

Abstract
Metastatic cancers account for more than 90% of cancer mortality. The metastasis of all cancers is critically mediated by enzymes that degrade extracellular matrix. Aggressive tumors are characterized by an imbalance between enzymes that degrade ECM and endogenous inhibitors of the enzymes. Matrix metalloproteinases (MMPs) make up the majority of ECM degrading enzymes implicated in cancer metastasis. The potent MMP inhibitory activities of tetracyclines, especially their chemically modified analogs, combined with their relatively well tolerated pharmacological profile, led several researchers to investigate their anticancer potential in a variety of cancers, including melanoma, lung, breast and prostate cancers. Chemically modified non-antibiotic tetracyclines (CMT, or COL) were tested using tumors of prostate, breast and melanomas. Some of these CMTs, notably, CMT-3 and CMT-308 significantly inhibited not only invasive potential and MMP activity, but also inhibited cell proliferation by inducing cell cycle arrest and apoptosis. CMT-3 and CMT-308 were significantly more potent than doxycycline or minocycline in inhibiting tumor cell-derived MMPs and inducing apoptosis in vitro and in vivo. CMT-3 (Col-3) showed potent inhibition of tumor growth in xenografts and in bone metastatic models of prostate cancer. Similar results were also reported in melanoma and breast cancer models. The mechanism by which CMTs kill tumor cells is via generation of hydroxyl free radicals ([OH]−) which permeate and depolarize mitochondria, which in turn activates caspase mediated apoptosis. Analysis of tumor tissues from CMT-3 treated rats demonstrated reduction in angiogenesis and increase in apoptosis; both emerged as mechanisms of CMT action. These observations led to testing the efficacy of CMT-3 in human clinical trials against several types of cancer with significant outcomes, which are described in the next chapter of this issue.
Keywords: prostate cancer, bone metastasis, Matrix metalloproteinase, Free radicals, apoptosis
1. Introduction
Recent estimates suggest that more than half a million patients in the United States of America will die of cancer in 2010 [1]. Despite all available treatments, cancer is the second most common cause of deaths in most countries. More than 90% of cancer deaths are due to metastasis of initially treated cancers. Invasion of healthy tissue and growth in that location, often distant from the initial site of the tumor is considered the most challenging aspect of treatment. Although we know a great deal about the mechanisms of invasion, many processes remain impervious to treatment. However, seminal discoveries in the last two decades on the molecular aspects of spread of the solid tumors, such as prostate, breast and lung cancers, have shown that matrix metalloproteinases (MMPs) play central roles in both the initial process of invasion and extravasations and in the metastatic growth of tumors, and also serve as activators of growth factors and of chemoattractant proteins [2–4]. While in normal tissue remodeling MMP activities are critical, such as collagen matrix remodeling in normal bone turnover, excessive activity has been linked to several disease processes as described in other chapters in this volume.
2. CMTs efficacy in prostate cancer models
Prostate cancer ranks second in incidence and death of cancer in men. In the United States, in 2009, an estimated 192, 280 men were diagnosed with prostate cancer and an estimated 27,360 died of this disease [1]. Although, over 70% of the patients are cured of this disease (> five-years of disease-free survival) upon primary therapy such as surgery or radiation, it does progress in more than 30% of the patients. The first sign of prostate cancer metastasis following radical prostate surgery, localized radiation (branchy therapy), or focused radiation, is resurgence of the prostate cancer marker, prostate specific antigen (PSA) in the circulation [5]. This initial increase in serum PSA is termed biochemical recurrence (clinically defined as an increase in serum PSA during two consecutive 6-month follow up visits [5–7]). The biochemical recurrence is ≥34% within five years after initial diagnosis and 46% after 10 years; thus prostate cancers progress slowly. Regardless of the low rate of recurrence, prostate cancer causes substantial morbidity and pain in patients who have failed treatment for recurrent disease, including hormonal therapy (total depletion of androgen by chemical means), local radiation or both [8]. The main source of pain, debilitation and ultimate death is metastasis of the disease to bone (lumbar, vertebral and even metastasis to skull) [9, 10].
