the growth factor, platelet-derived growth factor alpha polypeptide (PDGF-A), and cell cycle proteins, cyclin D1, cyclin B1 and Cdc2 12–14.
In addition to its role in regulating proliferation, a number of studies indicate that KLF5 may act as a mediator of external stress responses. KLF5 expression is shown to be induced in vascular smooth muscle cells following balloon injury in rat aorta 15. This induction is mediated through mitogen-activated protein kinase (MAPK) signaling by the early response gene, early growth response factor 1 (Egr1) 15, 16. Additional studies in heterozygous Klf5 knockout (Klf5+/−) mice reveal that KLF5 is a critical factor for tissue remodeling in injured vascular tissues by participating in proliferative and angiogenic responses 14. In intestinal epithelial cells (IECs), KLF5 is activated by the bacterial component, lipopolysaccharide, and enhances inflammatory responses in these cells through its effects on NF-κB signaling 17. Furthermore, in vivo studies show that KLF5 is induced by colonization of the mouse colon with the bacterial pathogen, Citrobacter rodentium, and that Klf5 mediates colonic epithelial hyperplasia activated by this infection 11. Taken together, these reports indicate a critical role for KLF5 in cellular responses to cardiovascular injury and suggest a similar function for KLF5 in intestinal tissues.
In the present study, we utilize the dextran sulfate sodium (DSS)-induced model of colitis to examine the role of Klf5 in promoting restoration of tissue homeostasis. WT and Klf5+/− mice were treated with the chemical irritant, DSS, and examined for the response and the ability to recover from DSS-induced injury. Results revealed that reduced levels of Klf5 heightened susceptibility to DSS-induced damage, indicating a protective role for Klf5 in response to chemical-induced injury. Moreover, Klf5+/− mice exhibited poor recovery from DSS treatment, indicating that KLF5 is important for restoration of intestinal epithelial homeostasis.
Materials and Methods
Mice
C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Klf5+/− mice on a C57BL/6 background were generated as described previously 11. Mouse strains were bred and housed in the Whitehead Animal Research Facility at Emory University. Animal care and procedures were conducted in compliance with Emory University Institutional Animal Care and Use Committee guidelines.
Cell Culture
Caco-2, HCT 116 and DLD-1 colon cancer cells were maintained at 37° C in 5% CO2. For inhibitor studies, Caco-2 cells were pretreated for 10 min prior to addition of DSS with U0126 at 50 μM (Cell Signaling Technology; Beverly, MA) or Bay 11-7082 at 20μM (Calbiochem; San Diego, CA).
Induction of DSS Colitis
Colitis was induced in 7- to 8-week old gender-matched WT and Klf5+/− mice by by addition of dextran sulfate sodium (DSS) (molecular weight, 35,000–50,000; MP Biomedicals, Solon, OH) to the drinking water at 3.5% (wt/vol) for 7 days. Controls received normal drinking water. For recovery experiments, WT and Klf5+/− mice were administered 3.5% or 2.5% DSS, respectively, for 7 days, followed by 5 days of recovery with normal dH2O.
Protein Isolation from Mouse Colon
Colon sections were flushed with phosphate-buffered saline (PBS), inverted and scraped to isolate mucosal protein. Scraped tissues were placed in Cell Extraction Buffer (Invitrogen, Carlsbad, CA) and homogenized with silicone beads using a Bullet Blender (Next Advance, Inc., Averill Park, NY) at 4° C for 5 min at Speed 5.
Antibodies
Antibodies are listed in Supplementary materials.
Histology
Colons were prepared as Swiss rolls for embedding in paraffin, and sections were stained with hemotoxylin and eosin (H&E). Images of tissue sections were captured using a Zeiss Axioskop2 plus microscope (Carl Zeiss MicroImaging, Inc., Thornwood, NY).
Immunohistochemistry
Immunohistochemistochemical staining was performed as described previously 11. Fluorescent images of Klf5 staining were analyzed for nuclear intensities using Metamorph Imaging software (Molecular Devices, Sunnyvale, CA).
Clinical and Histological Assessment of Colitis
Clinical and histological scores for measuring the degree of colitis were determined by the method described by Cooper et al. 18
MPO Activity
Neutrophil infiltration was quantified by measuring myeloperoxidase (MPO) activity. Colon tissue was homogenized in HTAB buffer (50 mM phosphate buffer, pH 6.0, 0.5% hexadecyltrimethyl ammonium bromide). Lysates (10 μl) were added to 1 mg/ml of o-dianisidine hydrochloride and 5 × 10−4% hydrogen peroxide, and the absorbance measured at 460 nm. The results were expressed as U activity per mg protein.
In Vitro Wounding Assays
HCT 116 cells or DLD-1 cells were plated in 6-well plates. Cells were transfected in triplicate wells with pCI-neo or pCI-neo-KLF5 expression vectors. At 24 hrs after transfection, confluent monolayers were wounded with a 10 μl pipette tip. Images were taken at wounding and 18 hrs later. Wound widths were determined by averaging 6 measurements per image.
Statistical Analysis
The data are presented as mean ± SEM. Equality of variances was tested by Levene’s test. Multigroup data were analyzed by ANOVA with multiple pair-wise comparisons made among groups using Tukey’s test. Data comparing 2 groups were analyzed using a 2-tailed t test. P values of < 0.05 were considered statistically significant.
