

Abstract
Synapse loss is the strongest correlate of cognitive decline in Alzheimer’s disease, and synapses are an attractive therapeutic target due to their plastic nature that allows for potential recovery with intervention. We have previously demonstrated in transgenic mice that form senile plaques that dendrites surrounding plaques become dystrophic and lose postsynaptic dendritic spines. Furthermore, we found strong evidence that plaque-associated dendritic changes are mediated by calcineurin, a calcium-dependent phosphatase involved in cell signaling, using in vitro models and genetically encoded inhibitors in mouse models. In this study, we pharmacologically inhibited calcineurin with FK506 treatment to test the hypothesis that calcineurin inhibition will allow recovery of plaque-associated synapse loss. We found that in plaque bearing transgenic mice, short term (1 week) FK506 treatment results in an amelioration of dendritic spine loss. We also observe an effect on spine morphology in wild-type mice with FK506 treatment. These data show that systemic FK506 administration, and hence calcineurin inhibition, may be neuroprotective for amyloid beta induced synaptic alterations.
Keywords: Alzheimer, dendritic spine, FK506, tacrolimus, synapse, transgenic
Introduction
Morphological alterations in neurons in the Alzheimer disease (AD) brain including neurite curvature, dystrophic neurite swelling, and synapse and dendritic spines loss are thought to contribute to cognitive decline. In both Alzheimer’s disease and mouse models, dendritic spine loss is particularly severe near amyloid plaques, which are composed largely of amyloid beta (Aβ) (Moolman et al., 2004; Spires et al., 2005). Simplification of the dendritic arbor has also been observed in AD mouse models (Alpar et al., 2006), which could contribute to dysfunction in learning and memory (Poirazi and Mel, 2001). Aβ is toxic to synapses and disrupts cognitive function (Cleary et al., 2005; Rowan et al., 2004), thus it may underlie dendritic spine loss near plaques. Snyder et al elegantly showed in cultured cortical neurons that Aβ promotes endocytosis of NMDA receptors, and that this requires α7-nAchRs and downstream pathways including calcineurin activation (Snyder et al., 2005). Although Aβ is a strong candidate for causing synapse loss around plaques, other plaque associated agents could also contribute including free radicals and microglial activation (Garcia-Alloza et al., 2006; Meyer-Luehmann et al., 2008).
We have recently observed that calcineurin mediates Aβ-induced morphological changes including spine loss and dendritic simplification in cultured neurons and that inhibition of calcineurin in AD model mice with a genetically encoded inhibitor can reverse plaque-associated pathologic alterations in dendrites and dendritic spines (Wu et al., 2010). Here we sought to test whether pharmacological inhibition of calcineurin with FK506, a commonly used, FDA approved drug, can also cause recovery of morphological alterations in AD model mice, since this would be a potential route for therapeutic intervention in AD. Our previous study injecting virus into the brain to inhibit calcineurin had beneficial effects but clearly is not a viable therapeutic administration route for humans. It has been reported previously that injection of FK506 has restored some cognitive function in AD mouse models (Dineley et al., 2007), which could be explained by recovery of dendritic spines. Calcineurin is also known to play critical roles in long term potentiation and long term depression phenomena in slice cultures, and manipulation of calcineurin impacts learning and memory in mouse models (Dineley et al., 2010; Klee et al., 1979; Winder and Sweatt, 2001).
Calcineurin inhibitor FK506 (tacrolimus), acts via the complex of FK506 and FK506-binding protein binding to calcineurin to prevent calcineurin-mediated dephosphorylation (Schreiber and Crabtree, 1992). It is a commonly used immunosuppressant to combat graft versus host disease after transplant surgery (1994); however, serious neurological side effects occur in over 10% of FK506 treated patients including tremor, aphasia, cortical blindness, hallucinations, and memory impairment (Lee et al., 2008; Wijdicks et al., 1994). Thus we also investigated the effects of FK506 on non-transgenic mice to ensure that any benefits seen in plaque-bearing mice would not come with the risk of serious side effects.
We find a recovery of dendritic spine density on cortical pyramidal neurons with one week of systemic FK506 administration, confirming a role for calcineurin in the synaptotoxic cascade associated with amyloid beta. We also observe changes in spine morphology in wild-type brain with this treatment, providing a morphological correlate to changes in learning and memory previously reported.
