

Abstract
Phosphodiesterase-5 (PDE-5) inhibitors including sildenafil and vardenafil induce powerful preconditioning-like cardioprotective effect against ischemia/reperfusion injury through opening of mitochondrial KATP channels in the heart. The goal of these studies was to demonstrate the protective effect of sildenafil and vardenafil on reperfusion injury and to compare it with the antianginal vasodilator nitroglycerin (NTG). In addition, we determined the role of mitochondrial KATP channels in protection. Adult male New Zealand white rabbits were anesthetized and subjected to ischemia by 30 min of coronary artery occlusion followed by 3 hrs of reperfusion. Seven groups were studied. 1- Controls; 2- Sildenafil (total dose: 0.71 mg/kg; iv) infused for 65 min starting 5 min before reperfusion; 3- Sildenafil+5-hydroxydecanoate (5-HD, blocker of mitochondrial KATP channel, total dose: 5 mg/kg) administered as 2 bolus injections; 4- Vardenafil (total dose: 0.014 mg/kg; iv) administered as in group 2; 5- Vardenafil+5-HD administered as in group 3; 6- 5-HD administered as two bolus injections and 7- Nitroglycerin (NTG, total dose: 2 μg.kg -1.min -1) administered as in group 2. Infarct size was reduced in sildenafil (19.19±1.3%) as well as vardenafil (17.0±2.0%) treated groups as compared to controls (33.8 ± 1.7%). However, NTG failed to confer similar cardioprotection (31.5±0.8%). 5-HD blocked the cardioprotective effects of sildenafil and vardenafil as shown by an increase in infarct size (34.0 ±1.1% and 28.3±1.9%, respectively). Both sildenafil and vardenafil protect the ischemic myocardium against reperfusion injury through a mechanism dependent on mitochondrial KATP channel opening. Future clinical trials are needed to exploit the potential utility of PDE-5 inhibitors in cardioprotection in patients with coronary artery disease.
Keywords: Phosphodiesterase inhibitors, Ischemia-reperfusion injury, Nitric oxide, cGMP, Potassium channel
Introduction
Phosphodiesterase-5 (PDE-5) inhibitors including sildenafil, vardenafil and tadalafil are a class of drugs that have been developed for treatment of erectile dysfunction (ED) (1) and more recently for pulmonary artery hypertension (2). A number of recent studies from our laboratory demonstrated powerful preconditioning-like cardioprotection with PDE-5 inhibitors against ischemia/reperfusion (I/R) injury in several animal models (3,4,5,6,7) that extends beyond their therapeutic capacity for treatment of ED. We showed that PDE-5 inhibition caused significant reduction of infarct size (3,4,7), inhibition of necrosis and apoptosis in adult cardiomyocytes (5) and attenuation of doxorubicin-induced cardiomyopathy in mice (8). We also demonstrated that the cardioprotective effect of PDE-5 inhibition is mediated through opening of mitochondrial KATP (mitoKATP) channel (1,6) and activation of KCa channel (9). In addition, we showed that nitric oxide (NO) generated from endothelial (eNOS) and inducible (iNOS) nitric oxide synthase (4) as well as activation of protein kinase C (PKC) (10) played a role in the cardioprotective effect of sildenafil. All of these studies were performed under experimental conditions where the animals or cardiomyocytes were treated with PDE-5 inhibitors prior to ischemia. However, it is not known whether these drugs would cause infarct size reduction when administered at reperfusion.
Nitroglycerin (glyceryl trinitrate, NTG) has been used to treat angina and heart failure for over 130 years. It is assumed that NTG is converted in vascular smooth muscle cells to NO or an NO congener (S-nitrosothiol, SNO), which activates guanylate cyclase and thus relaxes vascular smooth muscle (11,12). Studies have shown that pretreatment with NTG produces a delayed cardioprotective effect that improves tolerance to ischemia during coronary angioplasty (13). Also, there is ample evidence which suggests that exogenous nitric oxide (NO) that results from administration of a NO donor is cardioprotective (14,15,16). In the current study, we hypothesized that a similar protection against I/R would be possible if PDE-5 inhibitors or NTG were administered at the time of reperfusion. Our data suggest that both sildenafil and vardenafil, but not NTG, reduce infarct size in the rabbit heart following I/R when infused at reperfusion. The protective effect of PDE-5 inhibitors was blunted by treatment with 5- hydroxydecanoate (5-HD), a putative blocker of the mitoKATP channel (17).
Materials and Methods
Animals
Male New Zealand White rabbits (2.8-3.3 kg) were used for the studies. The care and use of the animals were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of Virginia Commonwealth University and the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals [DHHS Publication No. (NIH) 80-23; Office of Science and Health Reports, Bethesda, MD 20205].
