

Abstract
A robust analysis and discovery platform for antibiotics targeting the bacterial rRNA A-site has been developed by incorporating a new emissive U surrogate into the RNA and labeling the aminoglycosides with an appropriate fluorescence acceptor. Specifically, a 5-methoxyquinazoline-2,4(1H,3H)-dione-based emissive uracil analogue was identified to be an ideal donor for 7-diethylaminocoumarin-3-carboxylic acid. This donor/acceptor pair displays a critical Förster radius (R0) of 27 Å, a value suitable for an A-site-aminoglycoside assembly. Titrating the coumarin labeled aminoglycosides into the emissive A-site construct, labeled at position U1406, shows a decrease in donor emission (at 395 nm) and concurrent increase of the acceptor emission (at 473 nm). Titration curves, obtained by fitting the donor’s emission quenching or the augmentation of the acceptor’s sensitized emission, faithfully generate EC50 values. Titration of unlabeled ligands into the preformed FRET complex showed a continuous increase of the donor emission, with a concurrent decrease of the acceptor emission, yielding valuable data regarding competitive displacement of aminoglycosides by A-site binders. Detection of antibiotic binding is therefore not dependent on changes in the environment of a single fluorophore, but rather on the responsive interaction between two chromophores acting as a FRET pair, facilitating the determination of direct binding and competitive displacement events with FRET accuracy.
Introduction
The ribosomal decoding site, also known as the A-site, ensures high fidelity in protein synthesis by appraising codon–anticodon matching.1–3 Numerous naturally occurring potent antibiotics, particularly the aminoglycosides family, have evolved to meddle with this precise monitoring and corrupt bacterial protein production.4–8 Specifically, the aminoglycosides bind a small loop within the 16S rRNA and interfere with the conformational flexibility of A1492 and A1493, two key adenine residues (Figure 1a).9–12 The aminoglycosides stabilize an RNA conformation similar to the one induced by the cognate acyl-tRNA–mRNA complex, causing the ribosome to lose its ability to distinguish between the correct and incorrect anticodon–codon hybrids.13–18
Figure 1.
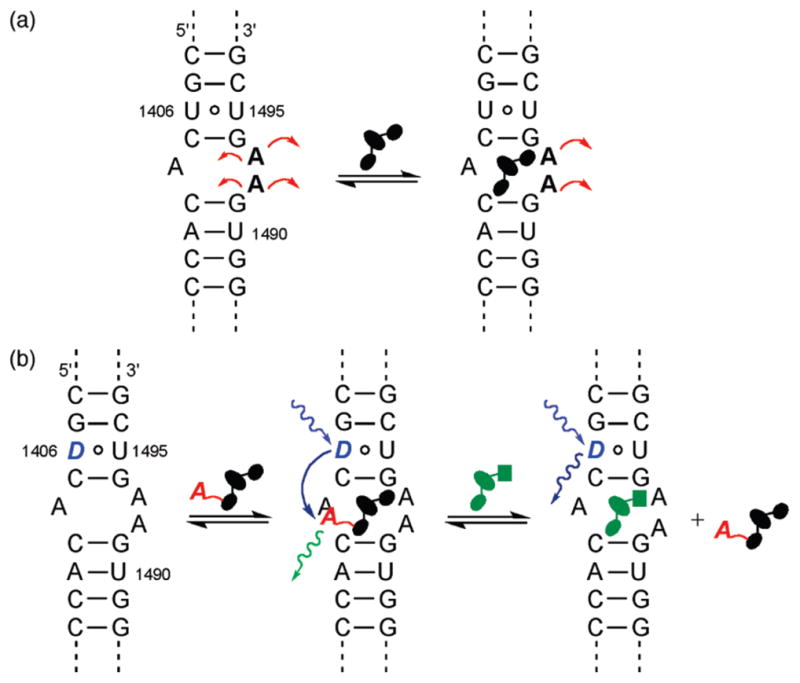
(a) Binding of aminoglycosides to the bacterial A-site impacts the placement and dynamics of the unpaired A1492 and 1493 residues. (b) By replacing one of the nucleosides in the A-site with an isosteric emissive nucleoside analogue as a donor (D) and tagging the antibiotics with an appropriate acceptor (A), binding and displacement events can be accurately monitored using FRET.
The A-site, the Achilles heel of the bacterial ribosome, has remained one of the most attractive targets for the discovery and development of new antibiotics.19–25 A number of tools have been developed to assess ligand binding to this unique RNA site.26,27 Fluorescent A-site constructs, which contain emissive and responsive nucleoside analogues, such as 2-aminopurine at positions 1492 or 1493, have shown great promise.28–31 Their fluorescence response is, however, antibiotic-dependent.28,32 To overcome this drawback and devise a robust analysis and discovery platform for A-site binders, we have envisioned an approach where detection of antibiotic binding is not dependent on changes in the environment of a single fluorophore, but rather on the interaction between two chromophores acting as a Förster Resonance Energy Transfer (FRET) pair (Figure 1b). In this fashion, we hypothesized that one could follow direct binding of appropriately labeled antibiotics and their displacement by competing binders with “FRET accuracy” without relying on a fluorescent nucleobase as the sole sensing moiety (Figure 1b).
To realize such a system, we have relied on two key features: (a) one of the native nucleobases in the A-site, proximal to the binding site, but not part of it, had to be replaced with an emissive isomorphic nucleobase analogue acting as a FRET donor, and (b) aminoglycosides, the cognate binders of the A-site, had to be labeled with an appropriate FRET acceptor in positions that are not essential for RNA binding (Figure 1b).33 By monitoring the interactions of ligands and their RNA targets based on distance and location, convoluting factors such as modes of binding can be eliminated when studying binding and displacement. Here we describe the design, assembly, and utility of such a FRET-friendly and minimally perturbed RNA construct, where a new fluorescent uridine analogue, serving as a fluorescent donor, is incorporated into the A-site and coumarin-labeled aminoglycosides act as FRET acceptors.
Results
Selection of the Donor and Acceptor
While diverse FRET donors and acceptors exist, small chromophores capable of serving as nonperturbing nucleobase surrogates are exceedingly rare.34 In pursuing such unique probes, we have synthesized, examined, and implemented new emissive nucleosides with absorption maxima between 290 and 350 nm and emission bands between 350 and 440 nm.35–39 We have identified 5-methoxyquinazoline-2,4-(1H,3H)-dione 1, an emissive uracil analogue, as an ideal donor for 7-diethylaminocoumarin-3-carboxylic acid 2 (Figure 2). The extinction coefficient of 2 at 320 nm, the absorption maximum of 1, is minimal, while the emission of 1, centered around 395 nm (φF = 0.16), overlaps perfectly with the absorption band of 2, which emits at 473 nm (φF = 0.83), suggesting excellent FRET pairing (Figure 3).40,41 Indeed, the critical Förster radius (R0) for the 1/2 pair was experimentally determined to be 27 Å, a suitable value for the proposed A-site assembly.42
Figure 2.

Structure of the donor 1 and acceptor 2, as well as 3 (the parent donor heterocycle) and 4 (an isomeric methoxy substituted heterocycle).40
Figure 3.
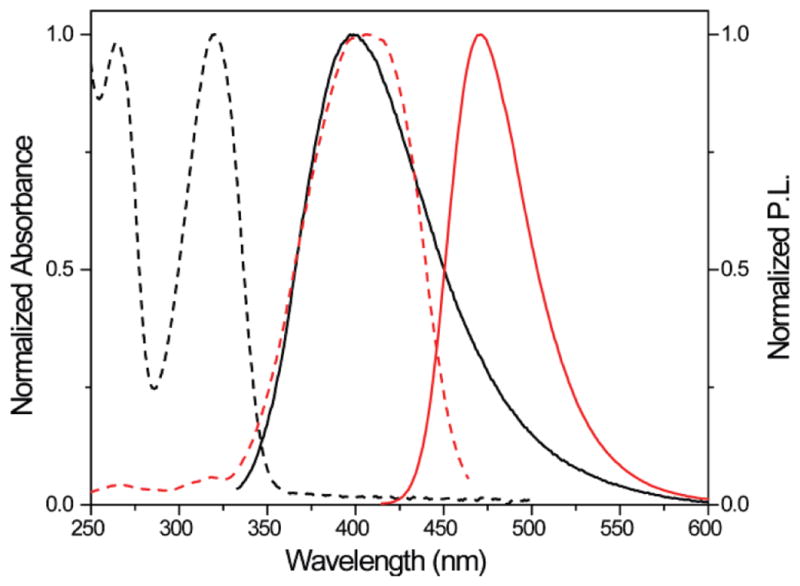
Normalized absorption (---) and emission (—) spectra of 1 (black) and 2 (red) in water.40
The presence and position of the methoxy group on the quinazoline-2,4-(1H,3H)-dione core impact the photophysical properties and are critical for the compatibility of the FRET partners, as evident by a comparison of the parent and to two isomeric methoxy substituted heterocycles. The emission profile of quinazoline-2,4-(1H,3H)-dione 3, the parent unsubstituted heterocycle, is hypsochromically shifted compared to the emission of 1, with a maximum at 370 nm (Figure 4). The emission of the isomeric 3-methoxyquinazoline-2,4-(1H,3H)-dione 4 is even further shifted to the blue with the emission maximum at 356 nm (Figure 4). Importantly, glycosylation of these heterocycles to yield the corresponding nucleosides negligibly impacts the photophysical properties.40
Figure 4.