Results
Induction of Klf5 Following DSS Treatment
The dextran sulfate sodium (DSS) mouse model of colitis exhibits many of the histological characteristics of human IBD, including disruption of crypt architecture, mucosal ulceration, and infiltration of neutrophils, monocytes and lymphocytes 19 To investigate whether KLF5 expression is altered in the setting of colitis, C57BL/6 (WT) mice were treated with DSS and examined for levels and cellular localization of Klf5. Mice were administered 3.5% DSS in the drinking water for seven days to induce colitis, after which they were euthanized and colonic tissues collected for protein analysis and immunohistochemistry. Immunohistochemical staining revealed that Klf5 was expressed at high levels in DSS-treated mice, particularly in epithelial cells contiguous to sites of ulceration. Intense KLF5 staining was seen in the crypts adjacent to ulcerated areas and at luminal surfaces covering the ulcers (Figure 1A; WT DSS). This staining pattern was in contrast to untreated WT mice in which staining of Klf5 was restricted to the proliferative zones in the lower two-thirds of the crypts, with no expression at the luminal surface (Figure 1A; WT H2O). Increased levels of Klf5 were confirmed by immunofluorescence staining, which showed a trend towards higher KLF5 intensity per cell as well as a higher percentage of Klf5-positive cells per crypt (Figures 3A–3C; WT H2O versus WT DSS). During a 5 day time course of DSS treatment, Western blot analysis indicated that Klf5 was induced in the colons of DSS-treated WT mice as early as 1 day after DSS treatment (Figure 1B). Increased levels of Klf5 began to decline at days 4 and 5, likely as epithelial cells were lost with progressive mucosal erosion.
Figure 1.
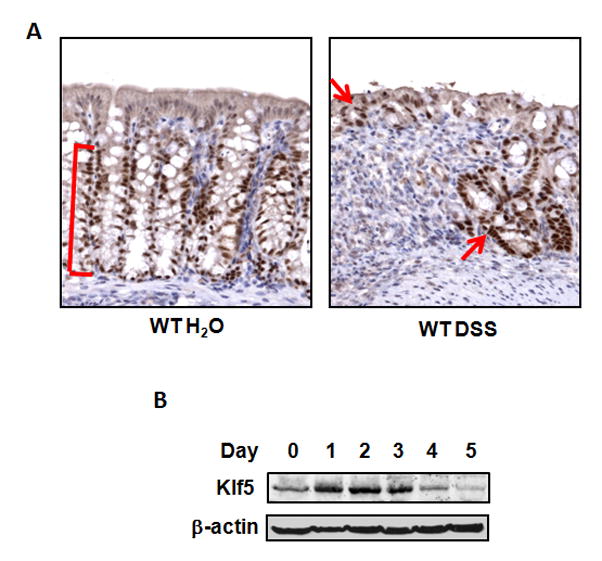
Klf5 is induced in wild-type (WT) mice treated with DSS. Eight week-old WT mice were given water or treated with 3.5% DSS (wt/vol). (A) Immunohistochemical staining of colon of Klf5 in WT mice untreated or treated with DSS for 7 days (brown, Klf5; blue; counterstain). Red bars and arrows indicate regions of Klf5 expression. (B) Western blots of lysates from WT mice treated with DSS over a 5 day time course. Lysates for each time point were pooled from 3 separate mice.
Figure 3.
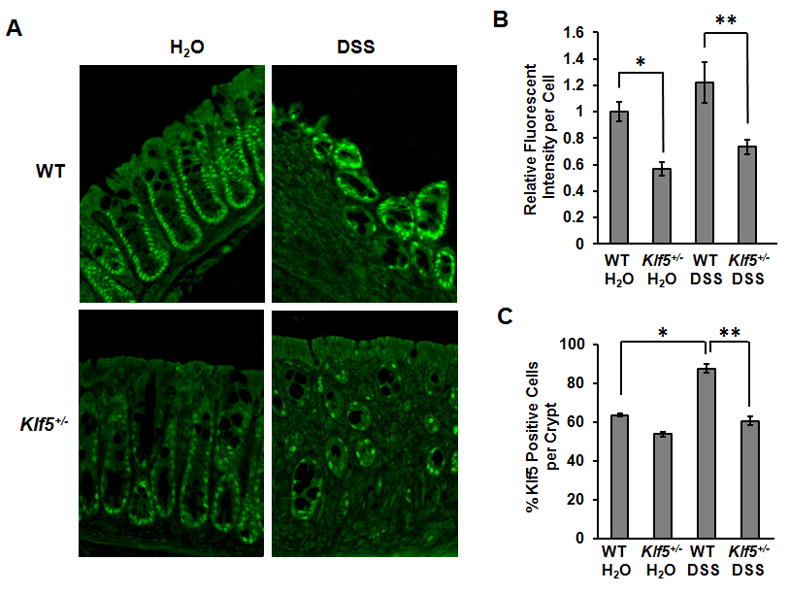
Klf5 expression is reduced at sites of ulceration in DSS-treated Klf5+/− mice. (A) Immunofluorescence staining of Klf5 in WT and Klf5+/− mice untreated or treated with DSS for 7 days. White arrows indicate Klf5 staining at luminal surface (B) Quantification of fluorescent intensities of Klf5 staining per cell. Data represent the mean ± SEM of 5 mice per group with at least 50 cells quantified per sample. * P = 0.01; ** P = 0.04. (C) Quantification of Klf5-positive cells per crypt in untreated and DSS-treated WT and Klf5+/− mice. Data represent the mean ± SEM of 3 mice per group, counting 5 crypts per mouse. * P < 0.001. ** P = 0.001.
To investigate the mechanism of Klf5 activation, two major signaling pathways were evaluated in response to DSS. MAPK signaling via activation of MAPK kinase (MEK) and extracellular-signal-regulated kinases 1 and 2 (ERK1/2) is known to induce Klf5 expression in several cell types 13, 16, 20. Phosphorylation of ERK1/2, which can occur in both epithelial and infiltrating immune cells, was increased at day 1 of DSS treatment and was induced further at days 4 and 5 (Figure 2A). NF-κB, which is a key regulator of inflammatory responses, also appeared to be activated by DSS treatment, as indicated by a decrease in levels of the NF-κB inhibitor IκB beginning at day 3. As both of these pathways are activated by DSS treatment in vivo, an in vitro system was used to examine the dependence of KLF5 induction on ERK1/2 and NF-κB signaling.
Figure 2.