Materials and Methods
FK506 treatment of mice
Mice expressing yellow fluorescent protein (YFP) in a subset of pyramidal neurons (Feng et al., 2000), and mice expressing AD-associated amyloid precursor protein and presenilin 1 (Jankowsky et al., 2001) as well as YFP (YFP-APP/PS1) from Jackson laboratories (Bar Harbor, ME) were group housed in standard rodent cages with ad libitum access to water and mouse chow. YFP-APP/PS1 mice were aged between 17 and 20 months and YFP mice were aged between 18 and 33 months. A bolus of 0.1 mL/ 10g of mouse of 10mg/mL FK506 (Sigma Aldrich, St. Louis, MO - dissolved in 10% ethanol with 1% tween 80 in PBS (Butcher et al., 1997)), vehicle (10% ethanol with 1% tween 80 in PBS without drug), or no treatment was administered i.p. daily for 7 days. We saw no difference between vehicle treated and untreated animals in any parameters measured so they are grouped together. We have four groups (two genotypes and two treatment paradigms). An average of 5 animals were treated per group with an average of 138 dendrites and 2414 spines analyzed per group (see table 1 for exact numbers in each group).
Table 1.
Numbers of animals, dendrites, and spines analyzed per group
| genotype | treatment | n animals | n dendrites | n spines |
|---|---|---|---|---|
| YFP | control | 4 | 88 | 1565 |
| YFP | FK506 | 3 | 76 | 1554 |
| YFP/APP | control | 8 | 221 | 3660 |
| YFP/APP | FK506 | 6 | 166 | 2876 |
Tissue Preparation, imaging, and image analysis
Mice were placed in a CO2 chamber until they stopped breathing then perfused using PBS followed by 4% paraformaldehyde and 0.1% Gluteraldehyde in PBS. Brains were kept in fixative solution at 4° C for 48 hours. Before cutting brains were switched to 30% sucrose for a further 48 hours. 50 µm frozen sections were cut on a Microm HM400 freezing microtome and mounted on glass slides. Plaques were stained with thioflavin S (Sigma) in 50% ethanol for 8 minutes followed by differentiation in 80% ethanol for 10 seconds. Image stacks (90µm × 90µm × 5–10µm with a z step 0.5 µm – 25× zoom 4, NA 0.8) of apical and basal dendritic segments from cortical layer II–III pyramidal neurons were acquired on a Zeiss LSM510 confocal/multiphoton microscope using laser excitation at 488nm for YFP, 2-photon excitation at 800nm for thioflavin S, and 543nm for smi312 staining with Cy3 conjugated secondary. Dendrite selection was made without regard to plaque proximity (in a different channel so plaques were not visible). Branches off of apical dendrites and basal dendrites were selected based on the ability to visualize more than 20 micron length of dendrite. Using Image J, the distance of each dendrite segment to the nearest plaque (if present) was measured. Neuron studio (Rodriguez et al., 2008) was used to automatically detect dendrites and dendritic spines (including length and morphological class) and an observer blind to treatment and genotype corrected any mis-detected spines. Linear spine density and spine length were calculated. Dystrophic neurites (swellings greater than 5 µm in diameter) were outlined in z-projections in image J and the area measured. The mean dystrophy area was calculated for each animal (n=3 animals, 283 dystrophies in FK506 treated mice and 5 animals, 451 dystrophies in vehicle treated mice).
Data analysis
For dendritic spine density and length data, normality of data was confirmed using a Shapiro-Wilkes test. 2-way ANOVAs with genotype and treatment as independent variables and post-hoc Tukey-Kramer tests were used to asses the effects of treatment on the two genotypes. Data are presented as means (of all dendrites per group in the case of spine density and all spines per group in the case of spine length) with associated standard deviations as previously published (Spires et al., 2005). Animals within groups did not significantly differ as assessed by one-way ANOVA. For spine morphology data, contingency table analyses were used to compare all spines in each treatment group.
