

Abstract

The highly enantioselective synthesis of 2-aryl- and 2-vinyl-piperidines has been accomplished through a catalytic dynamic resolution (CDR) of N-Boc-2-lithiopiperidine. The method has been applied to the synthesis of both enantiomers of the tobacco alkaloid anabasine.
Optically active 2-aryl- and 2-vinyl-piperidines are found in a variety of natural products and some have useful pharmacological properties.1–3 Previously, Dieter demonstrated that transmetalation of N-Boc-2-lithiopyrrolidine to its organocopper counterpart provides a convenient way to synthesize vinylated pyrrolidines.4,5 Using an asymmetric deprotonation methodology with the chiral base s-BuLi/(–)-sparteine6,7 to generate an enantioenriched organolithium, Dieter achieved enantioselective vinylations with er’s up to 93:7. Later, Campos used the asymmetric deprotonation methodology and transmetalation to an organozinc species, followed by a palladium-mediated Negishi coupling with aryl bromides, to prepare N-Boc-2-arylpyrrolidines in good yields and ≥96:4 er’s (Scheme 1).8–11
Scheme 1.

Campos conditions for enantioselective arylation of N-Boc-pyrrolidine.8
The remarkable chemical and configurational stability of the intermediate organozinc compound, even at 60 °C, and the high degree of tolerance for both electron rich and electron deficient aryl halides make this transformation very attractive.
Previous attempts to synthesize enantioenriched 2-aryl-piperidines via Negishi coupling conditions have been less successful. O’Brien recently reported an asymmetric deprotonation of N-Boc-piperidine using s-BuLi and O’Brien’s diamine12 (Figure 1), followed by trapping with 4-bromoveratrole, affording the arylated product in 33% yield and 82:18 er (S:R).11 Although the Coldham group successfully synthesized racemic members of this family via Negishi coupling,13 attempts to effect enantioselectivity by dynamic thermodynamic resolution (DTR) using stoichiometric amount of chiral ligand led to no arylation products.14 They reported two examples of enantioenriched 2-aryl-piperidines (er 82:18 (R:S)) obtained by transmetalation of an enantioenriched stannane to the organolithium under conditions that ensured configurational stability of the latter.
Figure 1.

Chiral ligands
We recently reported the highly enantioselective synthesis of 2-substituted piperidines by catalytic dynamic resolution (CDR) of N-Boc-2-lithiopiperidine 1 using diastereomeric ligands (S,S)-2 and (S,R)-2 (Figure 1).15 In our report, we utilized copper-mediated coupling to synthesize enantioenriched 2-allyl and 2-benzyl-piperidines (Scheme 2) via an organozinc reagent formed by transmetalation of the resolved N-Boc-2-lithiopiperidine.
Scheme 2.