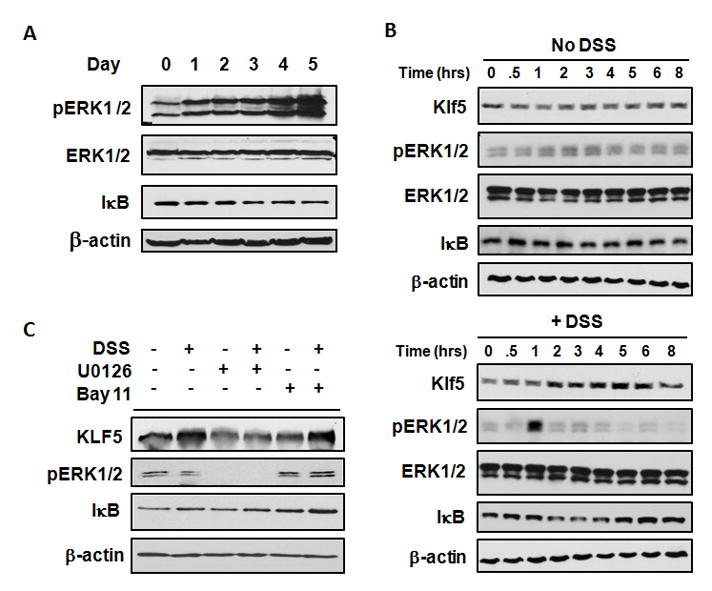
Induction of KLF5 with DSS treatment is dependent on ERK1/2 signaling. (A) Detection of ERK1/2 and NF-κB activation by Western blotting, using pooled lysates from WT mice treated with 3.5% DSS over a 5 day time course (n=3). (B) Caco-2 cells were treated with 3% DSS for up to 8 hrs, and lysates were examined by Western blotting. (C) Caco-2 cells were pretreated with inhibitors of MEK/ERK (U0126) or NF-κB (Bay 11-7082), and treated for 5 hrs with 3% DSS. Lysates were examined by Western blotting.
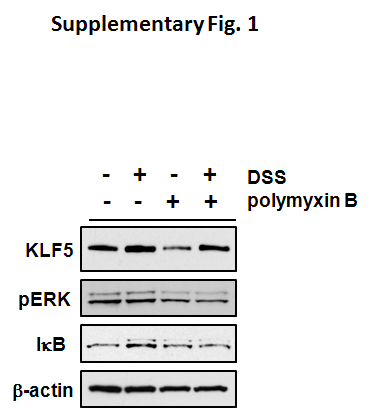
In Caco-2 colon cancer cells treated with 3% DSS during an 8 hour period, KLF5 levels increased two hours after treatment with DSS and reached maximal induction at five hours (Figure 2B). Since DSS preparations can be contaminated with lipopolysaccharide (LPS) 21, cells were treated with DSS and polymyxin B (a scavenger for LPS) to verify that the effects of DSS on KLF5 were not skewed by the presence of LPS. At the 5 hour time point, KLF5 levels continued to be induced by DSS in the presence of polymyxin B, indicating no involvement of LPS (Supplementary Figure 1). ERK1/2 showed acute activation one hour after addition of DSS, and, NF-κB was activated beginning at two hours after DSS (Figure 2B). To determine whether activation of these signaling pathways were required for induction of KLF5, Caco-2 cells were pretreated with inhibitors to MEK/ERK or NF-κB prior to the addition of DSS for 5 hours. KLF5 was induced by DSS in the absence of inhibitors as well as in the presence of Bay 11-7082 (Figure 2C). However, KLF5 induction was blocked by U0126, indicating that KLF5 induction is dependent on MEK/ERK signaling.
Reduced Levels of Klf5 in Regenerating Tissues of Klf5+/− Mice Treated with DSS
To examine the role of KLF5 in DSS-induced colitis, mice with heterozygous deletion of Klf5 (Klf5+/−) 11 were compared to WT mice for responses to DSS treatment (homozygous deletion of Klf5 is embryonic lethal 14). Klf5+/− mice develop normally and are fertile. No histological differences have been noted in the colonic mucosal architecture of Klf5+/− mice compared to WT mice, and epithelial barrier function is comparable between the two genotypes (Supplementary Figures 2A and 2B). Also, blood analysis revealed no abnormalities in the development of hematopoietic cells (Supplemental Figure 2C). WT and Klf5+/− mice were treated for 7 days with DSS, and Klf5 levels were analyzed in colon tissues by immunofluorescence. In untreated mice, basal levels of Klf5 in the heterozygous mice were reduced in intensity compared to WT mice, confirming haploinsufficiency of the Klf5 gene (Figures 3A and 3B). Upon treatment with DSS, Klf5 exhibited strong immunofluorescence staining in regenerative crypts and at the luminal surface of WT mice, with a trend towards increased Klf5 intensity per cell compared to untreated WT mice. In addition, WT mice treated with DSS had a higher percentage of Klf5-positive cells per crypt compared to untreated WT mice (Figure 3C). In Klf5+/− mice treated with DSS, Klf5 levels were significantly lower than in WT treated mice, with reduced intensity of Klf5 staining per cell and a lower percentage of Klf5-positive cells per crypt (Figures 3A–3C).