Results
To test the hypothesis that pharmacological inhibition of calcineurin ameliorates morphological degeneration of neurons associated with amyloid pathology, we treated mice that express YFP in a subset of pyramidal neurons and YFP mice that also express mutant human APP and presenilin 1 with FK506 daily for 7 days. FK506 treatment was not associated with morbidity. YFP filled neurons were examined near and far from plaques using confocal microscopy (figure 1), and dendritic spine density, length, and shape were measured in image stacks. We observe a 14% decrease in dendritic spine density in YFP-APP/PS1 mice treated with vehicle compared to YFP mice treated with vehicle (figure 2a, p=0.0005 Tukey-Kramer post-hoc comparison). This decrease in spines is exacerbated near plaques (figure 2b, significant correlation of 0.2678 of spine density with distance of the dendrite from a plaque vehicle p=0.0037 Spearman’s rho test). We have observed similar decreases of spine density in proximity to plaques in other mouse models (Spires-Jones et al., 2007; Spires-Jones et al., 2009; Spires et al., 2005). With FK506 treatment, spine density significantly increases by 8.5% in YFP-APP/PS1 mice compared to YFP-APP/PS1 treated with vehicle (p=0.0381 Tukey-Kramer post-hoc comparison) bringing spine density back close to control levels (FK506 treated YFP-APP/PS1 density is not significantly different from YFP vehicle treated or YFP FK506 treated spine density). There is still a significant correlation between plaque distance and spine density (0.2653 p=0.0089). Since dendritic spines form the postsynaptic element of the majority of excitatory synapses in cortex and they are lost in AD and mouse models, these results show that 7 days of FK506 treatment is beneficial in terms of reversing synapse loss.
Figure 1.

Dendritic spines were imaged in YFP mice (A, B) and YFP mice also expressing APP and PS1 transgenes that have senile plaques labeled blue with ThioS (C, D). Mice were treated with either vehicle (A, C) or FK506 (B, D) by i.p. injection for 7 days. Neuron studio (Rodriguez et al., 2008) was used to reconstruct dendrites and detect spines (bottom panels show reconstructions). Scale bars represent 20 µm in top panels, 10 µm in bottom panels.
Figure 2.

Analysis of dendritic spine density reveals that in vehicle treated animals, there is a 14% reduction in spine density in YFP-APP/PS1 mice compared to YFP mice without the APP and PS1 transgenes (A). This is rescued with 7 days of FK506 treatment. In both vehicle and FK506 treated groups, spine density correlated with distance from the nearest plaque (B), indicating the effects of FK506 are greater on dendrites farther from plaques, since many of the dendrites analyzed had no plaque within measurable distance (greater than 100 µm away). * p<0.05 post-hoc Tukey-Kramer test.
While the number of dendritic spines scales with synapse number, the morphology of dendritic spines affects the integration of synaptic inputs by controlling the passive flow of depolarization from the site of the synapse at the spine head into the parent dendrite. Thus we also examined dendritic spine morphology (length and shape classification) in FK506 treated mice (figure 3). We found that at baseline, YFP-APP/PS1 mice have shorter spines than YFP mice in the vehicle treated condition (p=0.0051 post-hoc Tukey-Kramer test). This agrees with an observed differential distribution of spine shapes in YFP-APP/PS1 mice with more of the shorter stubby spines and less of the longer thin and mushroom shaped spines (contingency table analysis Pearson’s test p=0.0004).
Figure 3.

Dendritic spines were classified by shape (A) as either mushroom (arrows), thin (arrowhead) or stubby (asterisk) based on the ratio of head to neck with. Length of dendritic spines was also measured from the base to tip (B) showing that spines are significantly shorter in YFP-APP/PS1 mice than in YFP cortical pyramidal neurons with vehicle treatment. Spine length was reduced by FK506 treatment in YFP mice compared to vehicle treated YFP mice resulting in lengths even shorter than YFP-APP/PS1 mice, indicating negative effects of this treatment on healthy brain. The shorter length of spines both with plaques and FK506 treatment appears to be due to a shift in the shapes of spines from longer thin and mushroom to shorter stubby spines (C). * p<0.005 0001 post-hoc Tukey-Kramer test in B, contingency table analysis Pearson’s test in C. Scale bar represents 2 µm.
FK506 treatment does not affect spine length in YFP-APP/PS1 mice. However, we detected a decrease in spine length in YFP mice without the APP and PS1 transgenes (p<0.0001 post-hoc Tukey-Kramer test). FK506 treatment also does not have an effect on the morphology of YFP-APP/PS1 spines but does significantly alter the YFP spines to have more stubby and fewer thin and mushroom spines (contingency table analysis Pearson’s test p<0.0001).
