

Abstract
Objective
Cell-based tissue engineering strategies are currently in clinical use and continue to be developed at a rapid pace for the repair of cartilage defects. Regardless of the repair methodology, chondrocytes within newly regenerated cartilage remain susceptible to the abnormal inflammatory and mechanical environments that underlie osteoarthritic disease, likely compromising the implant’s integration, function, and longevity. The present study investigates the use of parathyroid hormone-related peptide (PTHrP) overexpression for chondroprotection.
Design
Bovine articular chondrocytes were transfected with human PTHrP (hPTHrP) constructs (1-141 or 1-173) and subjected to injurious cyclic tensile strain (CTS; 0.5 Hz and 16% elongation) for 48 hours. mRNA expression of matrix remodeling, inflammatory signaling, hypertrophic, and apoptotic genes were examined with real-time reverse transcription polymerase chain reaction. Nitric oxide (NO) and prostaglandin E2 (PGE2) production were measured using the Griess assay and enzyme immunoassay, respectively.
Results
CTS induced an arthritic phenotype in articular chondrocytes as indicated by increased gene expression of collagenases and aggrecanases and increased production of NO and PGE2. Additionally, CTS increased collagen type X (Col10a1) mRNA expression, whereas overexpression of either hPTHrP isoform inhibited CTS-induced Col10a1 gene expression. However, hPTHrP 1-141 augmented CTS-induced NO and PGE2 production, and neither hPTHrP isoform had any significant effect on apoptotic genes.
Conclusions
Our results suggest that chondrocytes overexpressing PTHrP resist mechanical strain-induced hypertrophic-like changes. Therapeutic PTHrP gene transfer may be considered for chondroprotection applications in newly regenerated cartilage.
Keywords: articular cartilage, parathyroid hormone-related peptide, chondroprotection, tensile strain, hypertrophy
INTRODUCTION
A number of surgical and cell-based regenerative strategies have been evaluated for the repair of cartilage defects. Current surgical techniques include mechanical penetration of the subchondral bone (i.e., abrasion arthroplasty, Pridie drilling, or microfracture) and autologous transplantation of periosteum, perichondrium, or osteochondral grafts1. Autologous chondrocyte implantation (ACI) is in current clinical application to mitigate proression to disease2, and tissue engineering approaches that deliver a matrix seeded with chondrogenic cells and chondrogenic factors have also been evaluated experimentally. Regardless of the methodology, newly implanted chondrocytes within the defect remain susceptible to the abnormal inflammatory and mechanical environments that underlie osteoarthritic disease3, 4. Subsequent arthritis-associated changes and aberrant functioning of these chondrocytes are likely to eventually compromise the integration, function, and longevity of regenerated cartilage5. Thus, chondroprotection of regenerated cartilage from the underlying disease would result in a more effective and durable therapy.
Resident chondrocytes provide the essential function of maintaining cartilage matrix homeostasis. This activity is impaired after traumatic mechanical loads damage articular cartilage during post-traumatic arthritis and osteoarthritis (OA)6, 7. Derangement of the extracellular matrix (ECM) significantly alters chondrocyte behavior and phenotype, and the cells play a direct role in the degradation process by upregulation of matrix-degrading proteases, such as matrix metalloproteinases (MMPs) and a disintegrin and MMP with thrombospondin motifs (ADAMTSs), as well as inflammatory intermediaries, including nitric oxide (NO) and prostaglandins (PGs)8. These increased proteolytic activities are not sufficiently counterbalanced by an increase in chondrocyte anabolic activities, resulting in matrix erosion. Another interesting aspect in the arthritis disease process is the recapitulation of molecular mechanisms that occur during fetal skeletogenesis. In addition to changes in anabolic and catabolic events, there are changes in cellular phenotype similar to those found in endochondral ossification, including hypertrophic differentiation, matrix calcification, and apoptosis9. Previous studies have found increased expression of hypertrophic markers in OA cartilage, including type X collagen, Indian hedgehog (Ihh), MMP-13, and alkaline phosphatase (ALP)9–12. Expression of these markers, which are typically low to absent in normal healthy cartilage, suggest that the progression of the degenerative cascade may involve the loss of inhibitory control over terminal differentiation genes in osteoarthritic chondrocytes.
Parathyroid hormone-related protein (PTHrP) maintains the function of proliferating chondrocytes and inhibits chondrocyte differentiation toward hypertrophy in the growth plate13. The anti-hypertrophy activity has been shown to result from binding of the N-terminus of PTHrP to its cell surface receptor (PTH1R), activating Sox914, 15. PTHrP also stimulates proliferation of endochondral chondrocytes and inhibits apoptosis, partly via induction of Bcl-216, 17. Therefore, PTHrP may be a therapeutic option for the protection of articular chondrocytes. Additionally, previous studies have used gene transfer methods to provide sustained transgene expression for the purposes of cartilage repair and chondroprotection18. The efficacy of sustained PTHrP transgene expression by articular chondrocytes in preserving phenotypic homeostasis remains unknown.
The present study explored the use of human PTHrP (hPTHrP) overexpression by articular chondrocytes to suppress arthritic-like changes induced by mechanical trauma. Two different PTHrP isoforms, 1-141 and 1-173, were tested. An cyclic tensile strain (CTS) model was used to promote arthritic changes in articular chondrocytes in vitro.
MATERIALS AND METHODS
Chondrocyte isolation and culture
Chondrocytes were isolated from the knees of adult steers (2–3 years old) using previously described methods19. Briefly, cartilage slices were diced finely and digested in a stirring flask for 8–10 h with 0.2% collagenase type II (Sigma) in DMEM (Invitrogen) supplemented with antibiotics at 37°C under 5% CO2. After filtration through a 40 μm nylon mesh cell strainer, the cells were re-suspended at a density of 1.5 × 105 cells/cm2 in DMEM supplemented with 10% fetal bovine serum and antibiotics for two days, and frozen for later use in liquid nitrogen.