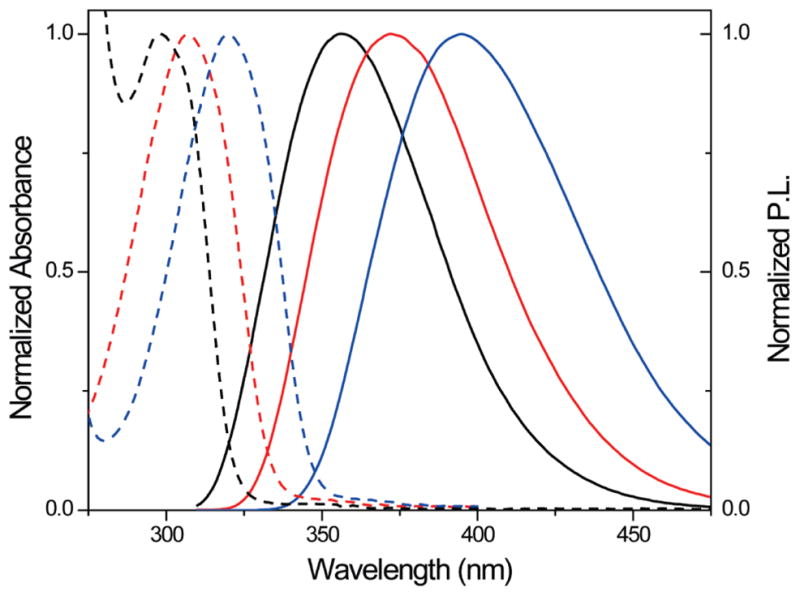
Normalized absorption (---) and emission (—) spectra of 4 (black), 3 (red), and 1 (blue) in water.
Synthesis and Photophysical Properties of Modified RNA
To modify the A-site RNA oligonucleotide, nucleoside 5 and its phosphoramidite 8 were prepared as shown in Scheme 1.40 The commercially available 2-methoxy-5-aminobenzoic acid was cyclized with sodium cyanate to yield the emissive heterocycle 1, which was glycosylated to provide the modified nucleoside 5 after saponification of all esters. Protection of the 5′-hydroxyl as the 4,4′-dimethoxytrityl (DMT) derivative and the 2′-hydroxyl as the (trisisopropylsiloxy)methyl (TOM) derivative, followed by phosphitylation of the 3′-hydroxyl, provided phosphoramidite 8 (Scheme 1).40
Scheme 1.
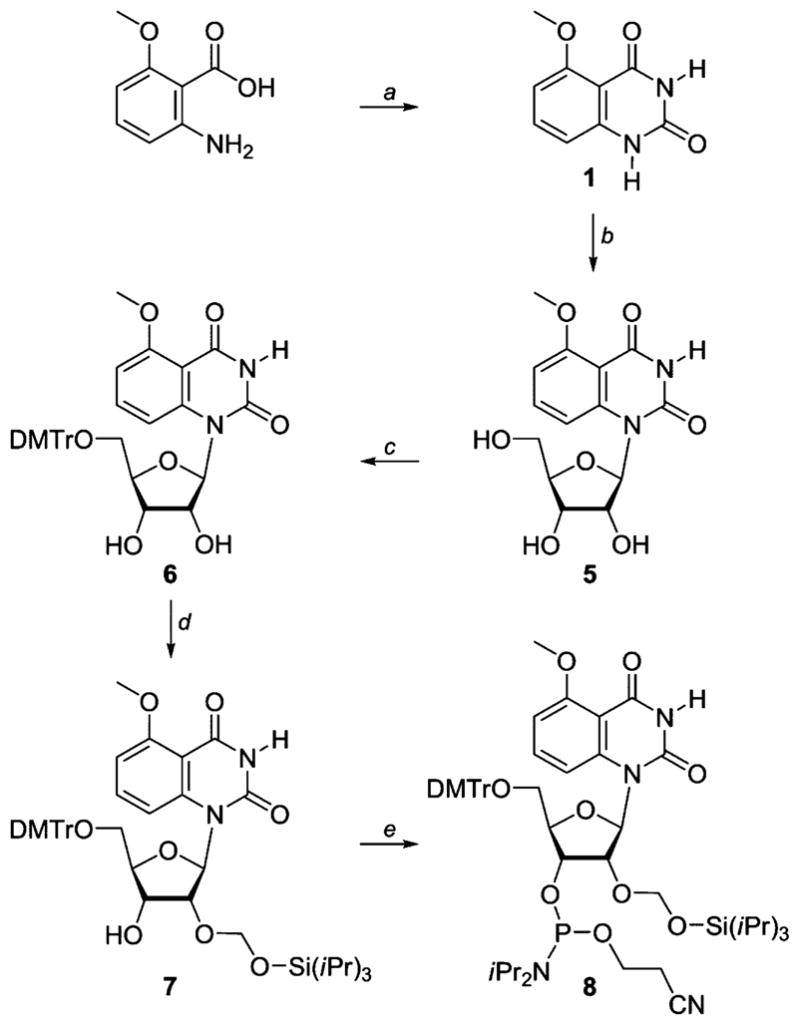
Synthesis of the Modified Nucleoside 5 and Its Phosphoramidite 8a
a Reagents: (a) NaOCN, NaOH, conc. HCl, water, 90%. (b) (i) N,O-bis(trimethylsilyl)acetamide, CF3SO3Si(CH3)3, β-D-ribofuranose 1-acetate 2,3,5-tribenzoate, CH3CN; (ii) conc. NH4OH, 81%. (c) DMTrCl, Et3N, pyridine, 85%. (d) iPr2NEt, nBu2SnCl2, (iPr3SiO)CH2Cl, ClCH2CH2Cl, 30%. (e) iPr2NEt, (iPr2N)P(Cl)O–CH2CH2CN, ClCH2CH2Cl, 60%.
Standard solid-phase oligonucleotide synthesis was utilized to prepare the 27-mer bacterial A-site model construct 10,43 where the fluorescent U analogue 5 replaces U1406 (Figure 5).44 The oligonucleotide was purified by PAGE, and MALDI-TOF mass spectrometry confirmed its full length and the presence of the intact emissive nucleoside 5.40 Thermal denaturation studies showed that the incorporated modified nucleobase had minimal impact on the stability of the folded RNA. The emissive RNA construct 10 displayed a Tm of 71 °C, while the unmodified control A-site construct 9 had a Tm of 72 °C (cacodylate buffer pH 7.0, NaCl 1.0 × 10−1 M).40 The emission profile of the emissive A-site construct 10, excited at 320 nm, resembled that of the parent nucleoside in water, with a maximum emission at 395 nm, albeit with a lower emission quantum yield (φF = 0.03).45
Figure 5.
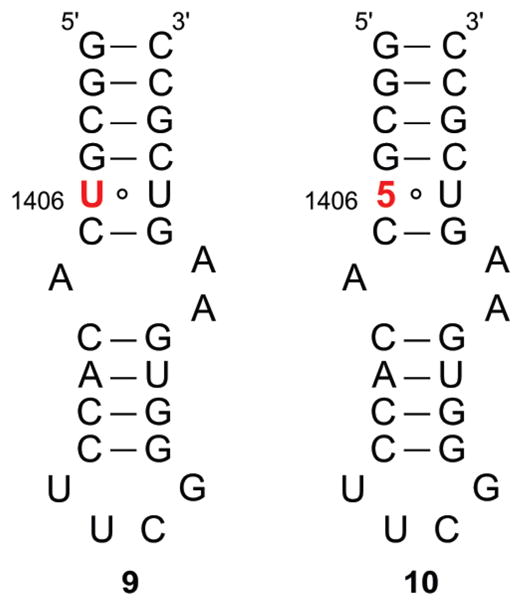
Unmodified 9 and modified 10 A-site constructs.