Our group [11] and Stearns et al [12] reported in 1993 that prostate tumor tissues and primary cultures of prostate cancer specimens as well as the established cell lines, produce increased quantities of MMPs, especially gelatinase A (MMP-2) and gelatinase B (MMP-9), with decreased levels of the tissue inhibitors of metalloproteinase (TIMP-1 and TIMP-2) [12, 13]. We used the classical MMP activity assay based on degradation of gelatin prepared from denatured, 3H- labeled rat tail collagen with zymography and reverse-zymography. These observations have been confirmed and extended in both animal models and human tissue samples [14, 15]. Our interest in the use of CMTs as anticancer agents resulted from these findings, which were stimulated by the discovery of the anticollagenase activity of CMTs and their application in treating periodontal diseases [16–18]. We proposed that tetracycline could be an ideal drug for use against metastasis of tumors to bone and other connective tissues, where the tumor cells establish a colony by extravasation from circulation, adhesion and eventually creation of angiogenic network to obtain nutrient supply [19]. An additional advantage of tetracyclines was their well-known affinity to bind to bone which would potentially provide a depot of the drug against invading or proliferating tumor cells [20].
2.1. Effect of CMTs on bone metastasis
Metastasis to bone is found in >95% autopsies of prostate cancer victims, indicating metastasis as the main cause of death [21]. Although prostate cancer metastasis to bone is very frequent, animal models of this cancer that spontaneously develop bone metastasis are few. Even highly aggressive transgenic prostate cancer models, such as Transgenic Adenocarcinoma of the prostate (TRAMP) and PTEN-conditional prostate knockout (prostate-PTEN KO) models, seldom form extensive bone metastasis spontaneously, although micro metastases are common [22,23]. However, several xenograft models using forced injection of tumor cells into femoral, tibial or tarsal bones have been used to test anti-bone metastasis drugs [24–26].
Accordingly, we used a classical approach to test the effect of CMTs and doxycycline for their potential inhibitory activity against bone metastasis. We observed extensive bone metastasis in a rat model of prostate cancer, the Dunning MAT Ly Lu, after intravenous injection of tumor cells while clamping the vena cava [27,28] in the male Copenhagen strain. This resulted in forced invasion of tumor cells into the lumbar vertebral venous plexus (Batson’s plexus) and extensive vertebral metastasis [29,30]. In addition, the Dunning MAT LyLu tumors also spread to lungs and form extensive lung metastasis [31].
The efficacy of CMT-3 was tested in this bone metastasis model by daily oral gavage of the drug (40 mg/kg) suspended in 2% carboxymethyl cellulose (vehicle), beginning 7 days before tumor cell injection (-7d). The end points of the study were the time needed to develop paraplegia due to lower vertebral metastasis and development of morbidity due to lung metastasis. Most animals developed acute pulmonary distress with or without paraplegia starting 12 days after injection of tumor cells. However, in the CMT-3 treated groups, we observed a significant delay in the development of pulmonary distress and paraplegia. Overall survival, beyond the time when all rats in the vehicle-only treated group died, was 10% to 30%, and depended on the timing of CMT-3 dosing; animals dosed beginning 2 days after tumor implant had only modest improvement in survival (10%) whereas animals predosed 7 days prior to tumor cell injection survived 30% longer (median survival 18 days, versus 13 days in vehicle only treated group). Interestingly, in this experiment, while 83% of the rats in the vehicle only group developed paraplegia at the time of euthanasia, only 17% developed paraplegia in the 7-day pretreatment group and 33% in post-tumor implant treatment group. Although, the rats gavaged with the vehicle developed extensive pulmonary metastasis, CMT-3 treatment groups had fewer lung metastases [27]. The inhibition of paraplegia by CMT-3 was >85%. One out of 7 rats developed paraplegia in the group dosed with CMT-3 starting 7 days before tumor cell injection. Similar increase in survival (percent of animals that are moribund) 12, 15 or 22 days after tumor cell injection was also observed in the CMT-3 gavaged animals versus vehicle (2% carboxy-methyl cellulose in sterile water) treated group. Furthermore, the incidence of lung tumors were also significantly reduced by ≥ 70% in this group, indicating that CMT-3 is effective in inhibiting bone metastasis in a rapidly metastasizing tumor model.
2.2. CMT-3 inhibits tumor-stroma interaction and inhibits tumor cell-induced stromal secretion of MMPs
Inhibition of bone metastasis and the reduction in the frequency of paraplegia in rats treated with CMT-3 was further examined by measuring the levels of biochemical markers of collagen degradation including the collagen cross link fragments, pyridinoline (PD) and deoxypyridinoline (DPD), in serum and urine. CMT-3 treatment produced a significant reduction in the level of PD (p≤0.03) in urine samples collected at the time of euthanasia (Gr. IV, Fig 1). The levels were elevated in the group of rats injected with tumor cells but only given the vehicle. There was an initial increase in the levels of DP in all groups, an increase of 54% ± 8.0 % when compared to naive animals. This level was reduced in the second and third weeks of treatment in CMT-3 treated group but not in those treated with vehicle alone. These results suggested that oral administration of the drug reduces bone matrix breakdown despite the tumors growing in the bone stroma.