Dystrophic swellings of axons and dendrites around senile plaques have been observed in AD and mouse models and can be reversed by removing Aβ with antibody treatment or by treatment with genetically encoded calcineurin inhibitors (Spires-Jones et al., 2009; Wu et al., 2010). We measured the cross-sectional area of YFP positive dystrophic swellings and found no difference in size of plaque-associated dystrophies with FK506 treatment (figure 4). Since YFP is expressed only in a subset of neurons, we used immunohistochemical staining with an axonal neurofilament marker to assess the number of dystrophic axonal swellings per plaque and found that this was also unchanged with FK506 treatment (figure 4). This could indicate that our treatment was not long enough to reverse dystrophic swelling or that the concentration of drug was not high enough for this effect.
Figure 4.
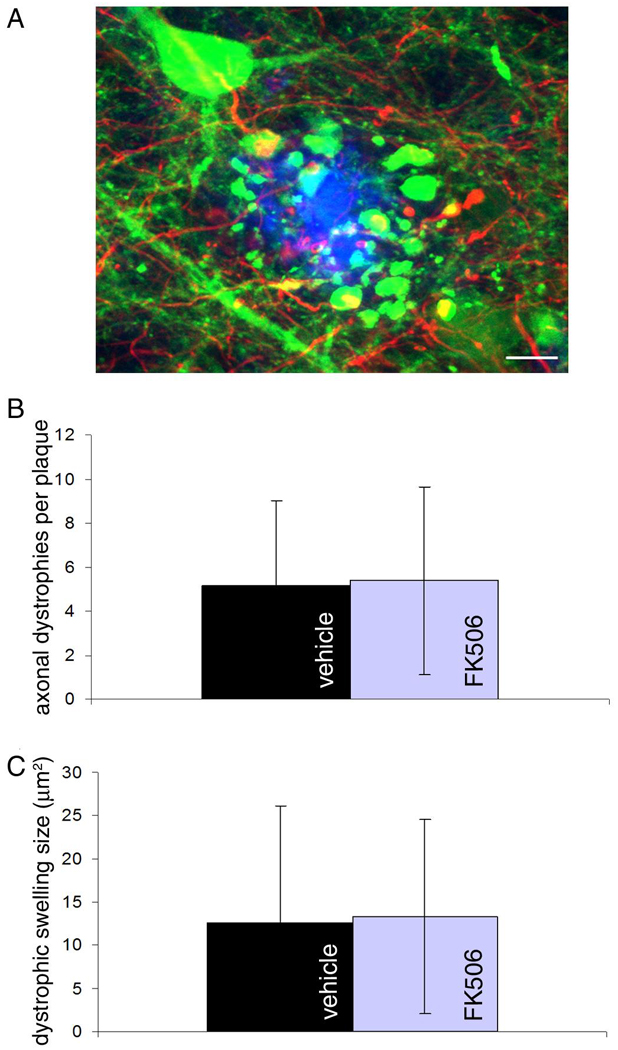
Dystrophic neurites filled with YFP (green) and axonal dystrophies stained with smi312 (red) were analyzed for size and number per plaque, respectively (A). FK506 treatment does not rescue either axonal dystrophy number per plaque (B) or YFP dystrophy size (C). Scale bar represents 5 µm.
Together these data indicate that 1 week of FK506 treatment is sufficient to have beneficial effects on dendritic spine density in APP/PS1 mice, particularly far from plaques; however, these benefits did not extend to spine length, spine morphology, or dystrophic swellings. Further, we observed effects of FK506 treatment on spine length and morphology in healthy mice.
Discussion
Despite the strong correlation between synapse loss and cognitive decline in AD, very few treatments have been found to cause recovery of synaptic loss. Removing amyloid β with immunotherapy causes rapid recovery of synaptic structural plasticity (Spires-Jones et al., 2009) longer-term increases in synaptic proteins (Rozkalne et al., 2009), and improvements in cognition (Lee et al., 2006) in mice with established plaque pathology, indicating that amyloid β contributes to synapse loss and that removing it allows synaptic recovery. We have previously observed that constitutively active calcineurin expressed in wild-type brain using gene transfer causes spine loss, neurite curvature, and dystrophic swellings, while expression of a genetically encoded calcineurin inhibitor reversed these changes in cultured neurons treated with Aβ and in APP/PS1 mice (Wu et al., 2010), indicating that calcineurin activation is downstream of Aβ in causing synapse loss. Here we sought to confirm these findings using a readily available pharmacological inhibitor of calcineurin, which would be more suitable than gene transfer for therapeutic administration in patients. We observe a loss of dendritic spines on cortical pyramidal neurons in plaque-bearing APP/PS1 mice which is exacerbated near plaques as has been previously reported in several different transgenic models (Alpar et al., 2006; Moolman et al., 2004; Spires-Jones et al., 2009; Spires et al., 2005; Tsai et al., 2004). Calcineurin inhibition with FK506 treatment for 7 days results in a rescue of spine density in APP/PS1 mice, confirming that calcineurin is an important downstream effector of Aβ mediated spine loss. Since dendritic spines are the post synaptic element of over 90% of cortical synapses and newly generated spines do form synapses (Knott et al., 2006), we interpret these data to mean calcineurin inhibition rapidly ameliorates synapse loss in AD model mice.