Increased Sensitivity of Klf5+/− Mice to DSS Treatment
The extent of colitis in both WT and Klf5+/− mice was determined after 7 days of DSS treatment using clinical and histological characterization. Weights of WT and Klf5+/− mice were recorded daily over the course of DSS treatment (Figure 4A). A significant difference was seen in the percentage weight loss between treated WT mice compared to treated Klf5+/− mice, with an average weight loss of 5% in WT animals versus an average of 13% in Klf5+/− mice (Figure 4A). H&E staining of colon from DSS-treated WT mice indicated moderate inflammation and ulceration, with some disruption of normal crypt architecture (Figure 4B). However, in Klf5+/− mice, the damage was more severe, with extensive infiltration of neutrophils and a complete loss of crypt architecture in large, continuous sections. Extent of neutrophil infiltration was determined by myeloperoxidase (MPO) activity, a lysosomal enzyme abundant in neutrophils (Figure 4C). MPO activity was approximately 3-fold higher in the Klf5+/− DSS-treated mice compared to WT-treated mice, correlating with an increased presence of granulocytes visualized by histology. As an indicator of colitis, colon lengths were significantly shorter in the Klf5+/− mice compared to WT mice, with lengths measuring 4.4 cm ± 0.2 and 5.4 cm ± 0.2, respectively (n = 7; P = 0.01). The extent of colitis was further quantified using a well-established method of clinical and histological scoring 18. Clinical scores, based on the parameters of weight loss, stool consistency and presence of fecal blood, indicated more extensive colitis in Klf5+/− mice (10.0 ± 0.6) compared to WT mice (5.9 ± 0.6; n = 7; P < 0.001) (Figure 4D). Similarly, histological scores, based on crypt destruction, mucosal damage, epithelial erosion and inflammatory infiltrate, mirrored the clinical findings, with Klf5+/− mice scoring significantly higher than WT mice (10.0 ± 0.3 versus 6.9 ± 0.5; n = 7, P < 0.001) (Figure 4E). Thus, the data support a protective role for Klf5 in responding to DSS-induced mucosal damage as indicated by increased sensitivity of Klf5+/− mice to DSS treatment.
Figure 4.
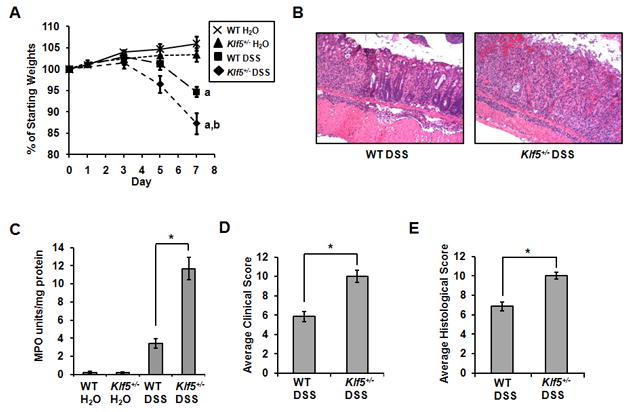
Klf5+/− mice exhibit increased susceptibility to colitis with DSS treatment. Eight-week old WT and Klf5+/− mice were treated with 3.5% DSS for 7 days and examined for clinical signs of colitis. Colon tissues were processed for enzymatic and histological analysis. (A) Weights of mice during 7 day treatment period; n = 9 mice per group, a: P < 0.001 versus control; b: P = 0.02 versus WT DSS. (B) Representative H&E staining of proximal colons from DSS-treated mice. (C) Myeloperoxidase (MPO) activity as a measure of neutrophil infiltration; n = 5, * P < 0.001. (D) Mean clinical scores of colitis with a maximum score of 12. n = 7, * P < 0.001. (E) Mean histological scores of colitis with a maximum score of 11. n = 7, * P < 0.001.
Poor Recovery of Klf5+/− Mice Following DSS Treatment
To determine whether recovery from DSS treatment is impaired in Klf5+/− mice, WT and Klf5+/− mice were examined during a recovery phase, in which the mice were returned to normal drinking water for 5 days following 7 days of DSS treatment. To begin the recovery phase with similar degrees of colitis in WT and Klf5+/− mice, different percentages of DSS were tested to identify treatment conditions that would yield similar effects in the two mouse strains. Consequently, Klf5+/− mice were treated with 2.5% DSS for 7 days whereas WT mice were treated with 3.5% DSS. While WT and Klf5+/− mice entered the 5 day recovery phase with similar degrees of colitis based on weights and colitis scores (Figures 5A, 5C and 5D, Day 7 DSS), Klf5+/− mice exhibited poorer recovery, with shorter colon lengths at the end of the 5 day recovery period (4.7 ± 0.2 cm versus 5.8 ± 0.2 cm in WT mice; n = 14, P < 0.001) and a continued decline in average body weight versus unchanged weight in the WT mice (Figure 5A). In addition, the Klf5+/− mice exhibited significantly higher mortality than the WT mice, with 36% of the mice reaching IACUC endpoints during the recovery phase (n = 14, P = 0.02) (Figure 5B). Clinical and histological scores also showed an inability of Klf5+/− mice to recover, as both measures indicated worsening of colitis during the recovery phase compared to stabilization or improvement in WT mice (Figures 5C and 5D). Therefore, Klf5+/− mice not only exhibited increased sensitivity to DSS compared to WT mice, but also failed to stabilize or improve during the recovery period, suggesting an impaired ability to heal injured mucosal tissues.
Figure 5.

Klf5+/− mice show impaired recovery during the DSS recovery phase. WT and Klf5+/− mice were treated with DSS (3.5% and 2.5%, respectively) for 7 days to achieve similar degrees of colitis prior to 5 day recovery phase. (A) Weights of WT and Klf5+/− mice after 7 days of DSS treatment and after 5 days of recovery (n = 14, * P = 0.02.) (B) Kaplan-Meier survival curve of WT and Klf5+/− mice during DSS treatment and recovery phase (n = 14; P = 0.02). (C&D) Clinical and histological scores after 7 days of DSS treatment or after 5 days of recovery. (C) n = 7; * P = 0.003; ** P = 0.05 (D) n = 7; * P < 0.001; ** P = 0.006.
Reduced Epithelial Proliferation and Migration in Klf5+/− Mice Treated with DSS
Because KLF5 is highly expressed in areas of ulceration, we wanted to determine whether KLF5 contributes to injury repair. To explore this possibility, two aspects of wound repair, proliferation and cell migration, were examined in colons of recovery mice. To determine relative rates of proliferation, actively dividing cells in WT and Klf5+/− recovery mice were pulse-labeled with the nucleoside, bromodeoxyuridine (BrdU), and numbers of proliferating cells per crypt were calculated in areas adjacent to ulcers (Figure 6A),. Whereas 80% of cells in regenerative glands of WT mice stained positive for BrdU, this percentage was reduced to 50% in Klf5+/− mice, indicating a slower rate of proliferation in regions of repair in Klf5+/− mice. To address the question of epithelial migration, dividing cells in the mice were labeled for 24 hrs with BrdU to track cell migration. Colonic tissues from recovery mice were co-stained for Ki67 and BrdU to distinguish between cells that were actively proliferating at the time of sacrifice (green cells expressing Ki67) and cells that had ceased to proliferate and were migrating into ulcerated areas (red cells stained for BrdU) (Figure 6B). Numerous cells in the WT mice were migrating vertically within the crypts and across the luminal surface, whereas only minimal cell migration was visible in Klf5+/− mice.