Myocardial infarction protocol
The rabbit model of I/R has been described previously (2). After the rabbits were anesthetized with ketamine HCl (35 mg/kg) and xylazine (5 mg/kg), a left thoracotomy was performed to expose the heart. Myocardial ischemia was induced by occlusion of the left descending coronary artery for 30 min, followed by reperfusion for 3 hr. After completion of ischemia-reperfusion protocol, 500 IU of heparin were injected and the heart was quickly removed and mounted on a Langendorff apparatus. The coronary arteries were perfused with 0.9% NaCl containing 2.5 mM CaCl2. After the blood was washed out, the suture around the coronary artery was retightened and ~2 ml of 10% Evans blue dye were injected as a bolus into the aorta until most of the heart turned blue. The heart was perfused with saline to wash out the excess Evans blue. Finally, the heart was removed, frozen, and cut into 8–10 transverse slices from apex to base of equal thickness (~1 mm). The slices were then incubated in a 1% triphenyltetrazolium chloride solution in an isotonic phosphate buffer (pH 7.4) at 37°C for 30 min. The areas of infarcted tissue, the risk zone, and the whole left ventricle were determined by computer morphometry using a Bioquant imaging software. Infarct size was expressed both as a percentage of the left ventricle and ischemic risk area.
Measurement of hemodynamics
Hemodynamic measurements included heart rate and mean arterial pressure. Rate-pressure product was calculated as the product of heart rate and peak arterial pressure.
Study protocol
All animals were subjected to an infarction protocol consisting of 30 min of sustained ischemia by occlusion of the coronary artery followed by 180 min of reperfusion. The effects of sildenafil and vardenafil were studied in the absence or presence of 5-HD, unlike in the case of NTG where 5-HD was not required. The rabbits were randomly assigned into one of the following groups (Figure 1A). In group I (saline control, n = 6), rabbits received 0.9% saline. In group II (sildenafil, n = 6), sildenafil powder (kindly provided by Pfizer, Inc.) was administered at a dose of 0.71 mg/kg; iv in a total volume of 55 ml saline infused at a constant rate of 50 ml/hr for 65 min starting at 5 min prior to the onset of reperfusion. In group III (sildenafil+5-HD), sildenafil was infused as in group II and 5-HD was administered as two bolus injections of 5 mg/kg, iv, the first 5 min prior to sildenafil infusion and the second at 30 min of reperfusion to ensure mitoKATP channel closure throughout the drug infusion. In group IV (vardenafil, n=9), vardenafil powder (kindly provided by Bayer Healthcare) was infused as sildenafil in group II. In group V (vardenafil+5-HD, n=6), both drugs were administered as in group III. In groups II-V, the doses of sildenafil and vardenafil were used based on our previous studies demonstrating the protective effect of these drugs when administered as bolus prior to initiating ischemia/reperfusion (2,7).
Figure 1.
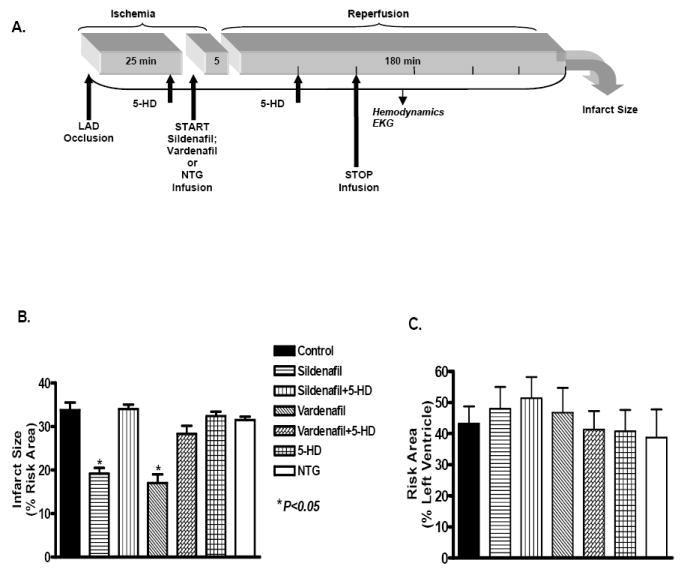
A. Experimantal protocol for ischemia and reperfusion. Arrows indicate the exact points of pharmacological drug administration for the various groups. B. Infarct size expressed as percent of the area at risk in the various experimental groups. C. The risk area expressed as percent of the left ventricle in the various groups. * denotes significant difference versus all other groups.