Synthesis and Photophysical Properties of Modified Aminoglycosides
To complete the FRET pair, neomycin B and tobramycin, two distinct 2-deoxystreptamine-based aminoglycosides, were conjugated to 7-diethylaminocoumarin-3-carboxylic acid to afford 11 and 12, respectively, by modifying their primary hydroxymethyl groups, previously shown not to be critical for RNA binding (Scheme 2).7,17,40,46 Using previously reported procedures, the Boc-protected aminoglycosides were activated at the primary hydroxymethyl group on the ribose by 2,4,6-triisopropylbenzenesulfonyl chloride (TIBS-Cl).47 Treatment with ammonia in MeOH provided the aminomethyl substituted product (e.g., 13, scheme 2). The newly installed amine was coupled to the coumarin carboxylic acid, using standard peptide coupling conditions. The resulting Boc-protected, coumarin-labeled aminoglycosides were treated with trifluoroacetic acid to remove the Boc groups, yielding 11 and 12 as their TFA salts. The emission profile of 11 and 12 resembled that of the parent heterocycle 2, displaying an emission maximum at 473 nm upon excitation at 400 nm, maintaining a quantum yield of 80%.
Scheme 2.

Structure of the Aminoglycosides Used for Binding and Displacement Studies (11 and 12) and the Synthesis of Coumarin-Labeled Neomycin 11a
a Reagents: (a) 7-(Et2N)coumarin-3-carboxylic acid, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide, 4-dimethylaminopyridine, iPr2EtN, Cl2CH2, 84%. (b) Trifluoroacetic acid, triisopropylsilane, CH2Cl2 82%.
Binding of Coumarin-Labeled Aminoglycosides to the Fluorescently Labeled A-Site
Titration of the coumarin-labeled neomycin B derivative 11 into the emissive A-site construct, excited at 320 nm, showed a continuous decrease of the donor emission at 395 nm, with a concomitant increase of the acceptor emission at 473 nm (Figure 6a).40 Similarly, titrating the coumarin-labeled tobramycin derivative 12 into the emissive A-site construct, excited at 320 nm, showed a decrease of the donor emission at 395 nm and a concurrent increase of the acceptor emission at 473 nm. Figure 7 graphically illustrates the titration curves generated by plotting the fractional fluorescence saturation of the donor and acceptor. Identical EC50 values of 0.84 (±0.03) × 10−6 M are obtained from curve fitting the quenching of the donor’s emission or the augmentation of the acceptor’s sensitized emission (Figure 7a). Analogous behavior is observed for the labeled tobramycin derivative 12 (Figure 7b), yielding a higher EC50 value of 2.3 (±0.2) × 10−6 M.
Figure 6.
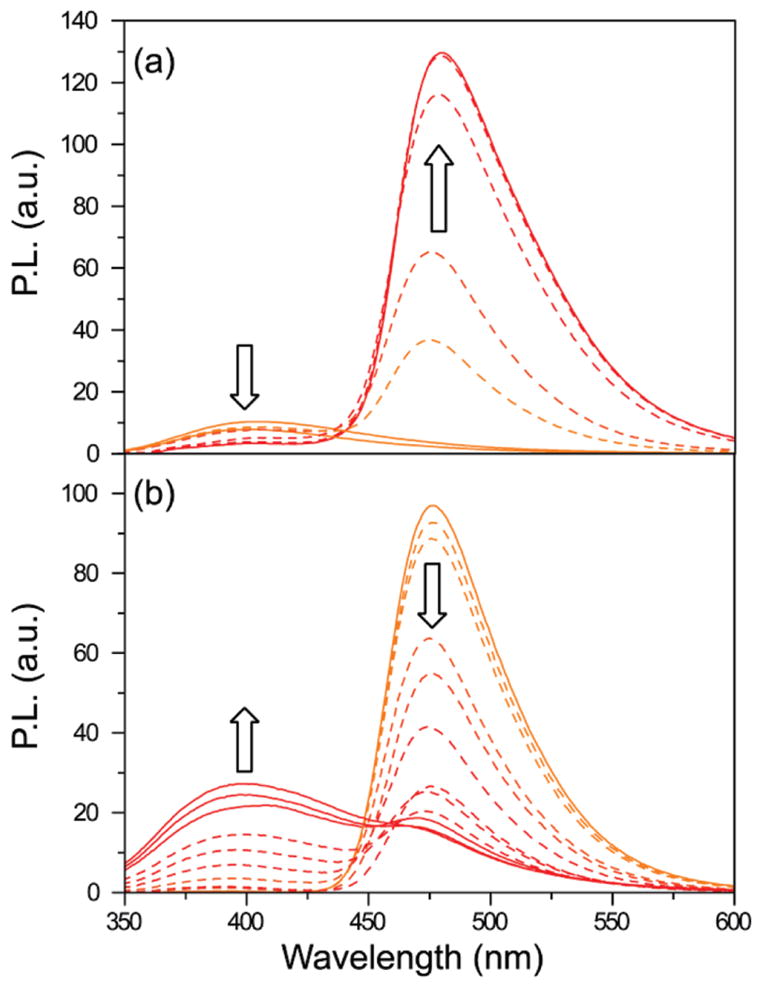
(a) Example of binding study. 11 is titrated into 10. (b) Example of displacement study. Tobramycin is titrated into 10 saturated with 12. Conditions: 10 (1 × 10−6 M), cacodylate buffer pH 7.0 (2.0 × 10−2 M), NaCl (1.0 × 10−1 M).
Figure 7.

Fractional fluorescence saturation of the donor (■) in the labeled A-site and the emissive acceptor (○) in the labeled aminoglycosides in the following experiments: (a) titration of 10 with 11; (b) titration of 10 with 12; (c) displacement of the A-site bound 11 with neomycin; (d) displacement of the A-site bound 12 with neomycin; (e) displacement of the A-site bound 11 with tobramycin; (f) displacement of the A-site bound 12 with tobramycin. Conditions: 10 (1.0 × 10−6 M), cacodylate buffer pH 7.0 (2.0 × 10−2 M), NaCl (1.0 × 10−1 M).
Displacement of A-site Bound Coumarin-Labeled Aminoglycosides with Unlabeled RNA Binders
Preformation of the FRET complex by saturating the modified A-site construct with the labeled neomycin or tobramycin derivatives facilitates the evaluation of the relative A-site affinity of other antibiotics by competition experiments, where the resonance energy transfer is disrupted by displacing the bound coumarin-labeled antibiotic.40 Titration of the unlabeled aminoglycosides into the preformed FRET complex, excited at 320 nm, showed continuous increase of the donor emission at 395 nm, with a concurrent decrease of the acceptor emission at 473 nm (Figure 6b). Titration curves were generated by plotting the fractional fluorescence saturation of the donor and acceptor against the concentration of the unlabeled aminoglycoside. Figure 7 (panels c–f) provides selected examples (see also Figures S4,S5). Competing off a bound neomycin, one of the strongest A-site binders, with other aminoglycosides (paromomycin, apramycin, hygromycin, kanamycin A, Figure 8), requires relatively high competitor concentrations (Table 1). In contrast, the same competitor antibiotics more easily displace the lower affinity tobramycin. Additionally, two synthetic amino–aminoglycosides (6″-amino-6″-deoxy-kanamycin A and 6″-amino-6″-deoxy-tobramycin prepared in our lab47a) are found to be rather potent A-site binders (Table 1). Importantly, spermidine, serving as a negative control polyamine, is unable to displace any of the bound aminoglycosides (up to 5 × 10−3 M).
Figure 8.

Aminoglycosides used for displacement studies.
Table 1.
IC50 Values of Aminoglycosides Competing off 11 and 12a
| Aminoglycosides | Displacement of 11b | Displacement of 12b |
|---|---|---|
| Neomycin | 0.03 ± 0.01 | 0.02 ± 0.01 |
| Tobramycin | 0.50 ± 0.02 | 0.16 ± 0.03 |
| Paromomycin | 1.14 ± 0.08 | 0.06 ± 0.03 |
| Apramycin | 3.00 ± 0.09 | 1.00 ± 0.05 |
| Hygromycin | 1.46 ± 0.06 | 1.00 ± 0.05 |
| Amikacin | 1.73 ± 0.07 | 0.56 ± 0.04 |
| Kanamycin A | 3.30 ± 0.09 | 1.61 ± 0.06 |
| Amino-Kanamycin A | 0.18 ± 0.03 | 0.05 ± 0.02 |
| Amino Tobramycin | 0.02 ± 0.01 | 0.01 ± 0.01 |
Conditions: 10 (1 × 10−6 M), cacodylate buffer pH 7.0 (2.0 × 10−2 M), NaCl (1.0 × 10−1 M).
Aminoglycoside concentration is given in 10−3 M.
Discussion
Among the RNA targets explored over the past two decades, the bacterial ribosomal decoding site (or A-site) holds a unique place. It is the only validated drug target.10,11,23,25,48–50 This conformational switch is the cognate binding site of aminoglycoside antibiotics, a diverse family of natural products evolved to interfere with the decoding process in bacteria.2 These highly effective bactericidal agents alter the conformational flexibility of two key residues, A1492 and A1493, inducing a conformation similar to the one found in the cognate acyl-tRNA–mRNA complex (Figure 1).51,52 This impairs the fidelity of ribosomal protein synthesis, ultimately leading to bacterial death.