Fig 1. CMT-3 Inhibition of experimental prostate cancer metastasizing to bone and lung in the Dunning MAT-LyLu model.
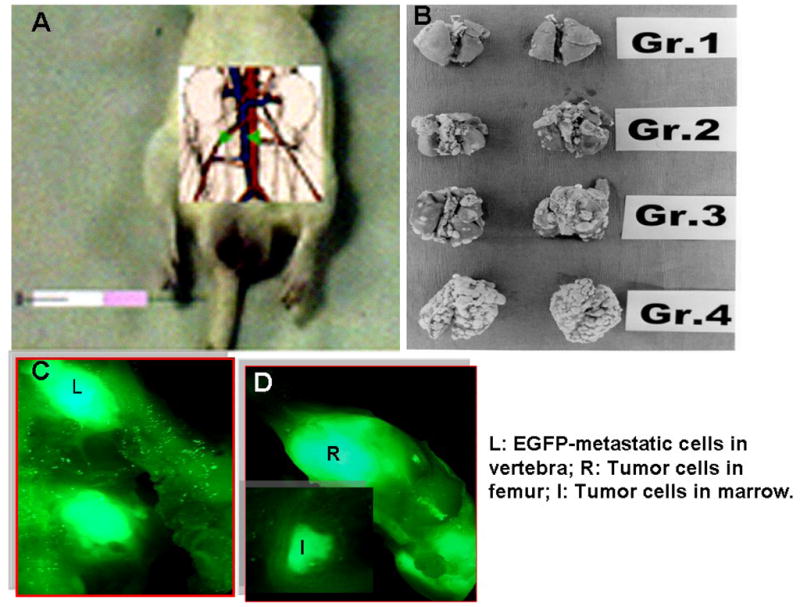
The MAT Ly Lu tumor cells were injected i.v following vena cava clamping with a mini surgical bulldog clamp. As described in the text, animals were orally dosed with CMT-3 or vehicle -7 days (Group 1),+ 1 day (Group 2), or +3 days (Group 3) and vehicle-only (Group 4) [Panel B]. In an independent experiment, MAT LyLU cells transfected with EGFP were injected as shown in Fig. 1A and were detected as dispersed in femoral bones (Fig. 1C and D). Cells isolated from marrow plugs were cultured to confirm tumor cells (data not shown).
This observation prompted us to determine whether the tumor cells induced the surrounding stromal cells to secrete collagenases. In fact, the MAT Ly Lu cells did induce the secretion of MMP-2 in endothelial cells as well as in bone marrow derived stromal cells [28]. This induction of MMP-2, which was increased by >200% over the basal levels, was effectively abolished by addition of CMT-3 (5μM) to the stromal cultures. These results demonstrated that CMT-3 can effectively inhibit bone metastasis in a highly metastatic model of prostate cancer (Dunning MAT LyLu).
2.3. CMTs inhibit tumor growth in vivo and cell proliferation, and also induce apoptotic cell death in tumor cells
In an effort to understand the broader role of CMTs as antitumor agents, we used the traditional antitumor activity assay in xenograft (subcutaneous tumor cell injection) models of prostate cancer. The antitumor activity of CMT-3 and doxycycline was tested in mice bearing subcutaneous xenografts of PC-3 tumors, a human castration-resistant prostate tumor model, and against the rat MAT LyLu tumors. Four treatment groups were used. In the first group the animals were orally gavaged with vehicle (2% CMC) while the second and third groups were pre-dosed with CMT-3 or doxycycline (DC), 40 mg/kg, respectively. The fourth and fifth groups were dosed, starting from the day of tumor cell injection. All dosing was stopped 28 days after tumor cell injection in rats or day 42 in mice, or the animals were euthanized due to morbidity. Tumor cells were implanted into lower flanks of male Copenhagen rats at 2 ×105 MAT Ly Lu tumor cells/site and 1 ×10 6 PC-3 cells/site in athymic nude mice.
We reported the results of these experiments in detail elsewhere [32, 34]. In brief, reductions in tumor incidence, growth rate and spontaneous metastases to lungs (MAT Ly Lu model only) were observed following CMT-3 therapy in both models. Specifically, predosing the rats was most effective; 50% (4/8) of the rats exhibited no tumor incidence in the treated animals. Although both DC and CMT-3 were effective in reducing tumor growth, the tumor burden was significantly lower in rats orally gavaged with CMT-3 compared to the vehicle-only control group.