Transfection procedure and plasmid DNA
Cells were thawed in DMEM with 10% FBS for two days before transfection. First, chondrocytes were treated with 40 U/ml bovine testicular hyaluronidase (Sigma) for 3 hours. Next, medium was replaced with a transfection solution containing Opti-MEM-I + GlutaMAX-I (Invitrogen), 4 U/ml hyaluronidase, FuGENE 6 (Roche), and plasmid DNA. Optimal transfection of bovine articular chondrocytes used a lipid/DNA ratio of 3:120. After 6 hours, the transfection solution was replaced with DMEM with 10% FBS. Transfection efficiency using this protocol was observed to be approximately 35% with GFP control plasmids.
hPTHrP 1-141 and 1-173 plasmids were generous gifts from Dr. Leonard J. Deftos, University of California, San Diego21. Both constructs were inserted downstream of a CMV promoter/enhancer in the pCI-neo expression plasmid vector (Promega). Control cells were simultaneously transfected with the blank pCI-neo vector. All plasmids were amplified and purified by MTR Scientific (Ijamsville, MD).
Application of cyclic tensile strain
Chondrocytes were trypsinized and cultured onto fibronectin-coated Bioflex plates (Flexcell) at a density of 40,000 cells/cm2 in DMEM with 1% FBS for 1 day. Bioflex plates were coated with 5 μg/ml bovine plasma fibronectin (Sigma) in 0.1M bicarbonate solution for 3 hours at 4°C, then replaced with DMEM with 10% FBS and incubated overnight to remove non-adherent fibronectin. Wells were rinsed with HBSS immediately before seeding. Cells were subjected to equibiaxial tensile strain using a custom manufactured vacuum-operated loading device. A pulsed waveform 16% cyclic tensile strain and 0.5 Hz frequency was applied. Seeding of cells onto the Bioflex plates and application of CTS were completed at 37°C under 5% CO2 and 5% O2. A 5.0 % ambient oxygen tension environment was maintained with a dual gas incubator (Forma II 3130, Thermo Scientific) and used to simulate normal physiologic conditions and reduce the production of reactive oxygen species relative to standard normoxic culture conditions. Cell viability at the end of experimentation was observed using LIVE/DEAD staining (Invitrogen) to detect live (green fluorescence) and dead (red fluorescence) cells.
RNA extraction and quantitative real-time PCR
Chondrocytes were homogenized in TRIzol reagent (Invitrogen) at 0, 12, 24, and 48 hours of CTS to extract total cellular RNA, which was further treated with Turbo DNase (Ambion) for 25 min at 37°C to remove genomic DNA. RNA concentration and purity were estimated spectrophotometrically on the basis of absorbance values at 260 nm and 280 nm. Reverse transcription of equal quantities of RNA (1 μg) from each sample was performed using SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen).
Real-time PCR was performed using the Stratagene Mx3000P instrument and Power SYBR Green PCR Master Mix (Applied Biosystems). Select PCR primers were designed using Primer3 v0.4.0. (Whitehead Institute for Biomedical Research) on the basis of bovine mRNA sequences (primer sequences available on request). The amplification program was as follows: denaturation, 95°C, 10 minutes; 40 amplification cycles, 95°C, 30 seconds; and annealing, 60°C, 1 minute. After amplification, melt analysis was performed by heating the reaction mixture from 60°C to 95°C at a rate of 0.2°C/second. Relative fold changes relative to the time zero control samples (24 hours post seeding but pre-CTS) were calculated using the comparative threshold cycle (CT) method (ΔΔCT) with glyceraldehyde-3-phosphate dehydrogenease (GAPDH) as the control gene. Four housekeeping genes were tested (GAPDH, HPRT, β2M, RPL13a), and GAPDH was found to be the most stable with a max fold change of 0.26 from baseline after 48 hours. Neither CTS nor PTHrP significantly affected expression of the housekeeping genes.
Enzyme immunoassay and biochemical analysis
Culture medium aliquots were stored at −80°C prior to biochemical analyses. Relative concentrations of NO were measured in the culture media using the Griess assay (Promega). The Griess reaction is based on the chemical diazotization reaction that uses sulfanilamide and N-1-napthylethylenediamine dihydrochloride (NED) under acidic (phosphoric acid) conditions. This system measures nitrite, which is one of two primary, stable and nonvolatile breakdown products of NO. PGE2 production was measured in the culture media using a high-sensitivity enzyme immunoassay (EIA) kit (Assay Designs). Total PTHrP production was measured in the culture media using an EIA kit (Bachem), which is designed to measure the 1-34 peptide sequence and detects both human and bovine PTHrP, but does not cross-react with PTH. All biochemical assays were normalized to number of viable cells, which was determined quantitatively using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega).
Statistical analysis
Each experiment was performed three times with different chondrocyte sources. Proportional fold changes are presented using the means (±95%CI) from a single replicate experiment, and statistical analyses was carried out by GraphPad Prism 5 using three replicate experiments. Two-tailed t-tests were used to compare gene expression differences between CTS and no CTS conditions. Two-way ANOVA with post hoc Bonferroni-corrected t-tests were used to compare differences between the three different PTHrP treatment conditions (control, 1-141, 1-173) for both CTS and no CTS conditions. Statistical significance was based on p<0.05.
RESULTS
Effects of 16% CTS on articular chondrocytes
When observed under light microscopy, unstrained cells continued to adhere and spread in monolayer during the experimental period. CTS caused most adherent cells to retain their round shape, while others took on a more stellate-shaped appearance. The majority of chondrocytes (>90%), regardless of their shape, remained viable after undergoing 48 hours of CTS (Figure 1). Furthermore, the number of viable cells between CTS and No CTS conditions, as measured by proliferation assays, was similar at all time points. The effects of CTS were ascertained by measuring the mRNA levels of multiple genes (Table 1). All MMP, ADAMTS, and TIMP gene levels were up-regulated by CTS. MMP-3 (stromelysin 1) and MMP-13 (collagenase 3) mRNA levels in strained chondrocytes were increased 3.31 and 3.73 times, respectively, over levels in unstrained chondrocytes after 48 hours (Figure 2A). ADAMTS-4 (aggrecancase 1) and ADAMTS-5 (aggrecancase 2) mRNA levels were increased in strained chondrocytes 10.63 and 1.14 times, respectively, over levels in unstrained cells after 48 hours (Figure 2B). In comparison, mRNA levels of these genes in unstrained cells did not change by more than 0.81-fold from time zero levels; changes in MMP-9 and MMP-13 mRNA levels were the only exceptions (−2.39 and −2.36, respectively). CTS decreased gene expression of Col2a1 by 0.37-fold from no CTS controls but did not alter expression of aggrecan over 48 hours (Figure 2C). The hypertrophy-associated genes, Col10a1 and Runx2, were significantly up-regulated 3.00-fold and 0.43-fold, respectively, by CTS over no CTS conditions after 48 hours (Figure 2D). Col10a1 gene expression was immediately increased with application of CTS, with significant increases at all measured time points. In comparison, Col10a1 mRNA levels increased by an average of only 0.93-fold over the 48 hour experimental period under no CTS conditions. Gene expression of inducible enzymes iNOS and COX-2 were up-regulated 5.46-fold and 1.06-fold, respectively, over no CTS controls after 48 hours of CTS. Secretion of their respective mediators, NO and PGE2, were significantly increased 5.17 and 4.50 times over controls after 48 hours of CTS (Figures 2E–F). NO and PGE2 levels in unstrained chondrocytes remained low and changed minimally over the course of the experimental period.