In group VI (5-HD, n=6), 5-HD was administered at the same time points as in group III. In group VII (NTG, n=7), NTG was administered at a clinically relevant dose of 2 μg.kg -1.min -1; iv by continuous infusion similar to sildenafil and vardenafil. The dose of NTG was chosen based on a previously published clinical study where patients received 2μg/kg/min NTG infusion (18). In addition, another study by Bolli and coworkers in rabbits reported that dilution of NTG with saline (to attain a dose of 2μg/kg/min) was the highest dose which caused no hemodynamic changes (19). Interestingly, these authors showed that such a hemodynamically inactive dose of NTG (and also another NO donor) alleviated myocardial stunning in rabbits.
Statistics
All measurements of infarct size and risk areas are expressed as group means ± SE. Changes in hemodynamics and infarct size variables were analyzed using two-way repeated-measures ANOVA to determine the main effect of time, group, and time-by-group interaction. If the global tests showed major interactions, post hoc contrasts between different time points within the same group or between different groups were performed using t-test. Statistical differences were considered significant if the P value was <0.05.
Results
Infarct size
The infarct size (% of risk area, mean ± SE, P < 0.05) decreased from 33.8 ± 1.7 in the saline-treated control group to 19.2 ± 1.3 and 17.0 ± 2.0 in the sildenafil and vardenafil -treated rabbits, respectively (Figure 1B). The infarct-limiting effect of sildenafil and vardenafil was abolished in animals treated with 5-HD as shown by significant increase in infarct size to 34.0 ± 1.1 and 28.3 ± 1.9, respectively. In contrast, NTG failed to mimic the protective effect of PDE-5 inhibitors as indicated by an infarct size of 31.5 ± 0.8. Control animals treated with 5-HD had an infarct size of 32.4 ± 1.0, which was not different from an infarct size of 33.8 ± 1.7 in the saline controls (P > 0.05). The risk areas expressed as a percentage of the left ventricle were not statistically different between the groups (Figure 1C). These data suggest that changes in the infarct size observed among various groups were not related to the percentage of the risk area of the left ventricle occluded by our surgical technique.
Hemodynamics
Administration of sildenafil, vardenafil and NTG caused a rapid but mild decrease in hemodynamics as indicated by the decline in systolic, diastolic, and mean arterial pressures, respectively (Figure 2A-C). Heart rate was not influenced following drug administration (Figure 2D). The hemodynamics remained reasonably stable, although they gradually decreased in all the groups during the experimental protocol. Except at the indicated time points, the mean values were not significantly different between the groups (Table 1).
Figure 2.
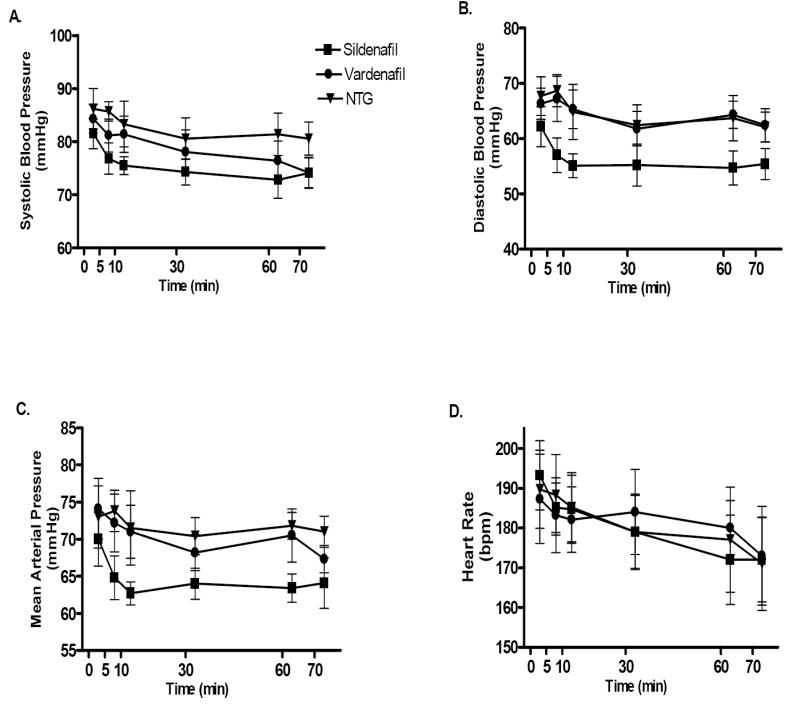
Changes in hemodynamics following drug administration in the various groups. A. Systolic blood pressure (mmHg); B. Diastolic blood pressure (mmHg); C. Mean arterial blood pressure (mmHg); D. Heart rate (bpm). P>0.05 within each of the experimental groups.
Table 1.