CMT-3 was also more effective than DC in inhibiting tumor growth in PC-3 tumors. Although there was a significant reduction in tumor incidence (35%, 3/7) in the CMT-3 treated group, this effect was not seen in DC treated rats. Moreover, there was a significant decrease in tumor incidence in the group that received CMT-3 pre-dosing, while there was no reduction in tumor incidence in the group pre-dosed with DC. When dosing was initiated after the tumor cells were injected neither CMT-3 or DC was effective in reducing tumor incidence. Tumor growth rate was also significantly decreased; 27% and 54% decreases were observed in tumor volumes following 42-days of oral gavage of DC or CMT-3, respectively. Since PC-3 tumor cells rarely form metastasis to lungs in athymic mice, following subcutaneous injections, we did not find consistent tumor foci in lungs or liver in either vehicle-only or drug gavaged mice.
Reduction in tumor growth prompted us to investigate the antiproliferative activities of CMT-3. We have performed extensive biochemical and cellular characterization of the mechanism of action of CMT-3 and DC in human prostate tumor cells. The results of these experiments are reported elsewhere [35, 36]. The following is a brief summary of our observations.
2.4. Mechanism of antitumor activities of CMTs and DC: CMTs inhibit cell proliferation and inhibit cell cycle progression
We tested the anti-proliferative and cytotoxic activities of CMTs (CMT-1 through CMT-8) and the nitro-derivatives of CMT-3 on several prostate cancer cell lines and in normal fibroblasts [36]. Among all the CMTs and Nitro-CMT3 analogs tested, CMT-3 was the most potent and specific inhibitor of cell proliferation in proliferating tumor cells and in fibroblasts, with 50% growth inhibition (GI50) at concentrations between 2.3 to 9.3 μM, depending on the proliferation activity of the cells. Slow growing cells such as primary prostate epithelial cells had higher GI50. This GI50 dose was nearly two to 5-fold lower than that of DC indicating that CMT-3 is a more potent inhibitor of cell proliferation than DC. Cell proliferation inhibition was also supported by experiments which measure the potential inhibition of cell cycle-progression. Cultures treated with CMT-3 showed increased accumulation in G1-S phase transition, suggesting that CMT-3 inhibits mitogenic signaling. We are unaware of any other publication that demonstrate the molecular mechanism of cell proliferation or cell cycle blockage by CMTs or other tetracycline compounds in mammalian cells.
2.4.2. CMTs induce apoptosis in tumor cells
We and others have reported the proapoptotic function of CMTs, especially CMT-3 and CMT-308 (9-amino-CMT-3), in a variety of human tumor cells. CMT-3 and to a significantly lesser extent, DC, are cytotoxic to tumor cells. At a given dose, above GI50 (e.g, ≥10 μM), the apoptotic cell death induced by CMT-3 was 5–10-fold higher than that of DC, indicating that CMT-3 is essentially a cytotoxic chemotherapeutic drug. The main thrust of most investigations addressing the effect of CMTs on cancer cells has focused on their potential antimetastatic activities. However, our studies on prostate cancer models and work by others on breast cancers, suggested that CMT-3 and CMT-308 are selectively cytotoxic to rapidly proliferating tumor cells. This led to us investigate the mechanism of their cytotoxicity. We determined whether tumor cells exposed to micromolar concentrations of CMTs inhibit cell proliferation by affecting cell cycle arrest, inducing programmed cell death (apoptosis).
We investigated the mechanism of CMT-3 induced apoptosis in prostate cancer. We found that both DC and CMT-3 were able to induce apoptosis in several prostate cancer cell lines although CMT-3 was more potent (fifty percent inhibition dose at 10 μM) than DC (IC50 = 20 μM). The process of induction of apoptosis involved a mitochondria-mediated intrinsic mechanism that resulted in activation of caspae-3 and caspase-9. Enhanced permeability of mitochondrial membranes preceded cell death as indicated by the release of free-nucleosomes. Furthermore, increased membrane permeability was mediated by rapid accumulation of peroxy/hydroxyl free radicals[32]. Onoda et al [37], using a human colon cancer cell line HT29, reported that CMT-3 (COL-3) causes both caspase-dependent and independent apoptotic cell death in colon cancer cells. Their report indicates that CMT-3 (COL-3) is twice as potent as DC in inducing apoptosis. COL-3 produced increased cytosolic cytochrome-C and loss of mitochondrial membrane potential in as little as 3h of exposure, compared to 24 h of exposure to DC. Furthermore, COL-3 and DC-induced apoptosis involved endonuclease G release and probably causes caspase-independent induction of apoptosis, as pretreatment with the pan-caspase inhibitor Z-VAD-FMK only partially reversed DC (36 %) and COL-3 mediated apoptosis (81%).