The utility of aminoglycosides has declined over the years due to their diminished potency in resistant bacteria, their adverse side effects, and, consequently, the availability of newer, potent, and safer drugs.23,25,27,53 The rapid emergence of resistant pathogens and the time-consuming and minimally productive development of new broad-spectrum antibiotics have created, however, alarming circumstances, where new antibiotics are needed to replace existing compromised drugs.54,55 These developments have generated a renewed interest in the A-site and triggered the development of new tools to assess ligand binding to this bacterial RNA site. In particular, fluorescent A-site constructs, which contain responsive nucleoside analogues such as 2-aminopurine, have proven useful,56–59 although their response is antibiotic-dependent, which is most likely due to the presence of distinct binding modes, which impact the emission readout. This could affect the ability of fluorescent nucleosides to accurately respond to ligand binding. To create a robust analysis and discovery platform for A-site binders, we have developed a FRET-enabled assembly, where the A-site serves as the donor and the aminoglycoside as the acceptor. The detection of antibiotic binding and competitive displacement is highly sensitive and is no longer dependent on arbitrary changes in the environment of a single fluorophore, but rather on the interaction between two matching chromophores acting as a FRET pair (Figure 1b).
In designing such chromophoric RNA–small molecule assemblies, two major issues need to be addressed. The first involves the position and type of modification of the RNA target with a donor, and the second, which is of related significance and is partially coupled, is the selection and placement of an appropriate chromophoric partner. The identification of the donor is the most challenging, as it must fulfill strict functional criteria: it needs to be small and structurally nonperturbing, while at the same time displaying useful photophysical characteristics that can be matched to a suitable and small acceptor. Due to its imposed small molecular footprint and high similarity to the native nucleobases, such a nucleobase analogue is likely to emit below or close to the visible range with a moderate emission quantum yield at best. This, in turn, impacts the selection of the acceptor to be placed on the aminoglycoside. In addition to being structurally and functionally nonperturbing (i.e., minimally impacting the recognition properties of the antibiotic), the acceptor needs to have a high degree of spectral overlap with the donor, while displaying intense emission in the visible range. As a FRET pair, the selected donor and acceptor need to have a critical Förster radius that matches the recognition phenomenon and the anticipated distances between the RNA and the bound antibiotic.
To confer useful emissive properties upon nucleic acids, while minimally perturbing their folded structure, we have been pursuing the development of isosteric/isomorphic nucleobase analogues.35–39 These heterocyclic surrogates can replace a native nucleobase without significantly altering the folding and recognition features of the native target, but with the added benefit of being fluorescent.60 Specifically, we have previously demonstrated that replacing the U residue at position 1406 with emissive nucleoside analogues retains the folding and antibiotic recognition properties of the A-site.36,61 Its proximity to the aminoglycoside binding site ensures adequate photophysical interaction with suitably labeled aminoglycoside, as shown in the modeled structure (Figure 9). Out of the possible motifs, we focused on a size-expanded U analogue with a methoxy group in the 5 position, which is a superior isostere to similar benzo[g]quinazoline nucleosides that have been previously utilized.62–64
Figure 9.
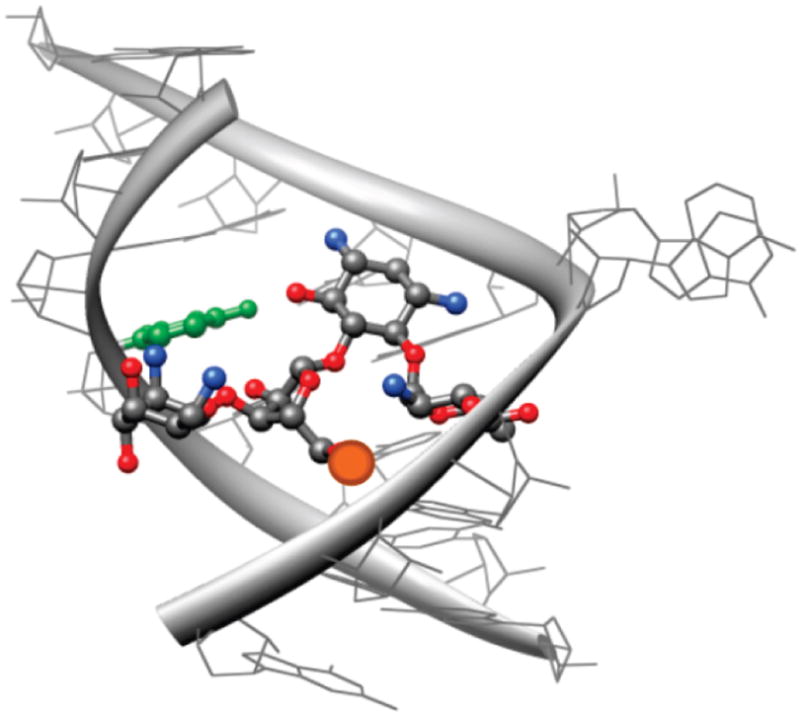
A model of neomycin bound to the A-site (PDB 2ET4). The distance from the center of U1406 (green) to the primary 5′ hydroxymethyl group on the ribose (orange) is less than 10 Å.
The emission profile of the fluorescent nucleobase 1 complements the absorbance of 7-diethylaminocoumarin (Figure 3), a commonly used fluorophore that can be conjugated to aminoglycosides at positions that do not significantly impact their binding to the A-site (Figure 9).65 Coumarin-based fluorophores are among the smallest organic chromophores displaying emission in the visible range with high quantum yields (φf > 0.5).66,67 Specifically, 7-diethylaminocoumarin lacks any significant molar absorptivity at 320 nm, which facilitates selective excitation of donor 1. It is worth noting that placing the electron-donating methoxy group on the quinazoline-2,4-(1H,3H)-dione skeleton is essential for maximizing the spectral overlap between the donor and acceptor. The absence of the methoxy group, as in the unsubstituted parent heterocycle 3, or its placement in an electronically related position, such as in the isomeric 4, causes a significant blue shift in the donors’ emission, diminishes the spectral overlap, and lowers FRET efficiency between the quinazoline and coumarin chromophores (Figure 4). We postulate that excited state stabilization (likely due to charge transfer from the methoxy to the conjugated carbonyl generating opposing dipoles in 1 only) leads to lower energy emission and optimal spectral overlap between 1 and 2.
In labeling the A-site, it was important to place 5 close to the recognition site without distorting the binding properties of the model RNA construct. Functional, isosteric nucleosides, such as furan-modified ribonucleosides, have been incorporated in place of native U1406 in the bacterial A-site for affinity assays to recognize RNA–small molecule interactions.36,61 These modifications have been found to be minimally perturbing and effective at monitoring binding events. Thus, we chose to replace U1406 with the fluorescent U analogue 5 for our labeled A-site model. According to thermal denaturation studies, the substitution of U1406 by 5 appeared to be insignificantly destabilizing, as the modified construct 10 and unmodified control construct 9 had essentially the same melting temperatures within error. The photophysical properties of 5 remained adequate in the A-site construct. The emission maximum of 10 was the same as that of fluorescent ribonucleoside 5 at 395 nm; however, the emission quantum yield dropped from 0.16 to 0.03. Such a diminished quantum yield upon incorporation into a RNA or DNA duplexes is not uncommon among fluorescent nucleosides.45 Nevertheless, with an almost perfect spectral overlap between 1 and 2, the “modified” Förster radius remains suitable for this assembly (R0 = 20 Å). As for the acceptors in our FRET pair, conjugating 7-diethylaminocoumarin 2 to the aminoglycosides had little effect on its photophysics; 11 and 12 resembled 2 in absorption and emission profiles and maintained a high emission quantum yield.
Titrating the labeled aminoglycosides into the donor-containing A-site construct yielded the expected spectral behavior, with continuous quenching of the donor’s emission and concomitant enhancement of the acceptor’s sensitized emission. As stated above, the ideal spectral overlap between 1 and 2 provides efficient energy transfer even with the reduced quantum yield of the donor’s emission within the RNA construct. Based on the efficiency of energy transfer at saturation, the calculated FRET distance between the bound coumarin-labeled neomycin 11 and tobramycin 12 to the modified RNA construct 10 are 14 ± 1 Å and 12 ± 2 Å, respectively. This is in good agreement with the distances predicted by examining the solid-state structures of these RNA–aminoglycoside complexes.40,42 Consequently, binding and competition experiments faithfully reproduce the expected trends in affinity of the diverse ligands tested and suggest that this FRET assembly is indeed a useful tool for the analysis of A-site binders. Furthermore, the discovery of two high RNA affinity amino–aminoglycoside derivatives, previously untested for A-site binding, demonstrates the potential utility of this tool for the discovery of new A-site binders.