Figure 1.
Cell viability at the end of experimentation was observed using LIVE/DEAD staining to detect live (green fluorescence) and dead (red fluorescence) cells. The majority of chondrocytes (>90%) remained viable after 48 hours of (A) No CTS and (B) CTS.
TABLE 1.
Gene expression changes in articular chondrocytes subjected to 48 hours of cyclic tensile strain
| Gene | CTS1 | No CTS1 | p-value2 |
|---|---|---|---|
| Col2a1 | −0.11 | 0.25 | 0.050 |
| Aggrecan | 0.15 | 0.17 | 0.148 |
| MMP-1 | 1.28 | 0.34 | 0.010 |
| MMP-3 | 2.49 | −0.81 | 0.080 |
| MMP-9 | 3.29 | −2.39 | 0.010 |
| MMP-13 | 1.37 | −2.36 | 0.049 |
| ADAMTS-4 | 9.92 | −0.72 | 0.087 |
| ADAMTS-5 | 0.34 | −0.81 | 0.074 |
| TIMP-1 | 0.23 | −0.20 | 0.067 |
| TIMP-2 | 0.36 | −0.10 | 0.073 |
| TIMP-3 | 2.21 | −0.31 | 0.005 |
| Col10a1 | 3.93 | 0.93 | 0.007 |
| ALP | −0.19 | 0.27 | 0.159 |
| Runx2 | 0.57 | 0.14 | 0.047 |
| bPTHrP | 3.72 | −0.56 | 0.000 |
| PTHR1 | −1.04 | 0.13 | 0.016 |
| CTGF | −0.62 | 0.01 | 0.010 |
| Sox9 | 0.51 | 0.18 | 0.044 |
| iNOS | 4.66 | −0.80 | 0.010 |
| COX-2 | 0.85 | −0.21 | 0.006 |
| Bcl-2 | 1.17 | −0.04 | 0.006 |
| Bcl-xL | 1.08 | −0.07 | 0.018 |
| Bax | 0.24 | −0.03 | 0.067 |
Proportional fold changes of mRNA levels over 48 hours, compared to time zero, of CTS and no CTS conditions presented as mean of N=3.
p-Values were calculated from two-tailed t-tests.
Col2a1 – collagen type IIa1; MMP – matrix metalloproteinase; ADAMTS – a disintegrin and metalloproteinase with thrombospondin motifs; TIMP – tissue inhibitors of metalloproteinases; Col10a1 – collagen type Xa1; ALP - alkaline phosphatase; Runx2 – runt-related transcription factor 2; bPTHrP – bovine parathyroid hormone-related peptide; PTHR1 – PTHrP receptor; CTGF – connective tissue growth factor; Sox9 – sex determining region Y box 9; iNOS – inducible nitric oxide synthase; COX-2 – cyclooxygenase-2; Bcl-2 – B-cell lymphoma 2; Bcl-xL – B-cell lymphoma – extra large; Bax – Bcl-2-associated X protein.
Figure 2.

Effects of 16% CTS on normal bovine articular chondrocytes CTS increased gene expression of (A) MMPs, (B) ADAMTSs, (D) hypertrophic genes, and (F) iNOS and COX-2 and decreased gene expression of (C) Col2a1. CTS increased secretion of (D) NO and PGE2, which correlates with (E). Error bars represent ±95% confidence intervals (N = 3). Solid lines = CTS; Dotted lines = No CTS. * = p<0.05 and ** = p<0.01 versus control. Specifically, MMP-13 48 hrs p = 0.0493, 24 hrs p = 0.0465; Col2a1 48 hrs p = 0.0498; Col10al p = 0.0072, 24 hrs p = 0.0124, 12 hrs p = 0.0122; Runx2 48 hrs p = 0.0471; iNOS 48 hrs p = 0.0097, 24 hrs p = 0.0139; COX-2 48 hrs p = 0.0059, 24 hrs p = 0.0130, 12 hrs p = 0.0075; NO 48 hrs p = 0.0397, 24 hrs p = 0.0301; PGE2 48 hrs p = 0.0147, 24 hrs p = 0.0066.
Total PTHrP secretion after transfection of articular chondrocytes
Chondrocytes transfected with either hPTHrP construct secreted more total PTHrP under both CTS and no CTS conditions (Figure 3A, CTS data shown). Under CTS conditions, chondrocytes expressing 1-141 and 1-173 secreted an average 1.59 and 1.39 times more total PTHrP than control cells, respectively, at 48 hours; under no CTS conditions, chondrocytes expressing 1-141 and 1-173 secreted an average of 1.27 and 1.12 times more total PTHrP than control cells, respectively, at 48 hours. Additionally, chondrocytes expressing hPTHrP 1-141 secreted more total PTHrP than those expressing hPTHrP 1-173 at all measured times, although the differences were not significant.
Figure 3.