COL-3 inhibited invasion is likely due to inhibition of MMP-2 and -9 activity and their synthesis
Several studies have shown anti-MMP activities of COL-3 and DC in various non-transformed cells. However, we were the first to demonstrate that COL-3 and DC are strong inhibitors of gelatinase in tumor cells (earlier studies by the Zucker group showed that minocycline was also a potent inhibitor of tumor cell gelatinases). To demonstrate the anti-gelatinolytic activity of DC and COL-3, we performed the gelatinolytic activity assay at low Ca2+ concentration (1mM) as well as the more traditional 10 mM Ca2+ concentration. As reported before [32], both compounds were 10-fold more potent at low concentrations of Calcium, a physiologically relevant calcium level (e.g., serum Ca level, 0.94 mM). At this Ca2+ concentration, the IC50 was 0.4 μM for COL-3 and 1 μM for DC compared to 1 μM and 10 μM, respectively, at 10 mM Ca2+. Thus, we propose that, because of the high potency of COL-3 at physiological concentrations of Calcium, the inhibition of lung and bone metastasis in the MAT LyLU model is likely due to potent inhibition of MMPs by this compound. The contribution of apoptotic and anti-proliferative activities of COL-3 may also contribute to significant inhibition of metastatic tumor colonies in lungs.
Several studies have shown that both DC and COL-3 can inhibit tumor cell invasion in vitro using the Matrigel coated Boyden chamber invasion assays [37–41]. This may be due to multiple mechanisms, including inhibition of cell motility, chemotactic motility and inability to degrade reconstituted basement membrane (Matrigel, BD BioSystems). However, it was also shown that COL-3 is capable of inhibiting chemotactic motility induced by laminin-5- gamma 2 chain fragments that act as chemotactic peptides in melanocytes [46]. In addition, we have shown that COL-3 is able to inhibit the synthesis of MMP-2 and MMP-9 at physiologically achievable concentrations [32]. Thus, COL-3 is a potent anti-metastatic and cytotoxic antitumor compound, capable of delivering a one-two punch against aggressive tumors, including melanoma, prostate and breast cancers.
Summary
These studies reflect a growing interest in the development of safe, bioavailable inhibitors of gelatinases that play multiple roles in tumor growth, angiogenesis and metastasis. Some of the non-antimicrobial, chemically modified tetracyclines (notably the CMT-3 compounds) have been found to be capable of inhibiting these cancer cell functions in several pre-clinical tumor models. These studies, directly or indirectly, have led to preliminary clinical trials in patients with advanced cancers, including breast, prostate and lung cancers and also in osteosarcomas and viral induced sarcomas (e.g. Kaposi sarcoma). These studies are described in the next article.
Fig. 2. A suggested model for CMT-3 induced tumor cell cytotoxicity.

CMT-3 induced cytotoxicity is due to both cell cycle arrest at G1/S interphase and free-radical induced-mitochondrial permeability changes and caspase activation. Both, cytosolic and ER mediated caspase activation.
Acknowledgments
This work was supported from CDMRP (DoD) Prostate cancer Idea development grant NO. DAMD 17–8–72; and PHS Grant numbers: NIH, 5R01 CA 63108, and R01 AT003544 over the last 10 years.