CDR of N-Boc-2-lithiopiperidine followed by copper-mediated allylation and benzylation.
We know (Scheme 2) that during transmetalation of lithium to zinc, then to copper, configurational stability is maintained. Based on Campos’ results (Scheme 1), we hoped that during transmetalation from zinc to palladium, configurational integrity would be retained.
Considering that we are able to resolve N-Boc-2-lithiopiperidine catalytically, we investigated a CDR in the Negishi arylations and vinylations. We herein expand the synthetic potential of our methodology to the direct enantioselective synthesis of 2-aryl and 2-vinyl-piperidines. Good yields and er’s are obtained with a 5% catalyst loading. Our method obviates the need for an enantioselective deprotonation or the asymmetric synthesis of a precursor stannane.
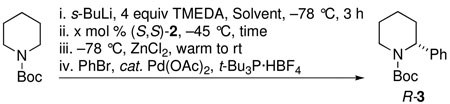
The conditions for the CDR of rac-1 were optimized as illustrated in Table 1, beginning with the previously optimized conditions.15 In oven-dried septum-capped flasks, rac-1 was generated by deprotonation of N-Boc-piperidine in either Et2O or MTBE at −78 °C[note 1] with s-BuLi/TMEDA, followed by addition of (S,S)-2, warming to −45 °C for 3 to 5 h, then cooling to −78 °C. A solution of ZnCl2 in THF was added prior to warming to room temperature and introduction of Pd(OAc)2, t-Bu3P·HBF4 and phenyl bromide sequentially (details are in the Supporting Information). After stirring for 3 h with 10 mol% of (S,S)-2 in the presence of Et2O, we were pleased to obtain R-3 in 74% yield and 90:10 er (entry 1). With MTBE, R-3 was obtained in 65% yield and 86:14 er (entry 2). After lowering the catalyst loading to 5 mol%, stirring at −45 °C for 3 h in Et2O improved the er to 93:7 (entry 3). Performing the CDR for an additional 2 h at −45 °C led to a further enhancement in the er of R-3 to 96:4 (entry 4). However, with MTBE, the same conditions afforded R-3 in 60% yield and 89:11 er (entry 5).
Table 1.
Optimization of the enantioselective arylation of N-Boc-2-lithiopiperidine by CDR.
 | |||||
|---|---|---|---|---|---|
| entry | (S,S)-2 (mol %) |
time (h) | solvent | yield (%) of 3 |
er of 3 (R:S) |
| 1 | 10 | 3 | Et2O | 74 | 90:10 |
| 2 | 10 | 3 | MTBE | 65 | 86:14 |
| 3 | 5 | 3 | Et2O | 70 | 93:7 |
| 4 | 5 | 5 | Et2O | 68 | 96:4 |
| 5 | 5 | 5 | MTBE | 60 | 89:11 |
| 6 | 100 | 3 | Et2O | 71 | 53:47 |
Two important findings emerged from our optimization: (i) the yields and er’s are lower in MTBE than in Et2O, (ii) the er’s are higher with lower loading of (S,S)-2. Intriguingly, when a DTR using a stoichiometric amount of (S,S)-2 was carried out, nearly racemic 3 was obtained (entry 6). The reason for the loss of enantioselectivity is not fully understood at this point.
Several other aryl and vinyl halides were evaluated under the optimized CDR conditions, with the results summarized in Table 2. In all cases, we obtained good yields and high er’s. Quenching with o-bromotoluene afforded R-4 in 63% yield and 92:8 er (entry 1) and S-4 of 6:94 er using (S,R)-2 (entry 2). Electrophilic quench with 4-bromoveratrole gave R-5 in 75% yield and 97:3 er (entry 3). Quenching with p-tert-butylbromobenzene afforded R-6 in 69% yield and 95:5 er (entry 4) while 2-bromomesitylene gave R-7 in 64% yield and 92:8 er (entry 5).
Table 2.
CDR of rac-1 and Negishi arylation or vinylation
 | ||||
|---|---|---|---|---|
| entry | electrophile | product | yield (%) |
er (R:S) |
| 1a | 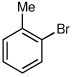 |
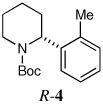 |
63 | 92:8 |
| 2a,b | 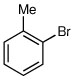 |
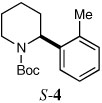 |
59 | 6:94 |
| 3a |  |
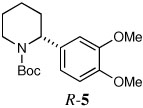 |
75 | 97:3 |
| 4a |  |
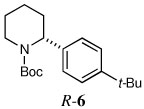 |