Hemodynamics during ischemia/reperfusion.
| Group | Baseline | 30-min Isch | 180-min Rep |
|---|---|---|---|
| Control (group I) | |||
| HR | 182 ± 6 | 172 ± 9 | 155 ± 8 |
| MAP | 86 ± 9 | 78 ± 8 | 71 ± 8 |
| RPP | 20,924 ± 2348 | 17,168 ± 2425 | 14,020 ± 2253 |
| Sildenafil (groupII) | |||
| HR | 198 ± 9 | 187 ± 8 | 161 ± 9a |
| MAP | 82 ± 10 | 66 ± 4 | 63 ± 5 |
| RPP | 19,408 ± 1439 | 14,960 ± 2764 | 13,236 ± 3112 |
| Sildenafil+5-HD (group III) | |||
| HR | 191 ± 8 | 186 ± 7 | 159 ± 14 |
| MAP | 84 ± 7 | 72 ± 6 | 67 ± 2 |
| RPP | 18,996 ± 1180 | 16,182 ± 1350 | 13,720 ± 1053a |
| Vardenafil (group IV) | |||
| HR | 188 ± 7 | 179 ± 12 | 146 ± 10a,b |
| MAP | 89 ± 4 | 71 ± 3a | 60 ± 3a,b |
| RPP | 19,740 ± 1563 | 15,380 ± 2170 | 10,804 ± 1235a |
| Vardeafnil+5-HD (group V) | |||
| HR | 181 ± 6 | 185 ± 8 | 160 ± 9 |
| MAP | 79 ± 4 | 74 ± 5 | 63 ± 7 |
| RPP | 17,652 ± 2080 | 16,650 ± 1460 | 12,320 ± 1833 |
| 5-HD (group VI) | |||
| HR | 190 ± 11 | 185 ± 8 | 160 ± 7 |
| MAP | 75 ± 6 | 67 ± 5 | 61 ± 3 |
| RPP | 17900 ± 1142 | 14,215 ± 2055 | 11,840 ± 1788 |
| Nitroglycerin (group VII) | |||
| HR | 178 ± 16 | 185 ± 8 | 160 ± 7 |
| MAP | 85 ± 5 | 77 ± 5 | 72 ± 7 |
| RPP | 18,154 ± 1912 | 16,617 ± 1462 | 13,360 ± 1743 |
Values are Means±SEM. HR – Heart rate (beats/min); MAP – Mean arterial blood pressure (mmHg); RPP – Rate pressure product (mmHg/min),
P < 0.05 vs. Baseline;
P < 0.05 vs. 30 min. Ischemia.
Discussion
Our results show that intravenous administration of sildenafil or vardenafil at reperfusion induced a significant cardioprotective effect as demonstrated by a reduction in infarct size when compared to the saline-treated controls. The cardioprotective effect of PDE-5 inhibitors at reperfusion was similar to their preconditioning-like effect (3,7), whereas NTG failed to express similar infarct-limiting effects. The protection was blocked by 5-HD suggesting that it was mediated by opening of mitoKATP channels. To our knowledge, this is the first study demonstrating (a) the cardioprotective effect of PDE-5 inhibition at reperfusion in vivo and (b) the involvement of mitoKATP channel in mediating this protection. In humans, PDE-5 inhibitors are known to cause a transient reduction in supine blood pressure within a few hours following treatment (20). Even though PDE-5 inhibitors or NTG are known to cause a decline in blood pressure when administered prior to I/R (3,7,11), we observed a much milder effect on hemodynamics with these drugs when administered in a slow infusion protocol with similar cumulative doses as in the pre-treatment protocol (Figure 2). We preferred this method of administration to avoid a detrimental decline in systemic blood pressure in the ischemic hearts. The dose of sildenafil used in the current study was based on the clinically relevant average dose of 50 mg pill used by men for management of ED. Since vardenafil is more potent than sildenafil (sildenafil- IC50 3.5 nM versus vardenfil IC50 0.14 nM), we decided to use a much lower dose of this drug. The lowest clinical dose for vardenafil is 2.5 mg for ED. In the present study, we used 0.014 mg/kg vardenafil which translates to an equivalent of 1 mg pill given to an average 70 kg patient. Thus the lack of effect on hemodynamics with vardenafil may well be due to the low dose infusion over an extended period of 65 minutes. In all the groups, we did not observe any significant change in pressures and heart rate after cessation of drug infusion (Table 1).