The results of FRET-monitored binding and displacement experiments reveal distinct advantages over traditional methods that rely on singly labeled RNA constructs. While the latter can monitor the binding of small molecules to RNA targets, multiple ligand binding modes might limit their accuracy and dependability. In fact, the popular 2-AP-modified A-site construct,56–59 while responding well to most aminoglycoside antibiotics, does not reliably detect the binding of neomycin, the strongest naturally occurring A-site binder.36 In addition, singly labeled RNA constructs are unable to consistently perform in evaluating competitive binding, as displacing a bound ligand with an equipotent one is unlikely to be “visible” to a probe only reporting the “bound” or “unbound” status of the RNA construct. Förster Resonance Energy Transfer between strategically placed donors and acceptors, on the other hand, can clearly overcome such limitations, as unequivocally demonstrated by our results. As FRET is distance dependent, when the ligand is bound, the donor and acceptor would be brought together, leading to the increased emission of the acceptor and the quenching of the donor. If another small molecule was added to the mix and competed off the original ligand, the donor and acceptor would be parted, decreasing the emission of the acceptor and simultaneously increasing the fluorescence of the donor. A FRET based system, therefore, unambiguously provides both association and competitive dissociation information that is independent of specific binding modes. Most importantly, while singly labeled RNA constructs might generate false positive signals due to remote binding at a nonfunctional state (which could alter the environment of a responsive probe), a FRET based technique, as described here, requires specific binding to the A-site pocket. A nonspecific RNA binder, for example, would not be able to displace an antibiotic from its cognate recognition site, nor would it generate an intense FRET signal in monitoring direct binding experiments.
Summary and Implications
We have constructed a useful and effective FRET pair system for the evaluation of antibiotics binding to the bacterial ribosomal decoding site. It relies on the incorporation of a new emissive uridine analogue into an A-site construct, which serves as a donor for a highly emissive coumarin acceptor that is attached to aminoglycosides. The strong dependence of FRET on the donor–acceptor distance is unambiguously used to detect the onset and offset of binding more effectively than a single chromophore system (which is also incapable of monitoring displacement and competition events). As a proof of concept, we have demonstrated binding of neomycin B and tobramycin, two A-site binding antibiotics displaying distinct affinity to their native rRNA target, and their competitive displacement by a variety of aminoglycoside antibiotics. It is worth noting here that antibacterial activity does not directly correlate with affinity to the A-site, which functions as a conformational switch.18 This further justifies the use of multiple probes that facilitate screening of potential new A-site binders with diverse affinity. We note that our design is not limited to the A-site and its binders; related FRET assemblies can be developed for other RNA-ligand recognition events. Additionally, further development of ideal FRET pairs, composed of isomorphic nucleosides and corresponding minimally perturbing FRET acceptors, will facilitate the analysis of other RNA targets with therapeutic potential.
Experimental Section
Materials
Unless otherwise specified, materials obtained from commercial suppliers were used without further purification. 2-Methoxy-5-aminobenzoic acid and spermidine were purchased from VWR. Neomycin, tobramycin, paromomycin, amikacin, apramycin, and hygromycin were purchased as their sulfate salts from Sigma-Aldrich and were converted into the corresponding neutral form by passing through DOWEX MONOSPHERE 550 Å (OH) anion exchange resin. The identities of the aminoglycosides were confirmed by mass measurements, 1H NMR, and 13C NMR. 7-(Diethylamino)coumarin-3-carboxylic acid was purchased from Sigma-Aldrich. Anhydrous pyridine, dichloroethane, and acetonitrile were obtained from Fluka. Anhydrous N,N-diisopropylethylamine and triethylamine were obtained from Acros. NMR solvents were purchased from Cambridge Isotope Laboratories (Andover, MA). The unmodified oligonucleotide was purchased from Thermo Scientific. Standard phosphoramidites and solutions necessary for solid phase RNA synthesis were purchased from Glen Research. Oligonucleotides were purified by gel electrophoresis and desalted on a Sep-Pak (Waters Corporation). Chemicals for preparing buffer solutions were purchased from Fisher Biotech (enzyme grade). Autoclaved water was used in all fluorescence titrations.
Instrumentation
NMR spectra were recorded on a Varian Mercury 400 MHz spectrometer. Mass spectra were recorded at the UCSD Chemistry and Biochemistry Mass Spectrometry Facility, utilizing either an LCQDECA (Finnigan) ESI with a quadrapole ion trap or an MAT900XL (ThermoFinnigan) FAB double focusing mass spectrometer. UV–vis spectra were recorded on an Agilent 8453 Diode Array Spectrometer. MALDI-TOF spectra were collected on a PE Biosystems Voyager-DE STR MALDI-TOF spectrometer in positive-ion, delayed-extraction mode. Reversed-phase HPLC (Vydac C18 column) purification and analysis were carried out using a Hewlett-Packard 1050 Series instrument. Steady-state fluorescence experiments were carried out in a microfluorescence cell with a path length of 1.0 cm (Hellma GmH & Co KG, Mullenheim, Germany) on a Jobin Yvon Horiba FluoroMax-3 luminescence spectrometer. A background spectrum (buffer) was subtracted from each sample. Modified oligonucleotides were synthesized on a Biosearch Cyclone Plus DNA synthesizer using a 0.2 μmol scale 500 Å CPG column. All hybridization and UV melting experiments were done with a Beckman-Coulter DU 640 spectrometer with a high performance temperature controller and micro auto six-cell holder.
Synthesis. 5-Methoxyquinazoline-2,4-(1H,3H)-dione (1)
Water (36 mL) and glacial acetic acid (0.7 mL) were added to 2-methoxy-5-aminobenzoic acid (1.00 g, 5.98 mmol). The slurry was stirred at 35 °C for 15 min. Sodium cyanate (0.97 g, 14.92 mmol) was dissolved in water (4 mL) and added slowly to the slurry. The reaction was stirred at 35 °C for 30 min. Sodium hydroxide (10.68 g, 267 mmol) was added slowly to the reaction. The reaction was cooled to room temperature. The pH was adjusted to 4 with concentrated hydrochloric acid. The white precipitate was collected and washed with water (200 mL). Product: white solid (1.03 g, 5.38 mmol, 90% yield). 1H NMR (400 MHz, DMSO-d6): δ 10.97 (s, NH, 1H), 10.87 (s, NH, 1H), 7.47 (t, J = 2.0 Hz, 1H), 6.68 (d, J = 14 Hz, 1H), 6.67 (d, J = 7.2 Hz, 1H), 3.79 (s, OCH3, 3H); 13C NMR (100 MHz, DMSO-d6): δ 161.54. 161.14, 150.81, 143.79, 136.05, 107.96, 105.57, 104.81, 56.53; ESI-MS calculated for C9H9N2O3 [M + H]+ 193.1, found 193.1.
5-Methoxyquinazoline-2,4-(1H,3H)-dione Ribonucleoside (5)
To a suspension of 1 (0.10 g, 0.52 mmol) in anhydrous acetonitrile (5 mL) N,O-bis(trimethylsilyl)acetamide (0.64 mL, 2.6 mmol) was added dropwise under argon. The reaction was stirred at 25 °C for 2 h. The reaction temperature was raised to 50 °C. TMSOTf (0.14 mL, 0.77 mmol) and β-D-ribofuranose-1-acetate-2,3,5-tribenzoate (0.26 g, 0.52 mmol) were added at the same time under argon. The reaction was stirred at 50 °C for 24 h. The reaction was cooled to room temperature and diluted with dichloromethane (10 mL). The solution was washed with saturated sodium bicarbonate and brine. The organic layer was dried over sodium sulfate. The solvent was removed under reduced pressure, and the crude product was dissolved in dioxane (5 mL) and transferred to a 200 mL pressure tube. Ammonium hydroxide (28%, 80 mL) was added. The reaction was stirred at 80 °C for 24 h. The solvent was removed was removed under reduced pressure, and the product was isolated by flash chromatography (88/12 dicholromethane/methanol). Product: white solid (0.14 g, 0.42 mmol, 81% yield over two steps). 1H NMR (400 MHz, DMSO-d6): δ 11.25 (s, NH, 1H), 7.55 (t, J = 8.4 Hz, 1H), 7.26 (d, J = 8.4 Hz, 1H), 6.88 (d, J = 8.8 Hz, 1H), 6.02 (d, J = 5.2 Hz, 1′-H, 1H), 5.22 (d, 2′–OH, 1H), 4.99 (b, 3′-OH, 1H), 4.94 (b, 5′-OH, 1H), 4.47 (t, J = 5.6 Hz, H-2′, 1H), 4.08 (t, J = 5.6 Hz, H-3′, 1H), 3.82 (s, OCH3, 3H), 3.66 (m, H-4′, 1H), 3.46–3.57 (m, H-5′, 2H); 13C NMR (100 MHz, DMSO-d6): δ 161.33, 160.08, 150.81, 142.99, 135.74, 109.25, 107.27, 106.47, 91.48, 85.02, 73.97, 69.47, 61.78, 56.74; ESI-MS calculated for C14H17N2O7 [M + H]+ 325.1, found 325.1.