Total PTHrP secretion by articular chondrocytes transfected with hPTHrP expression constructs. (A) Chondrocytes expressing hPTHrP constructs secrete more total PTHrP (bovine and human) under CTS and No CTS conditions (only CTS data shown). Error bars represent ±95% confidence intervals (N = 3). * = p<0.05 and ** = p<0.01 versus control. # denotes p = 0.0102 versus No CTS control. (B) Endogenous PTHrP (bPTHrP) gene expression, as measured by RT-PCR using a primer specific for the bovine gene, was up-regulated by CTS. Chondrocytes expressing hPTHrP 1-141 and 1-173 demonstrated feedback inhibition of endogenous bPTHrP mRNA levels. Error bars represent ±95% confidence intervals (N = 3). ** = p<0.01 control versus hPTHrP 1-141 and 1-173.
Several trends in PTHrP secretion by control cells were observed. When comparing CTS and no CTS conditions, CTS significantly increased PTHrP secretion by 1.31 times over that in no CTS cells at 24 hours (Figure 3A). Additionally, while total PTHrP secretion generally trended down during experimental period with CTS, endogenous PTHrP gene expression was significantly up-regulated by CTS as measured by real-time PCR (Figure 3B). After 48 hours of CTS, bPTHrP gene expression increased 4.28-fold over no CTS controls. However, chondrocytes expressing hPTHrP 1-141 and 1-173 demonstrated a 1.68- and 1.21-fold feedback inhibition, respectively, of endogenous bPTHrP mRNA levels.
hPTHrP overexpression inhibits CTS-induced Col10a1 gene expression
PTHrP overexpression suppressed both basal and CTS-induced Col10a1 gene expression. hPTHrP 1-141 and 1-173 expression suppressed CTS-induced Col10a1 mRNA levels during all measured time points (Figure 4A). Increases in Col10a1 gene levels by CTS were inhibited significantly by 2.39- and 1.86-fold, respectively, at 48 hours and by 0.85- and 0.92-fold, respectively, at 24 hours. In comparison, hPTHrP 1-141 and 1-173 suppressed basal Col10a1 mRNA levels significantly by 0.84- and 0.62-fold, respectively, at 48 hours in no CTS cells (Figure 4B). hPTHrP did not significantly affect expression of matrix remodeling genes.
Figure 4.

Inhibition of Col10a1 gene expression in articular chondrocytes by PTHrP overexpression. Expression of exogenous hPTHrP in chondrocytes suppressed (A) CTS-induced Col10a1 gene expression and (B) basal Col10a1 gene expression. hPTHrP overexpression had no significant effect on matrix remodeling genes (data not shown). Error bars represent ±95% confidence intervals (N = 3). * = p<0.05 and ** = p<0.01 versus hPTHrP 1-141 and 1-173.
Effects of hPTHrP overexpression on CTS-induced iNOS and COX-2 gene expression and NO and PGE2 production
Expression of hPTHrP 1-141 augmented CTS-induced iNOS gene expression at 24 hours by 1.99-fold, although the differences were not significant (Figure 5A). However, hPTHrP 1-141 augmented CTS-induced NO production significantly at all measured time points, with an average 1.81 times increased production versus control at 48 hours (Figure 5B). In addition, hPTHrP 1-141 augmented both COX-2 gene levels and PGE2 production by 1.20- and 1.78-fold over control levels at 48 hours (Figure 5C–D). Chondrocytes overexpressing hPTHrP 1-173 exhibited similar iNOS and COX-2 mRNA levels as control, with a maximum 0.61-fold change during the experimental period. NO and PGE2 production between 1-173 and control groups were almost identical as well, with a maximum 0.10-fold change during the experimental period. Under no CTS conditions, neither hPTHrP isoform affected iNOS and COX-2 mRNA levels or NO and PGE2 production, all of which remained low and showed minimal change during the experimental period.
Figure 5.

Effects of hPTHrP overexpression on CTS-induced iNOS and COX-2 gene expression and NO and PGE2 production in articular chondocytes. Overexpression of hPTHrP 1-141 augmented gene expression of inducible enzyme isoforms, (A) iNOS and (C) COX-2, and production of their respective inflammatory mediators, (B) NO and (D) PGE2. Chondrocytes overexpressing hPTHrP 1-173 did not show significant changes in iNOS and COX-2 gene expression or production of NO and PGE2 when compared to control. Error bars represent ±95% confidence intervals (N = 3). * = p<0.05 and ** = p<0.01 versus control.
Effect of hPTHrP overexpression on CTS-induced apoptotic gene expression
CTS increased Bcl-2 and Bax gene expression by 1.21- and 0.27-fold, respectively, over no CTS controls after 48 hours. Neither hPTHrP isoform had any significant effect on Bcl-2 or Bax mRNA levels (Figure 6A–B, CTS data shown). The Bcl-2/Bax mRNA ratio was calculated, which also did not change significantly upon hPTHrP overexpression when compared to control (Figure 6C). Finally, basal Bcl-2 and Bax expression levels in unstrained chondrocytes remained low and changed minimally during the experimental period and were not altered by hPTHrP overexpression (data not shown).
Figure 6.
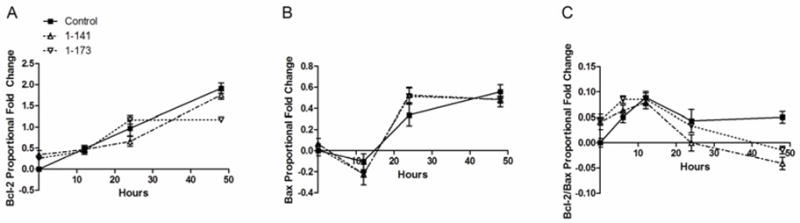
Effects of hPTHrP overexpression on CTS-induced apoptotic gene expression in articular chondrocytes. Chondrocytes overexpressing either exogenous hPTHrP isoform did not show any significant change in CTS-induced (A) Bcl-2 and (B) Bax gene expression levels. (C) hPTHrP overexpression also did not significantly affect Bcl-2/Bax ratio. Error bars represent ±95% confidence intervals (N = 3).