Footnotes
The author dedicates this article to Ms. Marie G. Selzer, who introduced him to the field of MMPs and to the pioneering work of Dr. Lorne M Golub on the non-antibiotic properties of tetracycline compounds.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010 Sep-Oct;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Liotta LA, Stetler-Stevenson WG. Metalloproteinases and cancer invasion. Semin Cancer Biol. 1990 Apr; 1(2):99-06 Liotta LA, Steeg PS, Stetler-Stevenson WG. Cancer metastasis and angiogenesis: an imbalance of positive and negative regulation. Cell. 1991 Jan 25;64(2):327–36. doi: 10.1016/0092-8674(91)90642-c. [DOI] [PubMed] [Google Scholar]
- 3.Bourboulia D, Stetler-Stevenson WG. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): Positive and negative regulators in tumor cell adhesion. Semin Cancer Biol. 2010 Jun;20(3):161–8. doi: 10.1016/j.semcancer.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010 Apr 2;141(1):52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stamey TA, Yang N, Hay AR, McNeal JE, Freiha FS, Redwine E. Prostate-specific antigen as a serum marker for adenocarcinoma of the prostate. N Engl J Med. 1987 Oct 8;317(15):909–16. doi: 10.1056/NEJM198710083171501. [DOI] [PubMed] [Google Scholar]
- 6.Oesterling JE. Prostate specific antigen: a critical assessment of the most useful tumor marker for adenocarcinoma of the prostate. J Urol. 1991 May;145(5):907–23. doi: 10.1016/s0022-5347(17)38491-4. [DOI] [PubMed] [Google Scholar]
- 7.Botchorishvili G, Matikainen MP, Lilja H. Early prostate-specific antigen changes and the diagnosis and prognosis of prostate cancer. Curr Opin Urol. 2009 May;19(3):221–6. doi: 10.1097/MOU.0b013e32832a2d10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Poppel H, Joniau S, Van Cleynenbreugel B, Mottaghy FM, Oyen R. Diagnostic evaluation of PSA recurrence and review of hormonal management after radical prostatectomy. Prostate Cancer Prostatic Dis. 2009;12(2):116–23. doi: 10.1038/pcan.2009.3. [DOI] [PubMed] [Google Scholar]
- 9.Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004 Apr 15;350(16):1655–64. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- 10.Kingsley LA, Fournier PG, Chirgwin JM, Guise TA. Molecular biology of bone metastasis. Mol Cancer Ther. 2007 Oct;6(10):2609–17. e XC, Choueiri M, Tu SM, Lin SH. Biology and clinical management of prostate cancer bone metastasis. Front Biosci. 2007 May 1;12:3273–86. doi: 10.2741/2311. [DOI] [PubMed] [Google Scholar]
- 11.Lokeshwar BL, Selzer MG, Block NL, Gunja-Smith Z. Secretion of matrix metalloproteinases and their inhibitors (tissue inhibitor of metalloproteinases) by human prostate in explant cultures: reduced tissue inhibitor of metalloproteinase secretion by malignant tissues. Cancer Res. 1993 Oct 1;53(19):4493–8. [PubMed] [Google Scholar]
- 12.Stearns ME, Wang M. Type IV collagenase (M(r) 72,000) expression in human prostate: benign and malignant tissue. Cancer Res. 1993 Feb 15;53(4):878–83. [PubMed] [Google Scholar]
- 13.Stearns ME, Stearns M. Autocrine factors, type IV collagenase secretion and prostatic cancer cell invasion. Cancer Metastasis Rev. 1993 Mar;12(1):39–52. doi: 10.1007/BF00689789. [DOI] [PubMed] [Google Scholar]
- 14.Dong Z, Nemeth JA, Cher ML, Palmer KC, Bright RC, Fridman R. Differential regulation of matrix metalloproteinase-9, tissue inhibitor of metalloproteinase-1 (TIMP-1) and TIMP-2 expression in co-cultures of prostate cancer and stromal cells. Int J Cancer. 2001 Aug 15;93(4):507–15. doi: 10.1002/ijc.1358. [DOI] [PubMed] [Google Scholar]
- 15.Nemeth JA, Yousif R, Herzog M, Che M, Upadhyay J, Shekarriz B, Bhagat S, Mullins C, Fridman R, Cher ML. Matrix metalloproteinase activity, bone matrix turnover, and tumor cell proliferation in prostate cancer bone metastasis. J Natl Cancer Inst. 2002 Jan 2;94(1):17–25. doi: 10.1093/jnci/94.1.17. [DOI] [PubMed] [Google Scholar]