5′-Dimethoxytrityl-5-methoxyquinazoline-2,4-(1H,3H)-dione Ribonucleoside (6)
Anhydrous pyridine (3 mL), anhydrous triethylamine (51 μL, 0.37 mmol), and 4,4′-dimethoxytrityl chloride (0.12 g, 37 mmol) were added to 3 (0.10 g, 0.31 mmol) over argon. The reaction was stirred at room temperature for 8 h and quenched with methanol (0.5 mL). The solvent was removed under reduced pressure, and the product was isolated by flash chromatography (1% triethylamine, 2% methanol, 97% dichloromethane). Product: white solid (0.16 g, 0.26 mmol, 85% yield). 1H NMR (400 MHz, CDCl3): δ 7.46–7.16 (m, 12H), 6.97 (t, J = 8.8 Hz, 1H), 6.75 (d, J = 8.8 Hz, 2H), 6.60 (d, J = 8.4 Hz, 1H), 6.32 (d, J = 5.6 Hz, 1H), 4.81 (t, J = 6.4 Hz, 1H), 4.57 (t, J = 6.4 Hz, 1H), 4.05 (br, 1H), 3.88 (s, 3H), 3.73 (s, 6H), 3.47–3.55 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 161.48, 160.50, 158.66, 150.60, 144.99, 142.68, 136.04, 135.97, 135.81, 130.48, 128.56, 128.02, 127.01, 113.30, 109.53, 106.56, 90.79, 86.48, 83.56, 69.96, 69.39, 63.42, 63.29, 56.58, 55.45, 53.13; ESI-MS calculated for C35H34N2NaO9 [M + Na]+ 649.2, found 649.2.
2′-(Trisisopropylsiloxy)methyl-5′-dimethoxytrityl-5-methoxyquinazoline-2,4-(1H,3H)-dione Ribonucleoside (7)
Anydrous dichloroethane (3 mL) and N,N-diisopropylethylamine (0.17 mL, 1.0 mmol) were added to 4 (0.20 g, 0.32 mmol). Dibutyltin dichloride (0.10 g, 0.33 mmol) was added to the reaction under argon and stirred at room temperature for 1 h. The reaction was placed into a 80 °C water bath and stirred for 10 min. (Triisopropylsiloxy)methyl chloride (87 μL, 38 mmol) was added, and the reaction was stirred at 80 °C for 15 min. The reaction was diluted with dichloromethane (10 mL) and poured into saturated sodium bicarbonate (15 mL). The mixture was stirred vigorously for 15 min. The organic layer was extracted, and the aqueous layer was washed with dichloromethane (5 mL). The organic layers were pooled and dried over sodium sulfate. The solvent was removed under reduced pressure, and the product was isolated by flash chromatography (1% triethylamine, 35% ethyl acetate, 64% hexanes). Product: white foam (0.078 g, 0.01 mmol, 30% yield). 1H NMR (400 MHz, CDCl3): δ 7.43–7.47 (m, 3H), 7.26–7.34 (m, 4H), 7.18–7.26 (m, 4H), 7.06 (t, J = 8.4 Hz, 1H), 6.78 (d, J = 8.8 Hz, 3H), 6.67 (d, J = 8.4 Hz, 1H), 6.35 (d, J = 5.2 Hz, 1H), 5.07 (d, J = 4.8 Hz, 1H), 4.90–4.95 (m, 2H), 4.61 (t, J = 6.4 Hz, 1H), 4.07 (dd, J1 = 4.0 Hz, J2 = 3.2 Hz, 1H), 3.95 (s, 3H), 3.77 (s, 6H), 3.51 (dd, J1 = 8.0 Hz, J2 = 2.4 Hz, 1H), 3.39 (dd, J1 = 6.4 Hz, J2 = 4.0 Hz, 1H), 3.28 (q, J = 7.2 Hz, 1H), 2.70 (q, J = 6.8 Hz, 1H), 1.07–1.00 (m, 21H); 13C NMR (100 MHz, CDCl3): δ 161.71, 160.02, 158.72, 149.92, 144.96, 143.91, 136.00, 135.94, 130.45, 128.53, 128.05, 127.08, 113.33, 109.71, 106.75, 106.13, 91.08, 88.99, 86.63, 83.70, 69.70, 63.44, 59.85, 56.71, 55.45, 29.95, 17.95, 12.02; ESI-MS calculated for C45H56N2NaO10Si [M + Na]+ 835.4, found 835.4.
3′-2-Cyanoethyldiisopropylphosphoramidite-2′-(trisisopropylsiloxy)methyl-5′-dimethoxytrityl-5-methoxyquinazoline-2,4-(1H,3H)-dione Ribonucleoside (8)
Anhydrous dichloromethane (0.6 mL) and N,N-diisopropylethylamine (0.13 mL, 0.75 mmol) were added to 5 (0.05 g, 0.062 mmol). The reaction was cooled on ice, and 2-cyanoethyl N,N-diisopropylchlorophosphoramidite (28 μL, 0.13 mmol) was added. The reaction was stirred at room temperature for 18 h. The solvent was removed under reduced pressure, and the product was isolated by flash chromatography (1% triethylamine, 15–30% ethyl acetate in hexanes). Product: white foam (0.038 g, 0.037 mmol, 60% yield). 1H NMR (300 MHz, CDCl3): δ 7.44–7.46 (m, 3H), 7.32–7.34 (m, 4H), 7.22–7.26 (m, 4H), 7.07 (t, J = 8.4 Hz, 1H), 6.78 (d, J = 8.1 Hz, 3H), 6.68 (d, J = 8.4 Hz, 1H), 6.35 (d, J = 5.4 Hz, 1H), 5.07 (d, J = 4.2 Hz, 1H), 4.91–4.94 (m, 2H), 4.62 (t, J = 6.4 Hz, 1H), 4.07 (b, 1H), 3.96 (s, 3H), 3.78 (s, 6H), 3.51 (m, 1H), 3.40 (m, 1H), 2.81 (q, J = 6.9 Hz, 1H), 2.01 (b, 1H), 1.20–1.15 (m, 8H), 1.01 (d, J = 6.8 Hz, 4H), 0.92–0.89 (m, 21H); 13C NMR (100 MHz, CDCl3): δ 171.37, 161.62, 160.19, 158.67, 158.65, 144.88, 136.10, 136.03, 135.88, 135.50, 130.47, 130.44, 128.67, 128.56, 127.99, 127.94, 127.05, 126.99, 117.94, 117.53, 113.23, 106.61, 106.23, 86.50, 86.46, 64.56, 60.60, 59.02, 56.66, 55.40, 55.35, 43.58, 43.45, 43.31, 43.19, 30.84, 29.90, 24.82, 24.75, 24.69, 21.24, 21.21, 19.23, 17.87, 17.81, 14.40, 13.92, 12.06, 12.01, 11.98; ESI-MS calculated for C54H73N4NaO11PSi[M + Na]+ 1035.5 and [M + K]+ 1051.4, found 1035.4 and 1051.4.
Boc6-Protected Coumarin-Labeled Neomycin (14)
Anhydrous dichloromethane (300 μL) and 7-diethylaminocoumarin-3-carboxylic acid (6.8 mg, 0.0263 mmol) were added to 13 (26.58 mg, 0.0219 mmol). To this, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (5.03 mg, 0.0262 mmol), N,N-diisopropylethylamine (8.62 μL, 0.048 mmol), and 4-(dimethylamino)pyridine (5.8 mg, 0.026 mmol) were added. The reaction was stirred for 18 h. The solvent was removed under reduced pressure, and the resulting solid was dissolved in ethyl acetate and washed with water and brine. The organic layer was dried over sodium sulfate, and the solvent was removed under reduced pressure. The product was isolated by flash chromatography (3% methanol in dichloromethane). Product: yellow powder (26.8 mg, 0.0184 mmol, 84% yield). 1H NMR (400 MHz, CD3OD): δ 8.66 (s, 1H), 7.45 (d, J = 10.5 Hz, 1H), 6.81 (d, J = 9 Hz, 1H), 6.56 (s, 1H), 5.34 (s, 1H), 5.12 (s, 1H), 5.02 (s, 1H), 4.28 (s, 3H), 4.09–4.06 (m, 1H), 4.01–3.98 (m, 1H), 3.90–3.87 (m, 4H), 3.82–3.79 (m, 2H), 3.76–3.71 (m, 4H), 3.61–3.44 (m, 24H), 3.34–3.15 (m, 2H), 2.63–2.57 (m, 1H), 1.99–1.89 (2H), 1.46–1.38 (m, 54H), 1.37 (t, J = 5 Hz, 6H); 13C NMR (125 MHz, CD3CN): δ 164.66, 163.53, 158.72, 158.21, 157.86, 157.62, 157.29, 156.55, 154.02, 149.22, 132.40, 111.34, 110.52, 109.87, 109.06, 101.22, 99.69, 97.06, 80.24, 79.90, 79, 81, 79.64, 79.56, 74.99, 73.82, 70.88, 68.43, 56.72, 53.13, 51.65, 45.70, 45.28, 42.54, 41.44, 28.69, 12.67, 12.21; ESI-MS calculated for C67H108N8O27 [M + Na]+ 1479.72, found 1479.71.