PDE-5 hydrolyzes the phosphodiester bond of cyclic cGMP (21) and therefore inhibition of this enzyme elevates the intracellular concentration of cGMP. In addition to its presence in the cavernosal tissue and penile arteries, PDE-5 is also available in all vascular smooth muscle cells and ventricular cardiomyocytes (5,22). Considering the importance of cGMP in inducing preconditioning by NO donors, PDE-5 inhibitors as well as natriuretic peptides (23,24), it was not surprising that PDE-5 inhibition at reperfusion and the consequent accumulation of cGMP would lead to a cardioprotective effect. Recent studies have also shown that the administration of atrial natriuretic peptide (ANP) at the beginning of reperfusion elicited a significant improvement in post-ischemic recovery of cardiac function in rat hearts undergoing 15 min of normothermic global ischemia (25). In addition, Yang et al (26) demonstrated that the administration of ANP at reperfusion limits infarct size in the rabbit heart through activation of protein kinase G (PKG), opening of mitoKATP channels and stimulation of downstream kinases. In vitro studies support these observations because adding purified PKG and cGMP caused opening of mitoKATP channels to a similar extent as the KATP channel openers including diazoxide and cromakalim (27). We have recently shown that adenoviral gene transfer of PKG induced protection against simulated ischemia/reoxygenation injury in cardiomyocytes by opening of mitoKATP channels (28). In these studies, the protective effect of PKGI-α was not inhibited by HMR1098, the selective blocker of sarcolemmal KATP channel thereby further supporting the role of mitoKATP channels in PKG-dependent protection. Also, it has been shown that cGMP-dependent signals activate sarcoplasmic reticulum Ca2+ pump (SERCA) via PKG-dependent phosphorylation of phospholamban (29). The increase in SERCA activity reduces peak intracellular Ca2+ as well as its oscillation during reoxygenation attenuates the excessive activation of the contractile machinery and thereby prevents hypercontracture. The possibility of such an effect of PDE-5 inhibition on SERCA activity at reperfusion needs to be investigated. Recent studies also suggest that activation of anti-apoptotic pro-survival kinase signaling cascades including phosphatidylinositol-3-OH kinase (PI3K)-Akt and p42/p44 extra-cellular signal-regulated kinases (Erk 1/2) confer protection against reperfusion-induced injury (30). A number of agents including insulin-like growth factor-1, transforming growth factor-ß1, cardiotrophin-1, urocortin, atorvastatin and bradykinin were shown to protect the heart at reperfusion by activating the PI3K-Akt and/or Erk 1/2 kinase cascades (30). In this respect, the possibility of cCMP dependent activation of RISK-pathway following treatment with PDE-5 inhibitors remains an attractive hypothesis and needs to be investigated.
Although NO generating system such as acidified nitrite has been shown to be protective when given before or during ischemia (16), our results show that NTG failed to reduce infarct size at reperfusion. The differences in these results may be attributed to the timing of infusion and the kinetics of cGMP accumulation following treatment with NTG or PDE-5 inhibitors. PDE-5 inhibition with the potent drugs including sildenafil and vardenafil causes a prompt accumulation of cGMP (by preventing its degradation) in the tissue (31). The higher availability of NO with NTG infusion would also increase cGMP by activating soluble guanylate cyclase in the presence of GTP but probably not as swiftly as selective inhibition of PDE-5. As a result, there may be insufficient availability of cGMP to prevent ischemic injury with NTG at the time of reperfusion. Another possible explanation could be the surge of superoxide radical that occurs at the onset of reperfusion (32, 33) which may interact with NO derived from NTG to form the peroxynitrite anion (ONOO-). ONOO- formation during the early period of reperfusion results in protein nitration and myocardial injury (34). Thus it appears that rapid up-regulation of cGMP by PDE-5 inhibition rather than increasing its synthesis through NO is an effective approach to prevent reperfusion injury in the heart.
In the present study, the interpretation for the involvement of mitoKATP channels in protection against reperfusion injury with PDE-5 inhibitors is solely based on the ability of 5-HD to block the protection. There are number of studies which have suggested the role of mitoKATP channels in ischemic and pharmacological preconditioning based on the ability of 5-HD to block cardioprotection (2,7,35,36). Moreover, the responses of flavoprotein fluorescence, which are frequently used to assay mitoKATP channel activity in diazoxide-induced K+ flux in the intact myocytes, were shown to be selectively blocked by 5-HD (37,38). These studies suggested that 5-HD was selective blocker of mitoKATP channels. Nevertheless, the reliability of 5-HD as a selective blocker has been questioned because this substituted fatty acid is activated and metabolized by the matrix ß-oxidation pathway (39,40). Furthermore, a recent study by Dröse et al (41) reported that decanoate, a medium chain fatty acid structurally related to 5-HD reversed diazoxide-induced flavoprotein oxidation. These authors speculated that the ability of 5-HD to reverse flavoprotein oxidation was due to switch from glucose to fatty acid metabolism rather than mitoKATP channel inhibition.