Figure 6.
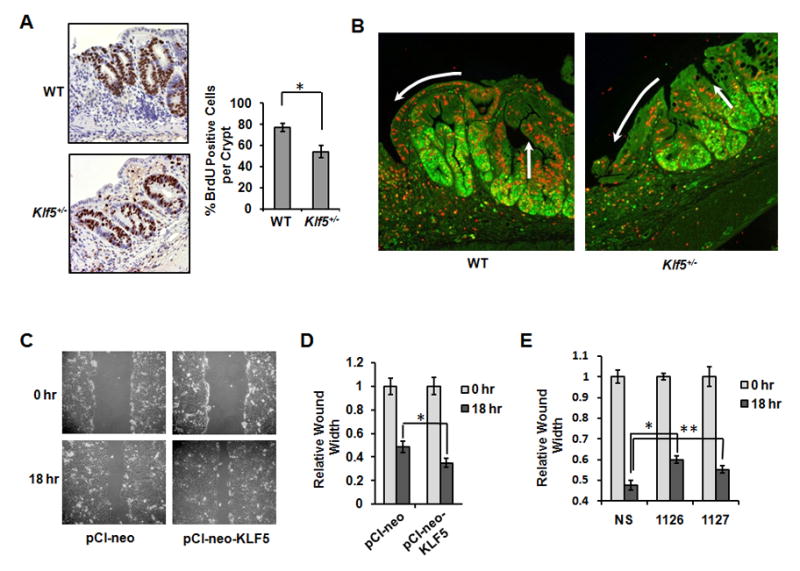
Epithelial proliferation and migration are reduced at sites of ulceration in Klf5+/− mice. Colon tissues were isolated after 5 days of recovery and subjected to immunohistochemical staining. (A) BrdU staining of tissues from mice injected with BrdU 4 hrs prior to sacrifice. Images show crypts in regions adjacent to ulcers. Cell counts are from 6 crypts per mouse, (*P = 0.009; n = 5). (B) Immunofluorescence staining of Ki67 (green) and BrdU (red) in colon tissues from mice injected with BrdU 24 hrs prior to sacrifice. White arrows indicate areas of migration. (C) Wounding assays in DLD-1 cells transfected with empty vector (pCI-neo) or an expression construct for KLF5 (pCIneo-KLF5). (D) Graphical data of relative wound widths from (C). *P = 0.01, n=3. (E) Wounding assays in DLD-1 cells transfected with non-specific siRNA (NS) or two different siRNAs specific for KLF5 (1126 or 1127). *P < 0.001; n=3. **P = 0.03; n=3.
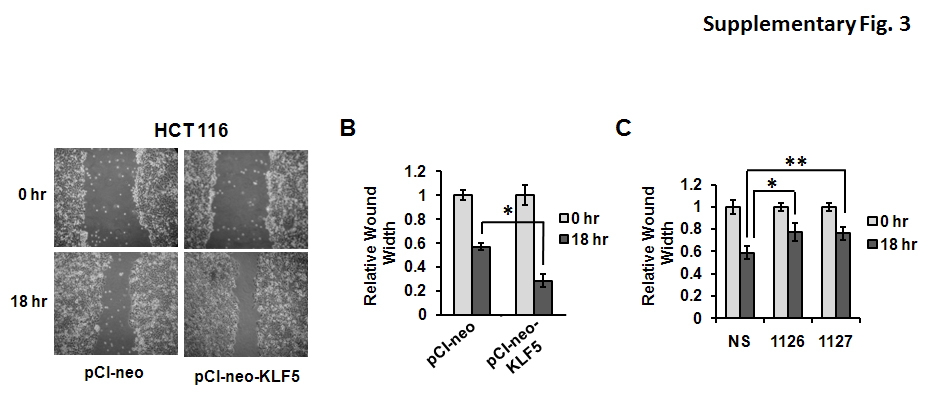
To examine further the effects of KLF5 on epithelial cell migration, scratch wound assays were used to measure differences in migration in colon cancer cells transfected with empty vector or with a vector for ectopic expression of KLF5. Confluent monolayers of DLD-1 cells were wounded with a pipette tip and examined for closure of the wound at 18 hrs post-wounding. Cells overexpressing KLF5 showed increased wound closure compared to cells transfected with empty vector (Figure 6C and 6D), indicating that KLF5 promotes cell migration into the wound site. In a reciprocal experiment, KLF5 expression was blocked by transfection of DLD-1 cells with two different siRNAs specific for KLF5. Cells transfected with KLF5-specific siRNAs exhibited reduced wound closure compared to cells transfected with a non-specific siRNA (Figure 6E). Similar results were obtained in HCT 116 colon cancer cells with overexpression and inhibition of KLF5 (Supplementary Figure 3). Thus, KLF5 promotes epithelial cell migration during wound repair.