DISCUSSION
Cell-based tissue engineering strategies are currently in clinical use and continue to be developed at a rapid pace for the repair of cartilage defects. Regardless of the repair methodology, newly created joint cartilage will still be susceptible to the abnormal inflammatory and mechanical environments that underlie osteoarthritic disease, potentially compromising its integration, function, and longevity. Chondroprotection measures must therefore be designed for the chondrocytes of regenerated cartilage, which are in close proximity to matrix proteases, bioactive extracellular matrix fragments, and inflammatory mediators responsible for degenerative OA. Previous reports have indicated that many of the biologic changes in articular chondrocytes during the progression of OA are similar to those in the cartilage growth plate during developmental endochondral ossification. Specifically, articular chondrocytes reinitiate a sequence of phenotypic changes that begin with hypertrophy and progress towards mineralization and apoptosis9, 10, 22, 23. Increased production of catabolic proteases, including MMP-1, MMP-3, MMP-9, and MMP-13, has been observed both in the growth plate and OA cartilage, resulting in matrix degradation in both processes12, 24–28.
Because the progression of the degenerative cascade is thought to involve the loss of inhibitory control over terminal differentiation genes, we hypothesized that an anti-hypertrophic therapy may protect healthy chondrocytes from arthritic disease. PTHrP is an important signaling factor in the regulation of endochondral ossification in the epiphyseal growth plate of long bones. Within cells, PTHrP is processed by members of the family of prohormone convertases to at least three fragments with different biologic functions29, 30. Several direct effects of these fragments make it an attractive option for chondroprotection therapy. Suppression of hypertrophy and terminal differentiation is mediated through parathyroid hormone receptor 1 (PTHR1). Activation of PTHR1 leads to subsequent activation of the PKA signaling pathway, increasing chondrocyte proliferation by decreasing p57 and Runx2 levels and increasing Sox9 phosphorylation31. Because the N-terminal regions of PTHrP and PTH are similar and share the same receptor (PTHR1), previous reports have shown the ability of these peptides to suppress hypertrophic changes and terminal differentiation of articular chondrocytes induced by T3 and 5-azacytidine32, 33. Additionally, PTHrP has a well-documented anti-apoptotic effect through its translocation to the nucleus via a mid-region bipartite nuclear targeting sequence (NTS)17, 34–36. Thus, PTHrP plays an important role in maintaining the survival and chondrogenesis of proliferating chondrocytes and suppressing terminal differentiation in the growth plate.
In the present study, the effects of PTHrP in suppressing early arthritic-like changes were ascertained in a mechanical CTS model. Because the half-life of the N-terminal fragment is only 6 to 8 min37, sustained delivery of PTHrP will eventually be needed to inhibit the effects of a chronic disease like OA to achieve optimal therapeutic results. Such a mechanism of sustained PTHrP delivery was tested by non-viral gene transfer of hPTHrP cDNA into the articular chondrocytes themselves. This study contributes several novel approaches and results. First, high magnitude CTS model induced an arthritic phenotype with a similar pattern of changes as OA chondrocytes, including increased gene expression of the collagenases and aggrecanases, as well as increased production of NO and PGE2. Additionally, high magnitude CTS was discovered to increase gene expression of Col10a1, a marker of chondrocyte hypertrophy, and this phenomenon may be mediated by Runx2 due to their concurrent increase. Endogenous bPTHrP gene expression increased with CTS, consistent with increased PTHrP secretion seen in human osteoarthritic cartilage38, 39 and thought to be a self-repair response to damaged cartilage40. Interestingly, gene expression of PTH1R, a marker of pre-hypertrophy, decreased initially for the first 12 hours with CTS but steadily increased for the next 36 hours (data not shown). This bimodal regulation may arise from CTS overriding the autocrine feedback down-regulation of receptor expression in cells producing PTHrP. However, cellular changes that occur during recovery from trypsinization may also modulate this response. Gene expression of ALP, a recognized marker for terminal differentiation, did not change significantly with 48 hours of CTS. It may be possible that only early hypertrophic genes were upregulated within the first 48 hours of CTS, while late hypertrophic genes would have been upregulated with a longer duration of CTS. Second, sustained PTHrP delivery to chondrocytes was achieved by means of FuGENE6-mediated transfection of hPTHrP constructs. As expected, chondrocytes expressing exogenous hPTHrP showed higher secreted levels of total PTHrP (bovine and human) than controls. Chondrocytes expressing isoform 1-141 generally secreted more total PTHrP than those expressing isoform 1-173, which is consistent with previous data showing that serial truncation of the C-terminal end increases the amount of secreted PTHrP21.
Third, overexpression of both hPTHrP isoforms inhibited CTS-induced and basal Col10a1 expression in articular chondrocytes. Although CTS significantly increased Runx2 mRNA levels, this change was so slight that whether inhibition of Col10a1 expression by hPTHrP was via Runx2 could not be ascertained. hPTHrP had no significant inhibitory effect on CTS-induced MMP-13 gene expression. MMP-13 is expressed by hypertrophic chondrocytes to degrade the cartilage matrix, to facilitate vascular invasion and matrix mineralization by osteoblasts. Because MMP-13 is expressed in only the most terminally differentiated hypertrophic chondrocytes41, the immediate MMP-13 up-regulation observed in this CTS model is most likely attributed to mechanotransduction rather than phenotype modulation. hPTHrP overexpression was not able to immediately inhibit the catabolic changes along with the hypertrophic-like changes, suggesting that the two pathways are regulated separately.