Reperfusion injury has been recognized for several decades although the clinically useful approaches to attenuate the injury have so far failed to materialize (42). Thus there is a compelling need to protect the ischemic myocardium in patients experiencing myocardial ischemia. In this respect, the treatment of reperfusion injury by PDE-5 inhibitors is quite attractive, considering their profound effect on infarct size reduction as shown in the present study. Research and clinical trial outcomes have already established the long-term safety and efficacy of PDE-5 inhibitors in treating ED. Although there are theoretical concerns of a reduced myocardial tolerance to ischemia or promoting cardiac arrhythmias with PDE-5 inhibitors, randomized trials and retrospective analyses do not support an increased cardiac risk with oral treatment (43). Therefore, these drugs may be excellent candidates for clinical trials to limit infarct size when given just before reperfusion.
Acknowledgments
This study was supported in part by grants from National Institutes of Health (HL51045, HL59469, and HL79424), Pfizer Inc. and Bayer Healthcare AG to RCK.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shabsigh R. Therapy of ED: PDE-5 Inhibitors. Endocrine. 2004;23(2-3):135–41. doi: 10.1385/ENDO:23:2-3:135. [DOI] [PubMed] [Google Scholar]
- 2.Kane LB, Klings ES. Present and future treatment strategies for pulmonary arterial hypertension : focus on phosphodiesterase-5 inhibitors. Treat Respir Med. 2006;5(4):271–82. doi: 10.2165/00151829-200605040-00005. [DOI] [PubMed] [Google Scholar]
- 3.Ockaili R, Salloum F, Hawkins J, Kukreja RC. Sildenafil (Viagra) induces powerful cardioprotective effect via opening of mitochondrial KATP channels in rabbits. Am J Physiol Heart Circ Physiol. 2002;283:H1263–H1269. doi: 10.1152/ajpheart.00324.2002. [DOI] [PubMed] [Google Scholar]
- 4.Salloum F, Yin C, Xi L, Kukreja RC. Sildenafil induces delayed preconditioning through inducible nitric oxide synthase-dependent pathway in mouse heart. Circ Res. 2003;92:595–597. doi: 10.1161/01.RES.0000066853.09821.98. [DOI] [PubMed] [Google Scholar]
- 5.Das A, Xi L, Kukreja RC. Phosphodiesterase-5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis: Essential role of NO signaling. J Biol Chem. 2005;280:12944–55. doi: 10.1074/jbc.M404706200. [DOI] [PubMed] [Google Scholar]
- 6.Das S, Maulik N, Das DK, Kadowitz PJ, Bivalacqua TJ. Cardioprotection with sildenafil, a selective inhibitor of cyclic 3’,5’-monophosphate-specific phosphodiesterase 5. Drugs Exp Clin Res. 2002;28:213–219. [PubMed] [Google Scholar]
- 7.Salloum FN, Ockaili RA, Wittkamp M, Marwaha VR, Kukreja RC. Vardenafil: a novel type 5 phosphodiesterase inhibitor reduces myocardial infarct size following ischemia/reperfusion injury via opening of mitochondrial KATP channels in rabbits. J Mol Cell Cardiol. 2006;40:405–11. doi: 10.1016/j.yjmcc.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 8.Fisher PW, Salloum F, Das A, Hyder H, Kukreja RC. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation. 2005;111:1601–10. doi: 10.1161/01.CIR.0000160359.49478.C2. [DOI] [PubMed] [Google Scholar]
- 9.Wang X, Fisher PW, Xi L, Takenoshita Y, Kukreja RC. Essential Role of Ca2+-Activated K+ Channels in Triggering and Mediating Sildenafil-Induced Delayed Cardioprotection: Evidence from Pharmacological Inhibition and in Vivo Gene Knockdown with Small Interfering RNA. Circulation. 2005;112:II–142. Suppl. Abstract. [Google Scholar]
- 10.Das A, Ockaili R, Salloum F, Kukreja RC. Protein kinase C plays an essential role in sildenafil-induced cardioprotection in rabbits. Am J Physiol Heart Circ Physiol. 2004;286:H1455–H1460. doi: 10.1152/ajpheart.01040.2003. [DOI] [PubMed] [Google Scholar]
- 11.Murad F, Mittal CK, Arnold WP, Katsuki S, Kimura H. Guanylate cyclase: activation by azide, nitro compounds, nitric oxide, and hydroxyl radical and inhibition by hemoglobin and myoglobin. Adv Cyclic Nucleotide Res. 1978;9:145–58. [PubMed] [Google Scholar]