Reduced EGFR Expression in Klf5+/− Mice Treated with DSS
Activation of EGFR is a key signaling event that drives cells to proliferate, migrate and cover wounds in several types of epithelia 22–24. EGFR is expressed at sites of ulcer healing in the gastrointestinal tract and is detected predominantly in the immature regenerative epithelium around ulcer margins 25. Previously, EGFR was reported to be a direct transcriptional target of KLF5 in primary esophageal keratinocytes by promoter analysis and chromatin immunoprecipitation assays 20. Given the participation of EGFR in wound healing, we examined whether EGFR was expressed differentially in WT and Klf5+/− mice treated with DSS. After five days of DSS treatment, WT mice showed positive staining of EGFR in areas of ulceration, particularly in regenerative glands and regions of epithelial restitution at the luminal surface (Figure 7A, panel 1). In contrast, Klf5+/− mice exhibited markedly reduced EGFR staining at comparable sites (Figure 7A, panel 2). Western blot analysis of lysates from DSS treated mice showed a pattern of induction of EGFR in WT mice that correlated with the induction of Klf5 (Figure 7B). In Klf5+/− mice, EGFR levels were induced to a lesser extent than in WT mice at each time point analyzed, suggesting that EGFR levels are dependent on Klf5 expression. Likewise, in recovery experiments, EGFR was detected in epithelial cells at the luminal surface of ulcers of WT mice (Figure 7A, panels 3 and 5), and this expression was reduced in Klf5 heterozygous animals (Figure 7A, panels 4 and 6). To examine directly whether KLF5 expression can regulate EGFR levels in colonic cells, we overexpressed or blocked expression of KLF5 in colon cancer cells. Suppression of KLF5 in DLD-1 cells with relatively high levels of endogenous KLF5 resulted in a marked reduction of EGFR protein in DLD-1 cells. Conversely, overexpression of KLF5 in HCT 116 cells with relatively low levels of endogenous KLF5 resulted in increased levels of EGFR. Taken together, these results indicate that EGFR levels are regulated by Klf5 expression in colonic tissues.
Figure 7.

EGFR levels are reduced at sites of ulceration in Klf5+/− mice treated with DSS. (A) EGFR staining in colons of mice after 5 days of DSS treatment (acute phase); panel 1, WT; panel 2, Klf5+/−. EGFR staining after 5 days of recovery from DSS treatment. Panel 3, WT (enlargement, panel 5); panel 4, Klf5+/− (enlargement, panel 6) Red arrows indicate luminal epithelial cells. (B) Western blots of colon lysates from WT and Klf5+/− mice treated with 3.5% DSS, days 0–5. Lysates are pooled from 3 separate mice. (C) Effects of Klf5 expression on EGFR levels in colon cancer cells. Western blot analysis with inhibition of KLF5 by siRNA in DLD-1 cells or overexpression of KLF5 in HCT 116 cells.
Discussion
The intestinal epithelium serves as a critical barrier against a broad spectrum of noxious and immunogenic substances within the intestinal lumen. When this barrier is disrupted, the integrity of the surface epithelium is quickly reestablished, even following severe damage, due to the remarkable regenerative capacity of the intestinal mucosa 26. However, in various intestinal disorders, including IBD, the mucosal barrier is repeatedly disrupted, allowing for increased exposure to infectious agents and toxins that can prolong the inflammatory condition 27. Indeed, barrier defects in IBD are thought to play a critical role in the pathobiology of disease, being intimately connected to chronic inflammatory responses 28. Thus, rapid repair of the mucosal barrier is critical for limiting inflammation and healing ulcerated lesions in chronic inflammatory diseases.
In this paper, we show that KLF5 serves a novel role in epithelial regeneration of DSS-damaged tissues by contributing both to proliferative and migratory processes essential for epithelial repair. The participation of KLF5 in proliferative responses to DSS damage is consistent with in vitro and in vivo studies which indicate a pro-proliferative role for KLF5 6–8, 11 However, the ability of KLF5 to promote proliferation can have pathogenic effects in the context of oncogenic mutations. Previously, we have reported that KLF5 mediates cell transformation during KRAS-induced intestinal tumorigenesis and that KLF5 is required for intestinal tumor initiation in the ApcMin/+ tumorigenesis model 9–10.
In addition to promoting proliferation in the setting of tissue repair, our studies provide the first evidence that KLF5 functions in epithelial cell migration in the colonic mucosa. Other KLF proteins have been implicated in regulating cell migration, including KLF2 in T cells and smooth muscle cells 29–31, KLF8 in immortalized human breast epithelial cells 32 and KLF6 in endothelial cells 33. Also, KLF5 was recently shown to enhance migration of esophageal keratinocytes through activation of the transcriptional target, integrin-linked kinase (ILK) 34. In our experiments, however, we saw no change in ILK levels with induction of Klf5 in the colons of mice treated with DSS (data not shown).
During ulcer healing, the mucosa at the ulcer margin forms a “healing zone”, in which the gastrointestinal glands become dilated, the cells lining the glands become de-differentiated and begin to express EGFR 35–37. This process is activated by growth factors that include epidermal growth factor (EGF), hepatocyte growth factor (HGF), platelet derived growth factor (PDGF) and basic fibroblast growth factor (bFGF), which stimulate proliferation and cell migration to restore mucosal surfaces and reconstitute mucosal glands 37. Several lines of evidence suggest that KLF5 may participate in intestinal wound healing through its effects on growth factor signaling. KLF5 has been shown to regulate expression of PDGF-A in vascular smooth muscle cells in response to vascular injury 14, 38. In addition, KLF5 has been reported to promote EGF signal amplification in primary esophageal cells through direct transcriptional activation of EGFR 20. Here, we show that EGFR levels correlate with changes in Klf5 levels in the DSS mouse injury model, and that EGFR expression is reduced in healing zones of Klf5+/− DSS-treated mice. In light of these results, future studies will explore the dependence of KLF5 on EGFR in promoting epithelial cell migration and wound healing.
In summary, these results demonstrate the importance of efficient epithelial repair in recovery from colonic injury. We show that KLF5 plays a critical role in epithelial repair following DSS treatment by promoting proliferation of colonic epithelial cells at sites of ulceration. In addition, we report a novel role for KLF5 in promoting epithelial cell migration as part of the restitution process.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Grant Support: This work was supported in part by grants from the National Institutes of Health (DK76742 to BBM, DK06411 and DK76825 to SVS, and DK52230, DK64399 and CA84197 to VWY).