Lastly, CTS increased NO and PGE2 production, which both act as strong catabolic signals in cartilage by altering chondrocyte function and enhancing chondrocyte apoptotic potential42. hPTHrP isoform 1-141 potentiated the production of these mediators with CTS by upregulating transcriptional levels of iNOS and COX-2. Because these effects were not observed with isoform 1-171, the underlying mechanism may be specific to the amount of secreted PTHrP or to the 140–141 region that differs between the two isoforms. Although PTHrP is known to stimulate NO release from endothelial cells for local regulation of vascular tone43, a possible mechanism for PTHrP-mediated NO release in chondrocytes has not been elucidated. Additionally, studies have found that PGE2 increases PTHrP production in chondrocytes44, 45, but the reverse has not been reported. Increased production of pro-inflammatory intermediaries is expected to promote apoptosis through regulation of Bcl-2 and Bax gene expression in chondrocytes. Bax forms heterodimers with Bcl-2, and when overexpressed, counters the anti-apoptotic effect of Bcl-2, causing accelerated cell death. Therefore, it is the ratio of Bcl-2 to Bax that determines the apoptotic fate of a cell. Our results show that Bcl-2 and Bax gene up-regulation by CTS follows a similar temporal pattern as hypertrophic chondrocytes in the growth plate16, 46. Anti-apoptotic Bcl-2 gene expression was immediately increased, possibly in a rescue attempt for survival, while pro-apoptotic Bax gene expression was not increased until 12 to 24 hours of CTS, when the ratio of the expression level of Bcl-2 and Bax gradually shifts in favor of Bax. Intracellular hPTHrP expression was expected to effect a pro-survival advantage to chondrocytes due to translocation of the NTS to the nucleus, as shown in previous work in the literature17, 34–36. However, hPTHrP overexpression had no significant effect on Bcl-2 and Bax gene expression. This result may be attributed to the short testing period. The intact viability of the majority of cells after 48 hours of CTS indicates that the only the initial stages of the apoptotic cascade were activated. Longer follow-up times may be needed to appreciate any apoptosis and cell death cause by CTS and thus, any anti-apoptotic effects of hPTHrP overexpression. Interestingly, Bcl-xL gene expression was upregulated with CTS and augmented by expression of hPTHrP 1-141 at 48 hours (data not shown).
Several studies have reported the use of inflammation-responsive promoters to regulate OA gene therapy47–49. Regulation of hPTHrP expression may be necessary to selectively activate targeted pathways because high concentrations of PTHrP can facilitate activation of other heterotrimeric G proteins, including the Gq family. The effect of Gq activation is opposite from that of Gs activation in that it mildly accelerates chondrocyte differentiation50. The purpose of these opposing actions may be to regulate the proliferation of chondrocytes based on their location, thus creating a functional gradient of PTHrP across the growth plate31. PTHrP overexpression may expose chondrocytes to concentrations that are too high, simultaneously activating Gs and Gq pathways. Therefore, we are currently testing the use of an inflammation-responsive, self-limiting promoter consisting of NF-κB repeat sequences to optimize PTHrP therapy.
This study demonstrates that PTHrP overexpression inhibits CTS-induced hypertrophic-like changes of articular chondrocytes. Several gene therapy approaches are currently being investigated for the repair of cartilage defects and the inhibition of OA progression. However, most of these approaches (i.e., MMP inhibitors) target only one of several players in the disease pathogenesis, and none have attempted to target the early hypertrophic-like changes seen in OA. Ultimately, a combination of these approaches may be needed for effective chondroprotection of regenerated cartilage. Because CTS only initiated early stage arthritic-like changes, the long-term protective effects of PTHrP may be even greater than indicated in this study. Future work will examine longer-term treatment regimens to better assess the effects of PTHrP overexpression on overall matrix metabolism and chondrocyte survival under mechanical loading conditions.
Acknowledgments
We thank Dr. Leonard J. Deftos for providing the hPTHrP constructs. This research was supported by the NIH Intramural Research Program (Z01 AR41131) and the Commonwealth of Pennsylvania Department of Health. D.W. was a Howard Hughes Medical Institute-NIH Research Scholar.
Footnotes
AUTHOR CONTRIBUTIONS
All authors contributed to the conception and design of the study, analysis and interpretation of data, and preparation and approval of the manuscript. In addition, D.W. and J.M.T. contributed to data acquisition, and D.W. (wangd@ccf.org) drafted the manuscript and takes responsibility for the integrity of the work.
COMPETING INTEREST STATEMENT
The authors had no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Steinert AF, Ghivizzani SC, Rethwilm A, Tuan RS, Evans CH, Noth U. Major biological obstacles for persistent cell-based regeneration of articular cartilage. Arthritis Res Ther. 2007;9:213. doi: 10.1186/ar2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brittberg M, Lindahl A, Nilsson A, Ohlsson C, Isaksson O, Peterson L. Treatment of deep cartilage defects in the knee with autologous chondrocyte transplantation. N Engl J Med. 1994;331:889–895. doi: 10.1056/NEJM199410063311401. [DOI] [PubMed] [Google Scholar]
- 3.Buckwalter JA, Mankin HJ. Instructional Course Lectures, The American Academy of Orthopaedic Surgeons - Articular Cartilage. Part II: Degeneration and osteoarthritis, repair, regeneration, and transplantation. J Bone Joint Surg Am. 1997;79:612–632. [Google Scholar]
- 4.Bullough PG. The pathology of osteoarthritis. In: Moskowitz RW, Goldberg VM, Hochberg MC, editors. Osteoarthritis. Philadelphia: Saunders; 1992. pp. 39–70. [Google Scholar]