Despite this positive effect on dendritic spine density, this exposure to FK506 treatment did not reduce dystrophic neurite size or number per plaque, and treatment did not rescue spine length and morphology changes seen in YFP-APP/PS1 mice. This contrasts with our previous observation that overexpressing a genetically encoded calcineurin inhibitor directly in neurons near plaques for 2–4 weeks could ameliorate plaque associated varicosities (Wu et al., 2010); we interpret this result to suggest that the genetically encoded calcineurin inhibitor was present continuously for a longer duration and likely at a higher local dose that we could achieve with daily systemic administration of FK506. Nonetheless, the recovery of spine density in APP/PS1 mice and even the changes observed in spine morphology in wild type mice suggest that (1) FK506 is able to access the brain in doses sufficient to impact dendritic spines, and (2) that calcineurin inhibition can improve Aβ–induced spine loss in an AD model. The latter point confirms the conclusion that calcineurin is an important mediator of Aβ–induced spine toxicity using a different mechanism of calcineurin inhibition (pharmacological rather than genetic) and systemic (rather than local) route of administration. These results provide an important proof of principle that calcineurin inhibition is a potent approach to ameliorating Aβ–induced synaptotoxicity.
Our data do not directly address whether FK506 induced morphological changes improve neural system function. The retention of PSD95 at the synapse depends to some extent on the shape of the spine (Gray et al., 2006) and calcium compartmentalization is thought to occur in spines with necks (excluding short stubby spines) (Bloodgood and Sabatini, 2007). Thus the increased proportion of stubby spines in APP/PS1 mice and in healthy brain with FK506 treatment may both change dynamics of synaptic proteins and allow more calcium to enter the parent dendrite. In fact, a loss of decoupling between spine and dendrite calcium as well as an increase in calcium concentration in a subset of dendrites associated with plaques has been observed using calcium imaging in APP/PS1 mice in vivo (Kuchibhotla et al., 2008). On the other hand, a single injection of FK506 (10 mg/kg, i.p.) 6 hours before behavioral testing has been shown to improve cognition in both APP overexpressing Tg2576 mice and wild-type mice with exogenously injected oligomeric Aβ-induced cognitive deficits (Dineley et al., 2007; Dineley et al., 2010). FK506 has also been reported to be neuroprotective in animal models of stroke, trauma, and glaucoma (Hailer, 2008; Huang et al., 2005; Macleod et al., 2005). Further studies directly comparing behavioral and morphological outcomes of calcineurin inhibition will be necessary to refine the relationship between these structural and functional markers of neuroprotection.
Acknowledgements
This work was supported by K99 AG033670-01A1, Alzheimer’s disease Drug Discovery Foundation/Association for Frontotemporal Dementias, P50 AG005134, AG08487.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- A comparison of tacrolimus (FK 506) and cyclosporine for immunosuppression in liver transplantation. The U.S. Multicenter FK506 Liver Study Group. N Engl J Med. 1994;331:1110–1115. doi: 10.1056/NEJM199410273311702. [DOI] [PubMed] [Google Scholar]
- Alpar A, et al. Different dendrite and dendritic spine alterations in basal and apical arbors in mutant human amyloid precursor protein transgenic mice. Brain Res. 2006;1099:189–198. doi: 10.1016/j.brainres.2006.04.109. [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Sabatini BL. Ca(2+) signaling in dendritic spines. Curr Opin Neurobiol. 2007;17:345–351. doi: 10.1016/j.conb.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Butcher SP, et al. Neuroprotective actions of FK506 in experimental stroke: in vivo evidence against an antiexcitotoxic mechanism. J Neurosci. 1997;17:6939–6946. doi: 10.1523/JNEUROSCI.17-18-06939.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JP, et al. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Dineley KT, et al. Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol Learn Mem. 2007;88:217–224. doi: 10.1016/j.nlm.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, et al. Amyloid-beta oligomers impair fear conditioned memory in a calcineurin-dependent fashion in mice. J Neurosci Res. 2010 doi: 10.1002/jnr.22445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Garcia-Alloza M, et al. Plaque-derived oxidative stress mediates distorted neurite trajectories in the Alzheimer mouse model. J Neuropathol Exp Neurol. 2006;65:1082–1089. doi: 10.1097/01.jnen.0000240468.12543.af. [DOI] [PubMed] [Google Scholar]