69 | 95:5 |
| 5a | 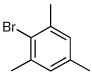 |
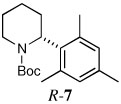 |
64 | 92:8 |
| 6a | 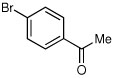 |
 |
60 | 91:9 |
| 7a,c | 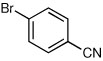 |
 |
66 | 90:10 |
| 8a | 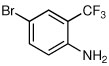 |
 |
60 | 93:7 |
| 9e | 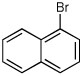 |
 |
67 | 97:3 |
| 10d | 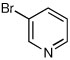 |
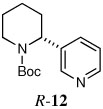 |
46 | 88:12 |
| 11b,d | 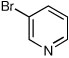 |
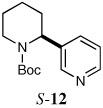 |
51 | 10:90 |
| 12d | 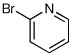 |
 |
50 | 93:7 |
| 13d | 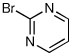 |
 |
53 | 85:15 |
| 14a | 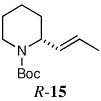 |
63 | 92:8 | |
| 15a,e | 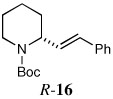 |
66 | 93:7 | |
stirred at rt for 18 h during step (iv),
CDR using (S,R)-2;
the aryl bromide contained a small amount of 2-bromobenzonitrile leading to formation of the ortho cyano constitutional isomer in 90:10 er;
stirred at 60 °C for 22 h during step (iv),
a small amount of the Z-alkene was obtained as a minor isomer in 92:8 er.
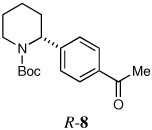
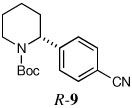
Some electron deficient aryl bromides were evaluated in order to highlight the high degree of tolerance of aryl coupling. With p-bromoacetophenone, R-8 was obtained in 66% yield and 91:9 er (entry 6). Electrophilic quench with 4-bromobenzonitrile gave R-9 in 66% yield and 90:10 er (entry 7).
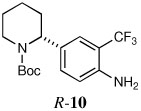
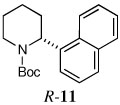
Quenching with 4-bromo-2-trifluoromethyl aniline afforded R-10 in 60% yield and 93:7 er (entry 8), thus illustrating that this Negishi arylation works even in the presence of an unprotected amine. Electrophilic quench with 1-bromonaphthalene gave R-11 in 67% yield and 97:3 er (entry 9).
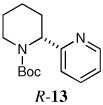
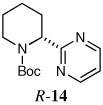
When heteroaryl halides were employed, the arylations occurred rather slowly and required mild heating to 60 °C. Quenching with 3-bromopyridine gave R-12 in 46% yield and 88:12 er (entry 10). When the CDR was carried out using (S,R)-2, S-12 was obtained in 51% yield and 90:10 er (entry 11). With 2-bromopyridine, R-13 of 50% yield and 93:7 er was obtained (entry 12). Electrophilic quench with 2-bromopyrimidine gave R-14 in 53% yield and 85:15 er (entry 13).
Vinylation was also investigated using the same reaction conditions. Thus, quenching with 1-bromo-1-propene afforded R-15 in 63% yield and 92:8 er (entry 14). With β-bromostyrene, R-16 was obtained in 66% yield and 93:7 er (entry 15).
We wondered whether the comparatively low er’s in the heterocycle arylations (entries 10–13) were due to slight loss of configurational stability at 60 °C or some unidentified process. Scheme 3 shows a control experiment in which the er was monitored at some key stages of the reaction. After performing a CDR at −45 °C for 5 h and then cooling to −78 °C, we quenched an aliquot with phenyl isocyanate,15 and found the product, R-17 to have 97:3 er. After adding ZnCl2 to the remaining mixture, warming to room temperature and introduction of the Pd(OAc)2, t-Bu3P·HBF4, and 2-bromopyrimidine, a second aliquot was allowed to stir for two days at room temperature. This aliquot showed that R-14 had 92:8 er. The remaining heterogeneous mixture was heated to 60 °C for 22 h, then cooled. The er from this sample was 86:14. Although the slight loss of er from 97:3 to 92:8 was anticipated, the significant loss of er to 86:14 reveals that the configurational stability with the piperidine system is compromised at 60 °C, in contrast with the pyrrolidines.8
Scheme 3.