- 5.Djouad F, Rackwitz L, Song Y, Janjanin S, Tuan RS. ERK1/2 activation induced by inflammatory cytokines compromises effective host tissue integration of engineered cartilage. Tissue Eng Part A. 2009 doi: 10.1089/ten.tea.2008.0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brandt KD, Myers SL, Burr D, Albrecht M. Osteoarthritic changes in canine articular cartilage, subchondral bone, and synovium fifty-four months after transection of the anterior cruciate ligament. Arthritis Rheum. 1991;34:1560–1570. doi: 10.1002/art.1780341214. [DOI] [PubMed] [Google Scholar]
- 7.Radin EL, Martin RB, Burr DB, Caterson B, Boyd RD, Goodwin C. Effects of mechanical loading on the tissues of the rabbit knee. J Orthop Res. 1984;2:221–234. doi: 10.1002/jor.1100020303. [DOI] [PubMed] [Google Scholar]
- 8.Goldring MB, Goldring SR. Osteoarthritis. J Cell Physiol. 2007;213:626–634. doi: 10.1002/jcp.21258. [DOI] [PubMed] [Google Scholar]
- 9.Kirsch T, Swoboda B, Nah H. Activation of annexin II and V expression, terminal differentiation, mineralization and apoptosis in human osteoarthritic cartilage. Osteoarthritis Cartilage. 2000;8:294–302. doi: 10.1053/joca.1999.0304. [DOI] [PubMed] [Google Scholar]
- 10.Tchetina EV, Squires G, Poole AR. Increased type II collagen degradation and very early focal cartilage degeneration is associated with upregulation of chondrocyte differentiation related genes in early human articular cartilage lesions. J Rheumatol. 2005;32:876–886. [PubMed] [Google Scholar]
- 11.Aigner T, Fundel K, Saas J, Gebhard PM, Haag J, Weiss T, et al. Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum. 2006;54:3533–3544. doi: 10.1002/art.22174. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Manner PA, Horner A, Shum L, Tuan RS, Nuckolls GH. Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthritis Cartilage. 2004;12:963–973. doi: 10.1016/j.joca.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 13.Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–622. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- 14.Bastepe M, Weinstein LS, Ogata N, Kawaguchi H, Juppner H, Kronenberg HM, et al. Stimulatory G protein directly regulates hypertrophic differentiation of growth plate cartilage in vivo. Proc Natl Acad Sci U S A. 2004;101:14794–14799. doi: 10.1073/pnas.0405091101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang W, Chung UI, Kronenberg HM, de Crombrugghe B. The chondrogenic transcription factor Sox9 is a target of signaling by the parathyroid hormone-related peptide in the growth plate of endochondral bones. Proc Natl Acad Sci U S A. 2001;98:160–165. doi: 10.1073/pnas.011393998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amling M, Neff L, Tanaka S, Inoue D, Kuida K, Weir E, et al. Bcl-2 lies downstream of parathyroid hormone-related peptide in a signaling pathway that regulates chondrocyte maturation during skeletal development. J Cell Biol. 1997;136:205–213. doi: 10.1083/jcb.136.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henderson JE, Amizuka N, Warshawsky H, Biasotto D, Lanske BM, Goltzman D, et al. Nucleolar localization of parathyroid hormone-related peptide enhances survival of chondrocytes under conditions that promote apoptotic cell death. Mol Cell Biol. 1995;15:4064–4075. doi: 10.1128/mcb.15.8.4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steinert AF, Noth U, Tuan RS. Concepts in gene therapy for cartilage repair. Injury. 2008;39 (Suppl 1):S97–113. doi: 10.1016/j.injury.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thirion S, Berenbaum F. Culture and Phenotyping of Chondrocytes in Primary Culture. In: Sabatini M, Pastoureau P, Ceuninck F, editors. Cartilage and Osteoarthritis: Volume 1: Cellular and Molecular Tools. New York: Humana Press; 2004. pp. 1–14. [Google Scholar]
- 20.Madry H, Trippel SB. Efficient lipid-mediated gene transfer to articular chondrocytes. Gene Ther. 2000;7:286–291. doi: 10.1038/sj.gt.3301086. [DOI] [PubMed] [Google Scholar]
- 21.Ditmer LS, Burton DW, Deftos LJ. Elimination of the carboxy-terminal sequences of parathyroid hormone-related protein 1-173 increases production and secretion of the truncated forms. Endocrinology. 1996;137:1608–1617. doi: 10.1210/endo.137.5.8612492. [DOI] [PubMed] [Google Scholar]
- 22.Pfander D, Swoboda B, Kirsch T. Expression of early and late differentiation markers (proliferating cell nuclear antigen, syndecan-3, annexin VI, and alkaline phosphatase) by human osteoarthritic chondrocytes. Am J Pathol. 2001;159:1777–1783. doi: 10.1016/S0002-9440(10)63024-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aigner T, Kim HA, Roach HI. Apoptosis in osteoarthritis. Rheum Dis Clin North Am. 2004;30:639–653. xi. doi: 10.1016/j.rdc.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 24.Ballock RT, Heydemann A, Wakefield LM, Flanders KC, Roberts AB, Sporn MB. TGF-beta 1 prevents hypertrophy of epiphyseal chondrocytes: regulation of gene expression for cartilage matrix proteins and metalloproteases. Dev Biol. 1993;158:414–429. doi: 10.1006/dbio.1993.1200. [DOI] [PubMed] [Google Scholar]
- 25.Tchetina E, Mwale F, Poole AR. Distinct phases of coordinated early and late gene expression in growth plate chondrocytes in relationship to cell proliferation, matrix assembly, remodeling, and cell differentiation. J Bone Miner Res. 2003;18:844–851. doi: 10.1359/jbmr.2003.18.5.844. [DOI] [PubMed] [Google Scholar]
- 26.Dean DD, Muniz OE, Berman I, Pita JC, Carreno MR, Woessner JF, Jr, et al. Localization of collagenase in the growth plate of rachitic rats. J Clin Invest. 1985;76:716–722. doi: 10.1172/JCI112026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cawston TE, Wilson AJ. Understanding the role of tissue degrading enzymes and their inhibitors in development and disease. Best Pract Res Clin Rheumatol. 2006;20:983–1002. doi: 10.1016/j.berh.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 28.Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, et al. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orloff JJ, Reddy D, de Papp AE, Yang KH, Soifer NE, Stewart AF. Parathyroid hormone-related protein as a prohormone: posttranslational processing and receptor interactions. Endocr Rev. 1994;15:40–60. doi: 10.1210/edrv-15-1-40. [DOI] [PubMed] [Google Scholar]