- 12.Ignarro LJ, Lippton H, Edwards JC, Baricos WH, Hyman AL, Kadowitz PJ, Gruetter CA. Mechanism of vascular smooth muscle relaxation by organic nitrates, nitrites, nitroprusside and nitric oxide: evidence for the involvement of S-nitrosothiols as active intermediates. J Pharmacol Exp Ther. 1981;218:739–749. [PubMed] [Google Scholar]
- 13.Leesar MA, Stoddard MF, Dawn B, Jasti VG, Masden R, Bolli R. Delayed preconditioning-mimetic action of nitroglycerin in patients undergoing coronary angioplasty. Circulation. 2001;103:2935–2941. doi: 10.1161/01.cir.103.24.2935. [DOI] [PubMed] [Google Scholar]
- 14.Lefer DJ, Nakanishi K, Johnston WE, Vinten-Johansen J. Antineutrophil and myocardial protecting actions of a novel nitric oxide donor after acute myocardial ischemia and reperfusion in dogs. Circulation. 1993;88:2337–2350. doi: 10.1161/01.cir.88.5.2337. [DOI] [PubMed] [Google Scholar]
- 15.Lochner A, Marais E, Genade S, Moolman JA. Nitric oxide: a trigger for classic preconditioning? Am J Physiol Heart Circ Physiol. 2000;279:H2752–H2765. doi: 10.1152/ajpheart.2000.279.6.H2752. [DOI] [PubMed] [Google Scholar]
- 16.Johnson G, 3rd, Tsao PS, Lefer AM. Cardioprotective effects of authentic nitric oxide in myocardial ischemia with reperfusion. Crit Care Med. 1991;9:244–52. doi: 10.1097/00003246-199102000-00021. [DOI] [PubMed] [Google Scholar]
- 17.Fryer RM, Hsu AK, Gross GJ. Mitochondrial K(ATP) channel opening is important during index ischemia and following myocardial reperfusion in ischemic preconditioned rat hearts. J Mol Cell Cardiol. 2001;33:831–4. doi: 10.1006/jmcc.2001.1350. [DOI] [PubMed] [Google Scholar]
- 18.Lobato EB, Janelle GM, Urdanta F, Martin TD. Comparison of milrinone versus nitroglycerin, alone and in combination, on grafted internal mammary artery flow after cardiopulmonary bypass: effects of alpha-adrenergic stimulation. J Cardiothorac Vasc Anesth. 2001;15:723–7. doi: 10.1053/jcan.2001.28316. [DOI] [PubMed] [Google Scholar]
- 19.Shinmura K, Tang XL, Takano H, Hill M, Bolli R. Nitric oxide donors attenuate myocardial stunning in conscious rabbits. Am J Physiol. 1999;277:H2495–503. doi: 10.1152/ajpheart.1999.277.6.H2495. [DOI] [PubMed] [Google Scholar]
- 20.Jackson G, Benjamin N, Jackson N, Allen MJ. Effects of sildenafil citrate on human hemodynamics. Am J Cardiol. 1999;83:13C–20C. doi: 10.1016/s0002-9149(99)00043-0. [DOI] [PubMed] [Google Scholar]
- 21.Burnett AL. Phosphodiesterase 5 mechanisms and therapeutic applications. Am J Cardiol. 2005;96:29M–31M. doi: 10.1016/j.amjcard.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 22.Senzaki H, Smith CJ, Juang GJ, Isoda T, Mayer SP, Ohler A, Paolocci N, Tomaselli GF, Hare JM, Kass DA. Cardiac phosphodiesterase 5 (cGMP-specific) modulates beta-adrenergic signaling in vivo and is down-regulated in heart failure. FASEB J. 2001;15:1718–1726. doi: 10.1096/fj.00-0538com. [DOI] [PubMed] [Google Scholar]
- 23.D’Souza SP, Yellon DM, Martin C, Schulz R, Heusch G, Onody A, Ferdinandy P, Baxter GF. B-type natriuretic peptide limits infarct size in rat isolated hearts via KATP channel opening. Am J Physiol Heart Circ Physiol. 2003;284:H1592–600. doi: 10.1152/ajpheart.00902.2002. [DOI] [PubMed] [Google Scholar]
- 24.Depre C, Vanoverschelde JL, Goudemant JF, Mottet I, Hue L. Protection against ischemic injury by nonvasoactive concentrations of nitric oxide synthase inhibitors in the perfused rabbit heart. Circulation. 1995;92:1911–8. doi: 10.1161/01.cir.92.7.1911. [DOI] [PubMed] [Google Scholar]
- 25.Sangawa K, Nakanishi K, Ishino K, Inoue M, Kawada M, Sano S. Atrial natriuretic peptide protects against ischemia-reperfusion injury in the isolated rat heart. Ann Thorac Surg. 2004;77:233–237. doi: 10.1016/s0003-4975(03)01493-0. [DOI] [PubMed] [Google Scholar]
- 26.Yang XM, Philipp S, Downey JM, Cohen MV. Atrial natriuretic peptide administered just prior to reperfusion limits infarction in rabbit hearts. Basic Res Cardiol. 2006;101:311–8. doi: 10.1007/s00395-006-0587-2. [DOI] [PubMed] [Google Scholar]