Abbreviations
- BrdU
bromodeoxyuridine
- BSA
bovine serum albumin
- DSS
dextran sulfate sodium
- EGFR
epidermal growth factor receptor
- FBS
fetal bovine serum
- H&E
hematoxylin and eosin
- IBD
inflammatory bowel disease
- KLF5
Krüppel-like factor 5
- MAPK
mitogen-activated protein kinase
- MPO
myeloperoxidase
- PBS
phosphate-buffered saline
- PDGF-A
platelet-derived growth factor alpha polypeptide
- WT
wild-type
Footnotes
Disclosures: The authors have no potential conflicts of interest.
Authors’ contributions
BBM designed and performed experiments, analyzed data and wrote the manuscript
SSK managed mouse strains and conducted mouse experiments and in vivo staining
ABB conducted in vitro wounding assays and Western blots
KY performed Western blots for time course experiments
SVS provided histological analysis and technical expertise and advised on study design
VWY advised on experimental design, analyzed data and edited the manuscript
All authors read and approved the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.de Lau W, Barker N, Clevers H. WNT signaling in the normal intestine and colorectal cancer. Front Biosci. 2007;12:471–91. doi: 10.2741/2076. [DOI] [PubMed] [Google Scholar]
- 2.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McConnell BB, Ghaleb AM, Nandan MO, Yang VW. The diverse functions of Kruppel-like factors 4 and 5 in epithelial biology and pathobiology. Bioessays. 2007;29:549–57. doi: 10.1002/bies.20581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagai RFSL, Kasuga M. The Biology of Kruppel-like Factors. Tokyo, Berlin, Heidelberg, New York: Springer; 2009. [Google Scholar]
- 5.Conkright MD, Wani MA, Anderson KP, Lingrel JB. A gene encoding an intestinal-enriched member of the Kruppel-like factor family expressed in intestinal epithelial cells. Nucleic Acids Res. 1999;27:1263–70. doi: 10.1093/nar/27.5.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bateman NW, Tan D, Pestell RG, Black JD, Black AR. Intestinal tumor progression is associated with altered function of KLF5. J Biol Chem. 2004;279:12093–101. doi: 10.1074/jbc.M311532200. [DOI] [PubMed] [Google Scholar]
- 7.Sun R, Chen X, Yang VW. Intestinal-enriched Kruppel-like factor (Kruppel-like factor 5) is a positive regulator of cellular proliferation. J Biol Chem. 2001;276:6897–900. doi: 10.1074/jbc.C000870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chanchevalap S, Nandan MO, Merlin D, Yang VW. All-trans retinoic acid inhibits proliferation of intestinal epithelial cells by inhibiting expression of the gene encoding Kruppel-like factor 5. FEBS Lett. 2004;578:99–105. doi: 10.1016/j.febslet.2004.10.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nandan MO, McConnell BB, Ghaleb AM, Bialkowska AB, Sheng H, Shao J, Babbin BA, Robine S, Yang VW. Kruppel-like factor 5 mediates cellular transformation during oncogenic KRAS-induced intestinal tumorigenesis. Gastroenterology. 2008;134:120–30. doi: 10.1053/j.gastro.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McConnell BB, Bialkowska AB, Nandan MO, Ghaleb AM, Gordon FJ, Yang VW. Haploinsufficiency of Kruppel-like factor 5 rescues the tumor-initiating effect of the Apc(Min) mutation in the intestine. Cancer Res. 2009;69:4125–33. doi: 10.1158/0008-5472.CAN-08-4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McConnell BB, Klapproth JM, Sasaki M, Nandan MO, Yang VW. Kruppel-like factor 5 mediates transmissible murine colonic hyperplasia caused by Citrobacter rodentium infection. Gastroenterology. 2008;134:1007–16. doi: 10.1053/j.gastro.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nandan MO, Chanchevalap S, Dalton WB, Yang VW. Kruppel-like factor 5 promotes mitosis by activating the cyclin B1/Cdc2 complex during oncogenic Ras-mediated transformation. FEBS Lett. 2005;579:4757–62. doi: 10.1016/j.febslet.2005.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nandan MO, Yoon HS, Zhao W, Ouko LA, Chanchevalap S, Yang VW. Kruppel-like factor 5 mediates the transforming activity of oncogenic H-Ras. Oncogene. 2004;23:3404–13. doi: 10.1038/sj.onc.1207397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shindo T, Manabe I, Fukushima Y, Tobe K, Aizawa K, Miyamoto S, Kawai-Kowase K, Moriyama N, Imai Y, Kawakami H, Nishimatsu H, Ishikawa T, Suzuki T, Morita H, Maemura K, Sata M, Hirata Y, Komukai M, Kagechika H, Kadowaki T, Kurabayashi M, Nagai R. Kruppel-like zinc-finger transcription factor KLF5/BTEB2 is a target for angiotensin II signaling and an essential regulator of cardiovascular remodeling. Nat Med. 2002;8:856–63. doi: 10.1038/nm738. [DOI] [PubMed] [Google Scholar]
- 15.Nagai R, Kowase K, Kurabayashi M. Transcriptional regulation of smooth muscle phenotypic modulation. Ann N Y Acad Sci. 2000;902:214–22. doi: 10.1111/j.1749-6632.2000.tb06316.x. discussion 222–3. [DOI] [PubMed] [Google Scholar]
- 16.Kawai-Kowase K, Kurabayashi M, Hoshino Y, Ohyama Y, Nagai R. Transcriptional activation of the zinc finger transcription factor BTEB2 gene by Egr-1 through mitogen-activated protein kinase pathways in vascular smooth muscle cells. Circ Res. 1999;85:787–95. doi: 10.1161/01.res.85.9.787. [DOI] [PubMed] [Google Scholar]