- 16.Golub LM, Ramamurthy N, McNamara TF, Gomes B, Wolff M, Casino A, Kapoor A, Zambon J, Ciancio S, Schneir M, et al. Tetracyclines inhibit tissue collagenase activity. A new mechanism in the treatment of periodontal disease. J Periodontal Res. 1984 Nov;19(6):651–5. doi: 10.1111/j.1600-0765.1984.tb01334.x. [DOI] [PubMed] [Google Scholar]
- 17.Golub LM, Lee HM, Lehrer G, Nemiroff A, McNamara TF, Kaplan R, Ramamurthy NS. Minocycline reduces gingival collagenolytic activity during diabetes. Preliminary observations and a proposed new mechanism of action. J Periodontal Res. 1983 Sep;18(5):516–26. doi: 10.1111/j.1600-0765.1983.tb00388.x. [DOI] [PubMed] [Google Scholar]
- 18.Rifkin BR, Vernillo AT, Golub LM, Ramamurthy NS. Modulation of bone resorption by tetracyclines. Ann N Y Acad Sci. 1994 Sep 6;732:165–80. doi: 10.1111/j.1749-6632.1994.tb24733.x. [DOI] [PubMed] [Google Scholar]
- 19.Bonfil RD, Chinni S, Fridman R, Kim HR, Cher ML. Proteases, growth factors, chemokines, and the microenvironment in prostate cancer bone metastasis. Urol Oncol. 2007 Sep-Oct;25(5):407–11. doi: 10.1016/j.urolonc.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 20.Sasaki T, Ramamurthy NS, Golub LM. Bone cells and matrix bind chemically modified non-antimicrobial tetracycline. Bone. 1994 May-Jun;15(3):373–5. doi: 10.1016/8756-3282(94)90303-4. [DOI] [PubMed] [Google Scholar]
- 21.Viadana E, Bross ID, Pickren JW. The metastatic spread of kidney and prostate cancers in man. Neoplasma. 1976;23(3):323–32. [PubMed] [Google Scholar]
- 22.Gingrich JR, Barrios RJ, Morton RA, Boyce BF, DeMayo FJ, Finegold MJ, Angelopoulou R, Rosen JM, Greenberg NM. Metastatic prostate cancer in a transgenic mouse. Cancer Res. 1996 Sep 15;56(18):4096–102. [PubMed] [Google Scholar]
- 23.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, Liu X, Wu H. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003 Sep;4(3):209–21. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 24.Nemeth JA, Harb JF, Barroso U, Jr, He Z, Grignon DJ, Cher ML. Severe combined immunodeficient-hu model of human prostate cancer metastasis to human bone. Cancer Res. 1999 Apr 15;59(8):1987–93. [PubMed] [Google Scholar]
- 25.Zhau HE, Li CL, Chung LW. Establishment of human prostate carcinoma skeletal kmetastasis models. Cancer. 2000 Jun 15;88(12 Suppl):2995–01. doi: 10.1002/1097-0142(20000615)88:12+<2995::aid-cncr15>3.3.co;2-p. [DOI] [PubMed] [Google Scholar]
- 26.Corey E, Quinn JE, Bladou F, Brown LG, Roudier MP, Brown JM, Buhler KR, Vessella RL. Establishment and characterization of osseous prostate cancer models: intra-tibial injection of human prostate cancer cells. Prostate. 2002 Jun 1;52(1):20–33. doi: 10.1002/pros.10091. [DOI] [PubMed] [Google Scholar]
- 27.Selzer MG, Zhu B, Block NL, Lokeshwar BL. CMT-3, a chemically modified tetracycline, inhibits bony metastases and delays the development of paraplegia in a rat model of prostate cancer. Ann N Y Acad Sci. 1999 Jun 30;878:678–82. doi: 10.1111/j.1749-6632.1999.tb07760.x. [DOI] [PubMed] [Google Scholar]
- 28.Zhu B, Block NL, Lokeshwar BL. Interaction between stromal cells and tumor cells induce chemoresistance and matrix metalloproteinase secretion. Ann N Y Acad Sci. 1999 Jun 30;878:642–6. doi: 10.1111/j.1749-6632.1999.tb07750.x. [DOI] [PubMed] [Google Scholar]
- 29.Shevrin DH, Kukreja SC, Ghosh L, Lad TE. Development of skeletal metastasis by human prostate cancer in athymic nude mice. Clin Exp Metastasis. 1988 Sep-Oct;6(5):401–9. doi: 10.1007/BF01760575. [DOI] [PubMed] [Google Scholar]
- 30.Geldof AA, Rao BR. Prostate tumor (Dunning R3327) skeletal metastasis. Prostate. 1990;16:279–290. doi: 10.1002/pros.2990160402. [DOI] [PubMed] [Google Scholar]
- 31.Isaacs JT, Isaacs WB, Feitz WF, Scheres J. Establishment and characterization of seven Dunning rat prostatic cancer cell lines and their use in developing methods for predicting metastatic abilities of prostatic cancers. Prostate. 1986;9(3):261–81. doi: 10.1002/pros.2990090306. [DOI] [PubMed] [Google Scholar]