- 27.Costa AD, Garlid KD, West IC, Lincoln TM, Downey JM, Cohen MV, Critz SD. Protein kinase G transmits the cardioprotective signal from cytosol to mitochondria. Circ Res. 2005;97:329–36. doi: 10.1161/01.RES.0000178451.08719.5b. [DOI] [PubMed] [Google Scholar]
- 28.Das A, Smolenski A, Lohmann SM, Kukreja RC. J Biol Chem. 2006 doi: 10.1074/jbc.M606142200. In press. [DOI] [PubMed] [Google Scholar]
- 29.Abdallah Y, Gkatzoflia A, Pieper H, Zoga E, Walther S, Kasseckert S, Schafer M. Mechanism of cGMP-mediated protection in a cellular model of myocardial reperfusion injury. Cardiovasc Res. 2005;66:123–31. doi: 10.1016/j.cardiores.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 30.Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc Res. 2004;61:448–60. doi: 10.1016/j.cardiores.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 31.Francis SH, Corbin JD. Phosphodiesterase-5 inhibition: the molecular biology of erectile function and dysfunction. Urol Clin North Am. 2005;32:419–29. doi: 10.1016/j.ucl.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 32.Zweier JL. Measurement of superoxide-derived free radicals in the reperfused heart Evidence for a free radical mechanism of reperfusion injury. J Biol Chem. 1988;263:1353–7. [PubMed] [Google Scholar]
- 33.Kukreja RC, Hess ML. The oxygen free radical system: from equations through membrane protein interaction to myocardial injury and protection. Cardiovasc Res. 1992;26:641–655. doi: 10.1093/cvr/26.7.641. [DOI] [PubMed] [Google Scholar]
- 34.Zweier JL, Fertmann J, Wei G. Nitric oxide and peroxynitrite in postischemic myocardium. Antioxid Redox Signal. 2001;3:11–22. doi: 10.1089/152308601750100443. [DOI] [PubMed] [Google Scholar]
- 35.Shultz Jo, Qian Y-Z, Gross GJ, Kukreja RC. 5-hydroxydecanoate blocks ischemic preconditioning in rat heart. J Mol Cell Cardiol. 1997;29:1055–1060. doi: 10.1006/jmcc.1996.0358. [DOI] [PubMed] [Google Scholar]
- 36.Ockaili R, Emani VR, Okubo S, Brown M, Krottapalli K, Kukreja RC. Opening of mitochondrial KATP channel induces early and delayed cardioprotective effect: role of nitric oxide. Am J Physiol. 1999;277:H2425–H2434. doi: 10.1152/ajpheart.1999.277.6.H2425. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y, Sato T, O’Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: Novel Effectors of Cardioprotection? Circulation. 1998;97:2463–2469. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- 38.Jabůrek M, Yarov-Yarovoy V, Paucek P, Garlid KD. State-dependent inhibition of the mitochondrial KATP channel by glyburide and 5-hydroxydecanoate. J Biol Chem. 1998;273:13578–13582. [PubMed] [Google Scholar]
- 39.Hanley PJ, Mickel M, Löffler M, Brandt U, Daut J. KATP channel-independent targets of diazoxide and 5-hydroxydecanoate in the heart. J Physiol (Lond) 2002;542:735–741. doi: 10.1113/jphysiol.2002.023960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lim KHH, Javadov SA, Das M, Clarke SJ, Suleiman M-S, Halestrap AP. The effects of ischaemic preconditioning, diazoxide and 5-hydroxydecanoate on rat heart mitochondrial volume and respiration. J Physiol (Lond) 2002;545:961–974. doi: 10.1113/jphysiol.2002.031484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drose S, Brandt U, Hanley PJ. K+-independent actions of diazoxide question the role of inner membrane KATP channels in mitochondrial cytoprotective signaling. J Biol Chem. 2006;281:23733–9. doi: 10.1074/jbc.M602570200. [DOI] [PubMed] [Google Scholar]
- 42.Baxter GF, Yellon DM. Current trends and controversies in ischemia-reperfusion research--meeting report of the Hatter Institute 3rd International Workshop on Cardioprotection. Basic Res Cardiol. 2003;98:133–6. doi: 10.1007/s00395-003-0393-z. [DOI] [PubMed] [Google Scholar]
- 43.Reffelmann T, Kloner RA. Pharmacotherapy of erectile dysfunction: focus on cardiovascular safety. Expert Opin Drug Saf. 2005;4:531–40. doi: 10.1517/14740338.4.3.531. [DOI] [PubMed] [Google Scholar]