- 17.Chanchevalap S, Nandan MO, McConnell BB, Charrier L, Merlin D, Katz JP, Yang VW. Kruppel-like factor 5 is an important mediator for lipopolysaccharide-induced proinflammatory response in intestinal epithelial cells. Nucleic Acids Res. 2006;34:1216–23. doi: 10.1093/nar/gkl014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–49. [PubMed] [Google Scholar]
- 19.Yan Y, Kolachala V, Dalmasso G, Nguyen H, Laroui H, Sitaraman SV, Merlin D. Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. PLoS One. 2009;4:e6073. doi: 10.1371/journal.pone.0006073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Y, Goldstein BG, Nakagawa H, Katz JP. Kruppel-like factor 5 activates MEK/ERK signaling via EGFR in primary squamous epithelial cells. FASEB J. 2007;21:543–50. doi: 10.1096/fj.06-6694com. [DOI] [PubMed] [Google Scholar]
- 21.Berndt BE, Zhang M, Chen GH, Huffnagle GB, Kao JY. The role of dendritic cells in the development of acute dextran sulfate sodium colitis. J Immunol. 2007;179:6255–62. doi: 10.4049/jimmunol.179.9.6255. [DOI] [PubMed] [Google Scholar]
- 22.Block ER, Matela AR, SundarRaj N, Iszkula ER, Klarlund JK. Wounding induces motility in sheets of corneal epithelial cells through loss of spatial constraints: role of heparin-binding epidermal growth factor-like growth factor signaling. J Biol Chem. 2004;279:24307–12. doi: 10.1074/jbc.M401058200. [DOI] [PubMed] [Google Scholar]
- 23.Repertinger SK, Campagnaro E, Fuhrman J, El-Abaseri T, Yuspa SH, Hansen LA. EGFR enhances early healing after cutaneous incisional wounding. J Invest Dermatol. 2004;123:982–9. doi: 10.1111/j.0022-202X.2004.23478.x. [DOI] [PubMed] [Google Scholar]
- 24.Zieske JD, Takahashi H, Hutcheon AE, Dalbone AC. Activation of epidermal growth factor receptor during corneal epithelial migration. Invest Ophthalmol Vis Sci. 2000;41:1346–55. [PubMed] [Google Scholar]
- 25.Abe S, Sasano H, Katoh K, Ohara S, Arikawa T, Noguchi T, Asaki S, Yasui W, Tahara E, Nagura H, Toyota T. Immunohistochemical studies on EGF family growth factors in normal and ulcerated human gastric mucosa. Dig Dis Sci. 1997;42:1199–209. doi: 10.1023/a:1018897922644. [DOI] [PubMed] [Google Scholar]
- 26.Dignass AU. Mechanisms and modulation of intestinal epithelial repair. Inflamm Bowel Dis. 2001;7:68–77. doi: 10.1097/00054725-200102000-00014. [DOI] [PubMed] [Google Scholar]
- 27.Sturm A, Dignass AU. Epithelial restitution and wound healing in inflammatory bowel disease. World J Gastroenterol. 2008;14:348–53. doi: 10.3748/wjg.14.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laukoetter MG, Nava P, Nusrat A. Role of the intestinal barrier in inflammatory bowel disease. World J Gastroenterol. 2008;14:401–7. doi: 10.3748/wjg.14.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sebzda E, Zou Z, Lee JS, Wang T, Kahn ML. Transcription factor KLF2 regulates the migration of naive T cells by restricting chemokine receptor expression patterns. Nat Immunol. 2008;9:292–300. doi: 10.1038/ni1565. [DOI] [PubMed] [Google Scholar]
- 30.Mack PJ, Zhang Y, Chung S, Vickerman V, Kamm RD, Garcia-Cardena G. Biomechanical Regulation of Endothelium-dependent Events Critical for Adaptive Remodeling. J Biol Chem. 2009;284:8412–20. doi: 10.1074/jbc.M804524200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carlson CM, Endrizzi BT, Wu J, Ding X, Weinreich MA, Walsh ER, Wani MA, Lingrel JB, Hogquist KA, Jameson SC. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature. 2006;442:299–302. doi: 10.1038/nature04882. [DOI] [PubMed] [Google Scholar]
- 32.Wang X, Zheng M, Liu G, Xia W, McKeown-Longo PJ, Hung MC, Zhao J. Kruppel-like factor 8 induces epithelial to mesenchymal transition and epithelial cell invasion. Cancer Res. 2007;67:7184–93. doi: 10.1158/0008-5472.CAN-06-4729. [DOI] [PubMed] [Google Scholar]
- 33.Das A, Fernandez-Zapico ME, Cao S, Yao J, Fiorucci S, Hebbel RP, Urrutia R, Shah VH. Disruption of an SP2/KLF6 repression complex by SHP is required for farnesoid X receptor-induced endothelial cell migration. J Biol Chem. 2006;281:39105–13. doi: 10.1074/jbc.M607720200. [DOI] [PubMed] [Google Scholar]
- 34.Yang Y, Tetreault MP, Yermolina YA, Goldstein BG, Katz JP. Kruppel-like factor 5 controls keratinocyte migration via the integrin-linked kinase. J Biol Chem. 2008;283:18812–20. doi: 10.1074/jbc.M801384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tarnawski A. Molecular mechanisms of ulcer healing. Drug News Perspect. 2000;13:158–68. doi: 10.1358/dnp.2000.13.3.858438. [DOI] [PubMed] [Google Scholar]
- 36.Tarnawski A, Stachura J, Durbin T, Sarfeh IJ, Gergely H. Increased expression of epidermal growth factor receptor during gastric ulcer healing in rats. Gastroenterology. 1992;102:695–8. doi: 10.1016/0016-5085(92)90123-g. [DOI] [PubMed] [Google Scholar]
- 37.Tarnawski AS. Cellular and molecular mechanisms of gastrointestinal ulcer healing. Dig Dis Sci. 2005;50 (Suppl 1):S24–33. doi: 10.1007/s10620-005-2803-6. [DOI] [PubMed] [Google Scholar]
- 38.Usui S, Sugimoto N, Takuwa N, Sakagami S, Takata S, Kaneko S, Takuwa Y. Blood lipid mediator sphingosine 1-phosphate potently stimulates platelet-derived growth factor-A and -B chain expression through S1P1-Gi-Ras-MAPK-dependent induction of Kruppel-like factor 5. J Biol Chem. 2004;279:12300–11. doi: 10.1074/jbc.M305025200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.