- 32.Lokeshwar BL, Selzer MG, Zhu BQ, Block NL, Golub LM. Inhibition of cell proliferation, invasion, tumor growth and metastasis by an oral non-antimicrobial tetracycline analog (COL-3) in a metastatic prostate cancer model. Int J Cancer. 2002 Mar 10;98(2):297–309. doi: 10.1002/ijc.10168. [DOI] [PubMed] [Google Scholar]
- 33.Dandekar DS, Lokeshwar BL. Inhibition of cyclooxygenase (COX)-2 expression by Tet-inducible COX-2 antisense cDNA in hormone-refractory prostate cancer significantly slows tumor growth and improves efficacy of chemotherapeutic drugs. Clin Cancer Res. 2004 Dec 1;10(23):8037–47. doi: 10.1158/1078-0432.CCR-04-1208. [DOI] [PubMed] [Google Scholar]
- 34.Dandekar DS, Lopez M, Carey RI, Lokeshwar BL. Cyclooxygenase-2 inhibitor celecoxib augments chemotherapeutic drug-induced apoptosis by enhancing activation of caspase-3 and -9 in prostate cancer cells. Int J Cancer. 2005 Jun 20;115(3):484–92. doi: 10.1002/ijc.20878. [DOI] [PubMed] [Google Scholar]
- 35.Lokeshwar BL, Escatel E, Zhu B. Cytotoxic activity and inhibition of tumor cell invasion by derivatives of a chemically modified tetracycline CMT-3 (COL-3) Curr Med Chem. 2001 Feb;8(3):271–9. doi: 10.2174/0929867013373516. [DOI] [PubMed] [Google Scholar]
- 36.Lokeshwar BL, Houston-Clark HL, Selzer MG, Block NL, Golub LM. Potential application of a chemically modified non-antimicrobial tetracycline (CMT-3) against metastatic prostate cancer. Adv Dent Res. 1998 Nov;12(2):97–102. doi: 10.1177/08959374980120012901. [DOI] [PubMed] [Google Scholar]
- 37.Onoda T, Ono T, Dhar DK, Yamanoi A, Nagasue N. Tetracycline analogues (doxycycline and COL-3) induce caspase-dependent and -independent apoptosis in human colon cancer cells. Int J Cancer. 2006 Mar 1;118(5):1309–15. doi: 10.1002/ijc.21447. [DOI] [PubMed] [Google Scholar]
- 38.Fife RS, Sledge GW., Jr Effects of doxycycline on in vitro growth, migration, and gelatinase activity of breast carcinoma cells. J Lab Clin Med. 1995 Mar;125(3):407–11. [PubMed] [Google Scholar]
- 39.Fife RS, Sledge GW., Jr Effects of doxycycline on cancer cells in vitro and in vivo. Adv Dent Res. 1998 Nov;12(2):94–6. doi: 10.1177/08959374980120012801. [DOI] [PubMed] [Google Scholar]
- 40.Fife RS, Rougraff BT, Proctor C, Sledge GW., Jr Inhibition of proliferation and induction of apoptosis by doxycycline in cultured human osteosarcoma cells. J Lab Clin Med. 1997 Nov;130(5):530–4. doi: 10.1016/s0022-2143(97)90130-x. [DOI] [PubMed] [Google Scholar]
- 41.Seftor RE, Seftor EA, De Larco JE, Kleiner DE, Leferson J, Stetler-Stevenson WG, McNamara TF, Golub LM, Hendrix MJ. Chemically modified tetracyclines inhibit human melanoma cell invasion and metastasis. Clin Exp Metastasis. 1998 Apr;16(3):217–25. doi: 10.1023/a:1006588708131. [DOI] [PubMed] [Google Scholar]
- 42.Saikali Z, Singh G. Doxycycline and other tetracyclines in the treatment of bone metastasis. Anticancer Drugs. 2003 Nov;14(10):773–8. doi: 10.1097/00001813-200311000-00001. [DOI] [PubMed] [Google Scholar]
- 43.Duivenvoorden WC, Popovi SV, Lhotak S, Seidlitz E, Hirte HW, Tozer RG, Singh G. Doxycycline decreases tumor burden in a bone metastasis model of human breast cancer. Cancer Res. 2002 Mar 15;62(6):1588–91. [PubMed] [Google Scholar]
- 44.Duivenvoorden WC, Hirte HW, Singh G. Use of tetracycline as an inhibitor of matrix metalloproteinase activity secreted by human bone-metastasizing cancer cells. Invasion Metastasis. 1997;17(6):312–22. [PubMed] [Google Scholar]
- 45.Duivenvoorden WC, Vukmirovi -Popovi S, Kalina M, Seidlitz E, Singh G. Effect of zoledronic acid on the doxycycline-induced decrease in tumor burden in a bone metastasis model of human breast cancer. Br J Cancer. 2007 May 21;96(10):1526–31. doi: 10.1038/sj.bjc.6603740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seftor RE, Seftor EA, Kirschmann DA, Hendrix MJ. Targeting the tumor microenvironment with chemically modified tetracyclines: inhibition of laminin 5 gamma2 chain promigratory fragments and vasculogenic mimicry. Mol Cancer Ther. 2002 Nov;1(13):1173–9. [PubMed] [Google Scholar]