- 30.Wysolmerski JJ, Stewart AF. The physiology of parathyroid hormone-related protein: an emerging role as a developmental factor. Annu Rev Physiol. 1998;60:431–460. doi: 10.1146/annurev.physiol.60.1.431. [DOI] [PubMed] [Google Scholar]
- 31.Kronenberg HM. PTHrP and skeletal development. Ann N Y Acad Sci. 2006;1068:1–13. doi: 10.1196/annals.1346.002. [DOI] [PubMed] [Google Scholar]
- 32.Jiang J, Leong NL, Mung JC, Hidaka C, Lu HH. Interaction between zonal populations of articular chondrocytes suppresses chondrocyte mineralization and this process is mediated by PTHrP. Osteoarthritis Cartilage. 2008;16:70–82. doi: 10.1016/j.joca.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 33.Chang JK, Chang LH, Hung SH, Wu SC, Lee HY, Lin YS, et al. Parathyroid hormone 1-34 inhibits terminal differentiation of human articular chondrocytes and osteoarthritis progression in rats. Arthritis Rheum. 2009;60:3049–3060. doi: 10.1002/art.24843. [DOI] [PubMed] [Google Scholar]
- 34.Aarts MM, Davidson D, Corluka A, Petroulakis E, Guo J, Bringhurst FR, et al. Parathyroid hormone-related protein promotes quiescence and survival of serum-deprived chondrocytes by inhibiting rRNA synthesis. J Biol Chem. 2001;276:37934–37943. doi: 10.1074/jbc.M105510200. [DOI] [PubMed] [Google Scholar]
- 35.Okoumassoun L, Averill-Bates D, Denizeau F, Henderson JE. Parathyroid hormone related protein (PTHrP) inhibits TNFalpha-induced apoptosis by blocking the extrinsic and intrinsic pathways. J Cell Physiol. 2007;210:507–516. doi: 10.1002/jcp.20892. [DOI] [PubMed] [Google Scholar]
- 36.Okoumassoun LE, Russo C, Denizeau F, Averill-Bates D, Henderson JE. Parathyroid hormone-related protein (PTHrP) inhibits mitochondrial-dependent apoptosis through CK2. J Cell Physiol. 2007;212:591–599. doi: 10.1002/jcp.21055. [DOI] [PubMed] [Google Scholar]
- 37.Henry JG, Mitnick M, Dann PR, Stewart AF. Parathyroid hormone-related protein-(1-36) is biologically active when administered subcutaneously to humans. J Clin Endocrinol Metab. 1997;82:900–906. doi: 10.1210/jcem.82.3.3811. [DOI] [PubMed] [Google Scholar]
- 38.Terkeltaub R, Lotz M, Johnson K, Deng D, Hashimoto S, Goldring MB, et al. Parathyroid hormone-related proteins is abundant in osteoarthritic cartilage, and the parathyroid hormone-related protein 1-173 isoform is selectively induced by transforming growth factor beta in articular chondrocytes and suppresses generation of extracellular inorganic pyrophosphate. Arthritis Rheum. 1998;41:2152–2164. doi: 10.1002/1529-0131(199812)41:12<2152::AID-ART10>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 39.Okano K, Tsukazaki T, Ohtsuru A, Osaki M, Yonekura A, Iwasaki K, et al. Expression of parathyroid hormone-related peptide in human osteoarthritis. J Orthop Res. 1997;15:175–180. doi: 10.1002/jor.1100150204. [DOI] [PubMed] [Google Scholar]
- 40.Gomez-Barrena E, Sanchez-Pernaute O, Largo R, Calvo E, Esbrit P, Herrero-Beaumont G. Sequential changes of parathyroid hormone related protein (PTHrP) in articular cartilage during progression of inflammatory and degenerative arthritis. Ann Rheum Dis. 2004;63:917–922. doi: 10.1136/ard.2003.008904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mattot V, Raes MB, Henriet P, Eeckhout Y, Stehelin D, Vandenbunder B, et al. Expression of interstitial collagenase is restricted to skeletal tissue during mouse embryogenesis. J Cell Sci. 1995;108 (Pt 2):529–535. doi: 10.1242/jcs.108.2.529. [DOI] [PubMed] [Google Scholar]
- 42.Goldring MB, Berenbaum F. The regulation of chondrocyte function by proinflammatory mediators: prostaglandins and nitric oxide. Clin Orthop Relat Res. 2004:S37–46. doi: 10.1097/01.blo.0000144484.69656.e4. [DOI] [PubMed] [Google Scholar]
- 43.Kalinowski L, Dobrucki LW, Malinski T. Nitric oxide as a second messenger in parathyroid hormone-related protein signaling. J Endocrinol. 2001;170:433–440. doi: 10.1677/joe.0.1700433. [DOI] [PubMed] [Google Scholar]
- 44.Yoshida T, Horiuchi T, Sakamoto H, Inoue H, Takayanagi H, Nishikawa T, et al. Production of parathyroid hormone-related peptide by synovial fibroblasts in human osteoarthritis. FEBS Lett. 1998;433:331–334. doi: 10.1016/s0014-5793(98)00940-5. [DOI] [PubMed] [Google Scholar]
- 45.Yoshida T, Sakamoto H, Horiuchi T, Yamamoto S, Suematsu A, Oda H, et al. Involvement of prostaglandin E(2) in interleukin-1alpha-induced parathyroid hormone-related peptide production in synovial fibroblasts of patients with rheumatoid arthritis. J Clin Endocrinol Metab. 2001;86:3272–3278. doi: 10.1210/jcem.86.7.7687. [DOI] [PubMed] [Google Scholar]
- 46.Cheung JO, Grant ME, Jones CJ, Hoyland JA, Freemont AJ, Hillarby MC. Apoptosis of terminal hypertrophic chondrocytes in an in vitro model of endochondral ossification. J Pathol. 2003;201:496–503. doi: 10.1002/path.1462. [DOI] [PubMed] [Google Scholar]
- 47.van de Loo FA, de Hooge AS, Smeets RL, Bakker AC, Bennink MB, Arntz OJ, et al. An inflammation-inducible adenoviral expression system for local treatment of the arthritic joint. Gene Ther. 2004;11:581–590. doi: 10.1038/sj.gt.3302182. [DOI] [PubMed] [Google Scholar]
- 48.Geurts J, Arntz OJ, Bennink MB, Joosten LA, van den Berg WB, van de Loo FA. Application of a disease-regulated promoter is a safer mode of local IL-4 gene therapy for arthritis. Gene Ther. 2007;14:1632–1638. doi: 10.1038/sj.gt.3303022. [DOI] [PubMed] [Google Scholar]
- 49.Rachakonda PS, Rai MF, Schmidt MF. Application of inflammation-responsive promoter for an in vitro arthritis model. Arthritis Rheum. 2008;58:2088–2097. doi: 10.1002/art.23598. [DOI] [PubMed] [Google Scholar]
- 50.Guo J, Chung UI, Kondo H, Bringhurst FR, Kronenberg HM. The PTH/PTHrP receptor can delay chondrocyte hypertrophy in vivo without activating phospholipase C. Dev Cell. 2002;3:183–194. doi: 10.1016/s1534-5807(02)00218-6. [DOI] [PubMed] [Google Scholar]