- Gray NW, et al. Rapid redistribution of synaptic PSD-95 in the neocortex in vivo. PLoS Biol. 2006;4:e370. doi: 10.1371/journal.pbio.0040370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailer NP. Immunosuppression after traumatic or ischemic CNS damage: it is neuroprotective and illuminates the role of microglial cells. Prog Neurobiol. 2008;84:211–233. doi: 10.1016/j.pneurobio.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Huang W, et al. Calcineurin cleavage is triggered by elevated intraocular pressure, and calcineurin inhibition blocks retinal ganglion cell death in experimental glaucoma. Proc Natl Acad Sci U S A. 2005;102:12242–12247. doi: 10.1073/pnas.0505138102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, et al. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Klee CB, et al. Calcineurin: a calcium- and calmodulin-binding protein of the nervous system. Proc Natl Acad Sci U S A. 1979;76:6270–6273. doi: 10.1073/pnas.76.12.6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott GW, et al. Spine growth precedes synapse formation in the adult neocortex in vivo. Nat Neurosci. 2006;9:1117–1124. doi: 10.1038/nn1747. [DOI] [PubMed] [Google Scholar]
- Kuchibhotla KV, et al. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, et al. Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem. 2006;281:4292–4299. doi: 10.1074/jbc.M511018200. [DOI] [PubMed] [Google Scholar]
- Lee SH, et al. Calcineurin inhibitor-mediated bilateral hippocampal injury after bone marrow transplantation. J Neurol. 2008;255:929–931. doi: 10.1007/s00415-008-0612-5. [DOI] [PubMed] [Google Scholar]
- Macleod MR, et al. Systematic review and metaanalysis of the efficacy of FK506 in experimental stroke. J Cereb Blood Flow Metab. 2005;25:713–721. doi: 10.1038/sj.jcbfm.9600064. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M, et al. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moolman DL, et al. Dendrite and dendritic spine alterations in alzheimer models. J Neurocytol. 2004;33:377–387. doi: 10.1023/B:NEUR.0000044197.83514.64. [DOI] [PubMed] [Google Scholar]
- Poirazi P, Mel BW. Impact of active dendrites and structural plasticity on the memory capacity of neural tissue. Neuron. 2001;29:779–796. doi: 10.1016/s0896-6273(01)00252-5. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, et al. Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One. 2008;3:e1997. doi: 10.1371/journal.pone.0001997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan MJ, et al. Mechanisms of the inhibitory effects of amyloid beta-protein on synaptic plasticity. Exp Gerontol. 2004;39:1661–1667. doi: 10.1016/j.exger.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Rozkalne A, et al. A single dose of passive immunotherapy has extended benefits on synapses and neurites in an Alzheimer's disease mouse model. Brain Res. 2009;1280:178–185. doi: 10.1016/j.brainres.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunol Today. 1992;13:136–142. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- Snyder EM, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Spires-Jones TL, et al. Impaired Spine Stability Underlies Plaque-Related Spine Loss in an Alzheimer's Disease Mouse Model. Am J Pathol. 2007;171:1304–1311. doi: 10.2353/ajpath.2007.070055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones TL, et al. Passive immunotherapy rapidly increases structural plasticity in a mouse model of Alzheimer disease. Neurobiol Dis. 2009;33:213–220. doi: 10.1016/j.nbd.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, et al. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai J, et al. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004;7:1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- Wijdicks EF, et al. FK506-induced neurotoxicity in liver transplantation. Ann Neurol. 1994;35:498–501. doi: 10.1002/ana.410350422. [DOI] [PubMed] [Google Scholar]
- Winder DG, Sweatt JD. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat Rev Neurosci. 2001;2:461–474. doi: 10.1038/35081514. [DOI] [PubMed] [Google Scholar]
- Wu H-Y, et al. Amyloid {beta} Induces the Morphological Neurodegenerative Triad of Spine Loss, Dendritic Simplification, and Neuritic Dystrophies through Calcineurin Activation. J. Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]