Coumarin-Labeled Neomycin (11)
Anhydrous dichloromethane (2 mL) and triisopropylsilane (200 μL) were added to 14 (26.8 mg, 0.0184 mmol). To this trifluoroacetic acid was added (2 mL), and the reaction was stirred at RT for 15 min. The reaction was diluted with toluene (5 mL), and the solvent was removed under reduced pressure. The resulting solid was dissolved in water and washed with dichloromethane. The aqueous layer was dried and concentrated under reduced pressure and further purified by reversed-phase HPLC using a gradient of 10–30% acetonitrile (0.1% TFA) in water (0.1% TFA) over 30 min, eluting at 14.78 min. Product: yellow powder (21.7 mg, 0.0151 mmol, 82% yield). 1H NMR (400 MHz, D2O): δ 8.65 (s, 1H), 7.62 (d, J = 9.2 Hz, 1H), 6.92 (d, J = 9.2 Hz, 1H), 6.67 (s, 1H), 5.96 (s, 1H), 5.36, (s, 1H), 5.29 (s, 1H), 4.49 (t, J = 5.5 Hz, 1H), 4.39 - 4.35 (m, 2H), 4.28 (t, J = 5 Hz, 1H), 4.21 (t, J = 3.5 Hz, 1H), 3.99–3.95 (m, 2H), 3.89–3.83 (m, 2H), 3.79 (s, 2H), 3.64 (t, J = 9.5 Hz, 1H), 3.59 (s, 1H), 3.56–3.51 (q, J1 = 6.5 Hz, J2 = 7.0 Hz, 4H), 3.33 (d, J = 4 Hz, 4H), 3.18–3.12 (m, 1H), 2.42–2.33 (m, 1H), 1.83–1.71 (m, 1H), 1.21 (t, J = 7 Hz, 6H); 13C NMR (125 MHz, D2O): δ 166.19, 164.44, 163.17, 162.99 (J1 = 27.8 Hz, J2 = 58.4 Hz), 157.78, 154.03, 149.05, 131.83, 116.25 (J = 231 Hz, J2 = 465 Hz), 115.08, 111.53, 110.43, 108.14, 106.39, 95.97, 95.44, 94.46, 79.69, 76.50, 73.24, 69.96, 67.52, 67.27, 50.7416, 48.49, 44.99, 40.32, 39.83, 11.44; ESI-MS calculated for C37H60N8O15 [M + 2H]2+ 429.21, [M + H]+ 857.43, and [M + Na]+ 879.41, found 429.35, 857.43, and 879.59.
Aminoglycoside Titrations
All titrations were performed with working solutions of 1.0 × 10−6 M 10 in 20 × 10−6 M calcodylate buffer (pH 7.0, 1.0 × 10−1 M NaCl, 5.0 × 10−4 M EDTA). The solutions were heated to 75 °C for 5 min, cooled to room temperature over 2 h, and placed on ice for 30 min prior to titrations. For binding studies, 10 was excited at 320 nm, and changes in emission upon titration with 11 or 12 were monitored at 395 and 473 nm. The concentrations of 11 and 12 were determined by UV absorbance at 400 nm (ε 20 000 M−1 cm−1). For competition studies, 11 or 12 was titrated into 10 until saturation. 10 was excited at 320 nm, and changes in emission upon displacement of 11 or 12 by aminoglycosides were monitored at 395 and 473 nm. EC50 and IC50 values were calculated using OriginPro 8 software by fitting a dose response curve (eq 1) to the fractional fluorescence saturation (Fs) plotted against the log of aminoglycoside (AG) concentration.
| (1) |
Fi is the fluorescence intensity at each titration point. F0 and F∞ are the fluorescence intensity in the absence of aminoglycoside or at saturation, respectively, and n is the Hill coefficient or degree of cooperativity associated with the binding.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health for their generous support (GM 069773), Mary Noé for her assistance with MALDI experiments, and the National Science Foundation (Instrumentation Grants CHE-9709183 and CHE-0741968).
Footnotes
Note Added after ASAP Publication. The fluorescence acceptor emission was incorrect in the abstract published ASAP November 12, 2009. The corrected version was published November 13, 2009.
Supporting Information Available: Additional synthetic details, thermal denaturation measurements, titration spectra, photophysical data, and MALDI-TOF MS spectrum. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Green R, Noller HF. Annu Rev Biochem. 1997;66:679–716. doi: 10.1146/annurev.biochem.66.1.679. [DOI] [PubMed] [Google Scholar]
- 2.Puglisi JD, Blanchard SC, Dahlquist KD, Eason RG, Fourmy D, Lynch SR, Recht MI, Yoshizawa S. Aminoglycosides Antibiotics and Decoding. In: Garrett RA, Douthwaite SR, Liljas A, Matheson AT, Moore PB, Noller HF, editors. The Ribosome: Structure, Function, Antibiotics, and Cellular Interactions. ASM Press; Washington, DC: 2000. pp. 419–429. [Google Scholar]
- 3.Rodnina MV, Wintermeyer W. Trends Biochem Sci. 2001;26:124–130. doi: 10.1016/s0968-0004(00)01737-0. [DOI] [PubMed] [Google Scholar]
- 4.Gale EF, Cundliffe E, Renolds PE, Richmond MH, Waring MJ. The Molecular Basis of Antibiotic Action. John Wiley & Sons; London: 1981. [Google Scholar]
- 5.Moazed D, Noller HF. Nature. 1987;327:389–394. doi: 10.1038/327389a0. [DOI] [PubMed] [Google Scholar]
- 6.Brodersen DE, Clemons WM, Carter AP, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Cell. 2000;103:1143–1154. doi: 10.1016/s0092-8674(00)00216-6. [DOI] [PubMed] [Google Scholar]
- 7.Harms JM, Bartels H, Schlünzen F, Yonath A. J Cell Sci. 2003;116:1391–1393. doi: 10.1242/jcs.00365. [DOI] [PubMed] [Google Scholar]
- 8.Wirmer J, Westhof E, Minoru F. Method Enzymol. 2006;415:180–202. doi: 10.1016/S0076-6879(06)15012-0. [DOI] [PubMed] [Google Scholar]
- 9.Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Nature. 2000;407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 10.Schlunzen F, Zarivach R, Harms J, Bashan A, Tocilj A, Albrecht R, Yonath A, Franceschi F. Nature. 2001;413:814–821. doi: 10.1038/35101544. [DOI] [PubMed] [Google Scholar]
- 11.Vicens Q, Westhof E. Chem Bio Chem. 2003;4:1018–1023. doi: 10.1002/cbic.200300684. [DOI] [PubMed] [Google Scholar]
- 12.Francois B, Russell RJM, Murray JB, Aboul N, Masquida B, Vicens Q, Westhof E. Nucleic Acids Res. 2005;33:5677–5690. doi: 10.1093/nar/gki862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Purohit P, Stern S. Nature. 1994;370:659–662. doi: 10.1038/370659a0. [DOI] [PubMed] [Google Scholar]
- 14.Fourmy D, Recht MI, Blanchard SC, Puglisi JD. Science. 1996;274:1367–1371. doi: 10.1126/science.274.5291.1367. [DOI] [PubMed] [Google Scholar]
- 15.Yoshizawa S, Fourmy D, Puglisi JD. EMBO J. 1998;17:6437–6448. doi: 10.1093/emboj/17.22.6437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vicens Q, Westhof E. Structure. 2001;9:647–658. doi: 10.1016/s0969-2126(01)00629-3. [DOI] [PubMed] [Google Scholar]
- 17.Vicens Q, Westhof E. Chem Biol. 2002;9:747–755. doi: 10.1016/s1074-5521(02)00153-9. [DOI] [PubMed] [Google Scholar]
- 18.Kaul M, Barbieri CM, Pilch DS. J Am Chem Soc. 2006;128:1261–1271. doi: 10.1021/ja056159z. [DOI] [PubMed] [Google Scholar]
- 19.Chow CS, Bogdan FM. Chem Rev. 1997;97:1489–1514. doi: 10.1021/cr960415w. [DOI] [PubMed] [Google Scholar]
- 20.Gallego J, Varani G. Acc Chem Res. 2001;34:836–843. doi: 10.1021/ar000118k. [DOI] [PubMed] [Google Scholar]
- 21.Tor Y. Chem Bio Chem. 2003;4:998–1007. doi: 10.1002/cbic.200300680. [DOI] [PubMed] [Google Scholar]
- 22.Hermann T, Tor Y. Expert Opin Ther Pat. 2005;15:49–62. [Google Scholar]
- 23.Sutcliffe JA. Curr Opin Microbiol. 2005;8:534–542. doi: 10.1016/j.mib.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 24.Hermann T. Biochimie. 2006;88:1021–1026. doi: 10.1016/j.biochi.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 25.Tor Y. Biochimie. 2006;88:1045–1051. doi: 10.1016/j.biochi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 26.Hofstadler SA, Griffey RH. Chem Rev. 2001;101:377–390. doi: 10.1021/cr990105o. [DOI] [PubMed] [Google Scholar]
- 27.Haddad J, Kotra LP, Llano-Sotelo B, Kim C, Azucena EF, Liu M, Vakulenko SB, Chow CS, Mobashery S. J Am Chem Soc. 2002;124:3229–3237. doi: 10.1021/ja011695m. [DOI] [PubMed] [Google Scholar]
- 28.Kaul M, Barbieri CM, Pilch DS. J Am Chem Soc. 2004;126:3447–3453. doi: 10.1021/ja030568i. [DOI] [PubMed] [Google Scholar]
- 29.Shandrick S, Zhao Q, Han Q, Ayida BK, Takahashi M, Winters GC, Simonsen KB, Vourloumis D, Hermann T. Angew Chem, Int Ed. 2004;43:3177–3182. doi: 10.1002/anie.200454217. [DOI] [PubMed] [Google Scholar]
- 30.Barbieri CM, Kaul M, Pilch DS. Tetrahedron. 2007;63:3567–3574. doi: 10.1016/j.tet.2006.08.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parsons J, Hermann T. Tetrahedron. 2007;63:3548–3552. [Google Scholar]
- 32.Chao PW, Chow CS. Bioorg Med Chem. 2007;15:3825–3831. doi: 10.1016/j.bmc.2007.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aminoglycosides have been fluorescently labeled before, but typically by modifying the amines that are also essential for specific RNA binding. See: Wang Y, Hamasaki K, Rando RR. Biochemistry. 1997;36:768–779. doi: 10.1021/bi962095g.Hamasaki K, Rando RR. Biochemistry. 1997;36:12323–12328. doi: 10.1021/bi970962r.Hamasaki K, Ueno A. Bioorg Med Chem Lett. 2001;11:591–594. doi: 10.1016/s0960-894x(01)00005-1.