Monitoring of the er during enantioselective arylation
Synthesis of anabasine enantiomers: The synthesis of either enantiomer of the tobacco alkaloid anabasine was accomplished as illustrated in Scheme 4. CDR of rac-1 using either (S,S)-2 or (S,R)-2, transmetalation, and Negishi coupling with 3-bromopyridine afforded R-12 or S-12 respectively. Hydrolysis of the enantiomeric carbamates with trifluoroacetic acid afforded (R)-anabasine in 46 % and 88:12 er; [α]D22 70.9 (c = 1.0, MeOH) or (S)-anabasine in 50 % and 90:10 er; [α]D22 −73.4 (c = 1.0, MeOH), lit.16 [α]D20 −80 (92% ee; c = 0.91, MeOH) in just two steps.
Scheme 4.

Preparation of (R) and (S)-anabasine16
In summary, the asymmetric arylation and vinylation of N-Boc-piperidine has been accomplished through catalytic dynamic resolution of N-Boc-2-lithiopiperidine followed by transmetalation and Negishi coupling. Most aryl and vinyl halides produce coupling products of the chiral organozinc intermediate with high enantioselectivity at room temperature. When somewhat elevated temperatures are required, such as with π-deficient heterocycles, some racemization occurs.
Supplementary Material
Acknowledgment
This work was supported by the National Science Foundation (CHE 1011788) and the Arkansas Biosciences Institute. Core facilities were funded by the National Institutes of Health (R15569) and the Arkansas Biosciences Institute. The authors are grateful to Sarah Pursley (University of Arkansas) for help in chromatographic separations.
Footnotes
Supporting Information Available Full experimental details and spectroscopic data. This information is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Amat M, Canto M, Llor N, Bosch J. Chem. Commun. 2002:526. doi: 10.1039/b200020m. [DOI] [PubMed] [Google Scholar]
- 2.Castro A, Ramirez J, Juarez J, Teran JL, Orea L, Galindo A, Gnecco D. Heterocycles. 2007;71:2699. [Google Scholar]
- 3.Horton DA, Bourne GT, Smythe ML. Chem. Rev. 2003;103:893. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- 4.Dieter RK, Li S. J. Org. Chem. 1997;62:7726. [Google Scholar]
- 5.Dieter RK, Topping CM, Chandupatla KR, Lu K. J. Am. Chem. Soc. 2001;123:5132. doi: 10.1021/ja0156587. [DOI] [PubMed] [Google Scholar]
- 6.Kerrick ST, Beak P. J. Am. Chem. Soc. 1991;113:9708. [Google Scholar]
- 7.Hoppe D, Hintze F, Tebben P. Angew. Chem. Int. Ed. Engl. 1990;29:1422. [Google Scholar]
- 8.Campos KR, Klapars A, Waldman JH, Dormer PG, Chen C. J. Am. Chem. Soc. 2006;128:3538. doi: 10.1021/ja0605265. [DOI] [PubMed] [Google Scholar]
- 9.O'Brien P, Bilke JL. Angew. Chem., Int. Ed. 2008;47:2734. doi: 10.1002/anie.200704539. [DOI] [PubMed] [Google Scholar]
- 10.Klapars A, Campos KR, Waldman JH, Zewge D, Dormer PG, Chen C-y. J. Org. Chem. 2008;73:4986. doi: 10.1021/jo8006804. [DOI] [PubMed] [Google Scholar]
- 11.Stead D, Carbone G, O'Brien P, Campos KR, Coldham I, Sanderson A. J. Am. Chem. Soc. 2010;132:7260. doi: 10.1021/ja102043e. [DOI] [PubMed] [Google Scholar]
- 12.Dearden MJ, Firkin CR, Hermet J-PR, O'Brien P. J. Am. Chem. Soc. 2002;124:11870. doi: 10.1021/ja027774v. [DOI] [PubMed] [Google Scholar]
- 13.Coldham I, Leonori D. Org. Lett. 2008;10:3923. doi: 10.1021/ol801579r. [DOI] [PubMed] [Google Scholar]
- 14.Coldham I, Raimbault S, Whittaker DTE, Chovatia PT, Leonori D, Patel JJ, Sheikh NS. Chem. Eur. J. 2010;16:4082. doi: 10.1002/chem.200903059. [DOI] [PubMed] [Google Scholar]
- 15.Beng TK, Gawley RE. J. Am. Chem. Soc. 2010;132:12216. doi: 10.1021/ja105772z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Felpin FX, Girard S, Vo-Thanh G, Robins RJ, Villieras J, Lebreton J. J Org Chem. 2001;66:6305. doi: 10.1021/jo010386b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.