- 34.See, however: Martí AA, Jockusch S, Li Z, Ju J, Turro NJ. Nucleic Acids Res. 2006;34:e50. doi: 10.1093/nar/gkl134.Borjesson K, Preus S, El-Sagheer AH, Brown T, Albinsson B, Wilhelmsson LM. J Am Chem Soc. 2009;131:4288–4293. doi: 10.1021/ja806944w.
- 35.Greco NJ, Tor Y. J Am Chem Soc. 2005;127:10784–10785. doi: 10.1021/ja052000a. [DOI] [PubMed] [Google Scholar]
- 36.Srivatsan SG, Tor Y. J Am Chem Soc. 2007;129:2044–2053. doi: 10.1021/ja066455r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sinkeldam RW, Greco NJ, Tor Y. Chem Bio Chem. 2008;9:706–709. doi: 10.1002/cbic.200700714. [DOI] [PubMed] [Google Scholar]
- 38.Srivatsan SG, Greco NJ, Tor Y. Angew Chem, Int Ed. 2008;47:6661–6665. doi: 10.1002/anie.200802199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Srivatsan SG, Weizman H, Tor Y. Org Biomol Chem. 2008;6:1334–1338. doi: 10.1039/b801054d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.See Supporting Information for additional details.
- 41.Note the red-shifted absorption of 1 compared to the native nucleobases (λmax 250–270 nm) and the intense blue emission of the acceptor.
- 42.Based on crystal structures (PDB 2ET4 and 1LC4), the estimated distance between U1406 and the fluorophore on 11 or 12 would be less than 15 Å.
- 43.The minimal A-site construct has been demonstrated to be an autonomous RNA domain capable of mimicking the function and antibiotics recognition features of the 16S rRNA. See ref 13 and 24.
- 44.Modification of U1406 with isosteric nucleosides has been demonstrated to have a minimal detrimental effect on the A-site construct’s folding and antibiotic recognition properties. See ref 36.
- 45.Such a decrease in the quantum yield is common among fluorescent nucleosides upon incoporation into an RNA or DNA duplex. See ref 34–39 and Hawkins ME, Pfleiderer W, Balis FM, Porter D, Knutson JR. Anal Biochem. 1997;244:86–95. doi: 10.1006/abio.1996.9879.Hawkins ME. Cell Biochem Biophys. 2001;34:257–281. doi: 10.1385/CBB:34:2:257.
- 46.Wang H, Tor Y. J Am Chem Soc. 1997;119:8734–8735. [Google Scholar]
- 47.(a) Wang H, Tor Y. Angew Chem, Int Ed. 1998;37:109–111. [Google Scholar]; (b) Michael K, Wang H, Tor Y. Bioorg Med Chem. 1999;7:1361–1371. doi: 10.1016/s0968-0896(99)00071-1. [DOI] [PubMed] [Google Scholar]; (c) Boer J, Blount KF, Luedtke NW, Elson-Schwab L, Tor Y. Angew Chem, Int Ed. 2005;44:927–932. doi: 10.1002/anie.200461182. [DOI] [PubMed] [Google Scholar]
- 48.Schluenzen F, Tocilj A, Zarivach R, Harms J, Gluehmann M, Janell D, Bashan A, Bartels H, Agmon I, Franceschi F, Yonath A. Cell. 2000;102:615–623. doi: 10.1016/s0092-8674(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 49.Yoshizawa S, Fourmy D, Puglisi JD. Science. 1999;285:1722–1725. doi: 10.1126/science.285.5434.1722. [DOI] [PubMed] [Google Scholar]
- 50.O’Connor M, Brunelli CA, Firpo MA, Gregory ST, Lieberman KR, Stephen Lodmell J, Moine H, Ryk DIV, Dahlberg AE. Biochem Cell Biol. 1995;73:859–868. doi: 10.1139/o95-093. [DOI] [PubMed] [Google Scholar]
- 51.Vicens Q, Westhof E. Structure. 2001;9:647–658. doi: 10.1016/s0969-2126(01)00629-3. [DOI] [PubMed] [Google Scholar]
- 52.Ogle JM, Brodersen DE, Clemons WM, Jr, Tarry MJ, Carter AP, Ramakrishnan V. Science. 2001;292:897–902. doi: 10.1126/science.1060612. [DOI] [PubMed] [Google Scholar]
- 53.Vakulenko SB, Mobashery S. Clin Microbiol Rev. 2003;16:430–450. doi: 10.1128/CMR.16.3.430-450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smolinski MS, Hamburg MA, Lederberg J. Microbial Threats to Health: Emergence, Detection, and Response. National Academy of Sciences; Washington DC: 2003. [PubMed] [Google Scholar]
- 55.Wax RG, Lewis K, Salyers A, Taber H. Bacterial Resistance to Antimicrobials. 2. CRC Press; Boca Raton, FL: 2007. [Google Scholar]
- 56.Ward DC, Reich E, Stryer L. J Biol Chem. 1969;244:1228–1237. [PubMed] [Google Scholar]
- 57.Kawai M, Lee MJ, Evans KO, Nordlund TM. J Fluorescence. 2001;11:23–32. [Google Scholar]
- 58.Jean JM, Hall KB. Proc Natl Acad Sci USA. 2001;98:37–41. doi: 10.1073/pnas.011442198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rachofsky EL, Osman R, Ross JBA. Biochemistry. 2001;40:946–956. doi: 10.1021/bi001664o. [DOI] [PubMed] [Google Scholar]
- 60.Tor Y. Tetrahedron. 2007;63:3425–3426. [Google Scholar]
- 61.Srivatsan SG, Tor Y. Nat Protocols. 2007;2:1547–1555. doi: 10.1038/nprot.2007.222. [DOI] [PubMed] [Google Scholar]
- 62.Liu H, Gao J, Kool ET. J Org Chem. 2005;70:639–647. doi: 10.1021/jo048357z. [DOI] [PubMed] [Google Scholar]
- 63.Lee AHF, Kool ET. J Am Chem Soc. 2006;128:9219–9230. doi: 10.1021/ja0619004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Godde F, Toulm JJ, Moreau S. Biochemistry. 1998;37:13765–13775. doi: 10.1021/bi9811967. [DOI] [PubMed] [Google Scholar]
- 65.According to crystal structures PDB 2ET4 and 1LC4, the positions of modification on the aminoglycosides are not involved in binding to the RNA.
- 66.Gold H. Fluorescent brightening agents. In: Venkataraman K, editor. The Chemistry of Synthetic Dyes. V. Academic Press; New York: 1971. pp. 535–542. [Google Scholar]
- 67.Baindur N, Triggle DJ. Med Res Rev. 1994;14:591–664. doi: 10.1002/med.2610140603. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.