

Abstract
Suppressors of cytokine signaling (SOCSs) are negative regulators of both innate and adaptive immunity via inhibition of signaling by cytokines such as type I and type II IFNs. We have developed a small peptide antagonist of SOCS-1 that corresponds to the activation loop of JAK2. SOCS-1 inhibits both type I and type II IFN activities by binding to the kinase activation loop via the kinase inhibitory region of the SOCS. The antagonist, pJAK2(1001–1013), inhibited the replication of vaccinia virus and encephalomyocarditis virus in cell culture, suggesting that it possesses broad antiviral activity. In addition, pJAK2(1001–1013) protected mice against lethal vaccinia and encephalomyocarditis virus infection. pJAK2(1001–1013) increased the intracellular level of the constitutive IFN-β, which may play a role in the antagonist antiviral effect at the cellular level. Ab neutralization suggests that constitutive IFN-β may act intracellularly, consistent with recent findings on IFN-γ intracellular signaling. pJAK2(1001–1013) also synergizes with IFNs as per IFN-γ mimetic to exert a multiplicative antiviral effect at the level of transcription, the cell, and protection of mice against lethal viral infection. pJAK2(1001–1013) binds to the kinase inhibitory region of both SOCS-1 and SOCS-3 and blocks their inhibitory effects on the IFN-γ activation site promoter. In addition to a direct antiviral effect and synergism with IFN, the SOCS antagonist also exhibits adjuvant effects on humoral and cellular immunity as well as an enhancement of polyinosinic-polycytidylic acid activation of TLR3. The SOCS antagonist thus presents a novel and effective approach to enhancement of host defense against viruses.
Viruses are a heterogeneous group of intracellular infectious agents that depend in varying degrees on the host synthetic machinery for replication. The poxviruses are large dsDNA viruses that are assembled in the cytoplasm of infected cells involving complex replication mechanisms (1). They are responsible for some of the most devastating pandemics in the history of humankind and have been estimated to cause ~500 million deaths globally in the past century alone (2). Modern medicine has responded to smallpox by global immunization, but this has been discontinued for many years, which has left an entire generation without immunity to the natural or deliberate reintroduction of this family of viruses (3). Attachment, internalization, and disassembling of poxviruses precedes the initiation of three waves of mRNA synthesis. The early wave codes for virus growth factors and decoy cytokine receptors. Decoy receptors for both type I and type II IFNs are produced during early protein synthesis in poxvirus-infected cells, thus blunting perhaps the most important innate host defense system against viral infections (2, 4). A well-known example of this is the B8R protein of vaccinia virus, which is a homolog of the extracellular domain of the IFN-γ receptor (5). The decoy receptor along with other evasion factors make these viruses very adept at neutralizing innate and adaptive host defense mechanisms against viruses.
Encephalomyocarditis virus (EMCV) is a small ssRNA picornavirus of the plus strand orientation with a wide host range (6). EMCV infection can cause myocarditis, leading to arrhythmias, heart failure, and death (7). Also, during cardiac transplantation and valve replacement, infection by EMCV has been implicated in the development of cardiomyopathy (8), which makes the development of effective therapies against this virus particularly important. In mice, EMCV infection is lethal, but is quite susceptible to IFN-γ or an IFN-γ mimetic treatment at early stages of infection (9). The IFN-γ mimetic is also effective against vaccinia virus infection even in the presence of B8R decoy receptor (10, 11). The IFN-γ mimetic is a small peptide corresponding to the C terminus of IFN-γ that functions intracellularly and thus does not interact with the extracellular domain of the IFN-γ decoy receptor of the virus (10).
The IFN-γ mimetic is also effective against another large dsDNA virus called HSV-1 that replicates in the cell nucleus (12). Close relatives include the varicella zoster virus and CMV (13). The broad spectrum of antiviral activity of IFN-γ mimetics is unique in that we are unaware of any other small antiviral that exhibits strong activity against poxviruses, picornaviruses, and herpesviruses.
The IFN system is regulated by an inducible endogenous tyrosine kinase inhibitor called suppressor of cytokine signaling 1 (SOCS-1) (14–18). SOCS-1 is a member of a family of inducible proteins that negatively regulate IFN and other cytokine signaling via inhibition of JAK/STAT signaling (14). There are currently eight members of the SOCS family, SOCS-1 to SOCS-7 and cytokine-inducible Src homology 2 (SH2) protein. SOCS-1 has distinct regions or domains that define the mechanism by which it inhibits the function of JAK tyrosine kinases such as JAK2 that are involved in activation of STAT transcription factors (14). The N terminus of SOCS-1 contains an SH2 domain, and N-terminal to it is an extended SH2 sequence adjacent to a kinase inhibitory region (KIR) (14). These regions or domains of SOCS-1 bind to the activation and catalytic regions of JAK2 and block its function. The C terminus of SOCS-1 contains a domain called the SOCS box, which is involved in proteasomal degradation of JAK2. We have shown that the KIR sequence of SOCS-1 binds to a peptide corresponding to the activation loop of JAK2, pJAK2 (1001–1013), and demonstrated that pJAK2(1001–1013) blocked SOCS-1 activity in cells (19). Specifically, pJAK2(1001–1013) enhances suboptimal IFN activity, blocks SOCS-1 induced inhibition of STAT3 activation, enhances IFN-γ activation site (GAS) promoter activity, and enhances Ag-specific proliferation. It is thus a potential antiviral.
There is a dynamic interaction between the JAK kinases that mediate the antiviral effects of IFNs and SOCS such as SOCS-1 in regulation of IFN activity, in which induced SOCS-1 prevents unregulated IFN activity (10–14). Along with the well-known induction of IFN in cells, there is also the constitutive presence of IFN-β, which interacts in complex ways with other IFNs in the induction of antiviral states and other functions (20–22). We reasoned that the peptide antagonist of SOCS-1 would either induce or enhance an antiviral state by limiting the ability of SOCS-1 to modulate constitutive or added IFN antiviral activity. Further, we reasoned that such an effect would have broad antiviral activity. We have previously shown that pJAK2(1001–1013) antagonized the effect of SOCS-1 in HSV-1–infected keratinocytes (12). We present data in this paper on pJAK2(1001–1013) inhibition of the infectivity of vaccinia virus and EMCV in cells and in protection of mice against lethal infection. Such inhibition would suggest that the SOCS-1 antagonist possesses broad antiviral activity.
Materials and Methods
Cell culture and virus
BSC-40, L929, WISH, or RAW264.7 cells were obtained from American Type Culture Collection (Manassas, VA) and propagated on DMEM with 10% FBS (BSC-40, L929, WISH) or RPMI 1640 with 10% FBS (RAW264.7). All cells were grown at 37–C in humidified atmosphere with 5% CO2. Vaccinia virus Western Reserve strain was a kind gift from Dr. Richard Condit (University of Florida, Gainesville, FL). Vaccinia virus was grown, purified on sucrose gradient, and titrated on BSC-40 cells as described (10). EMCV was grown and titrated on L929 cells as described (9).
Peptides
The sequence of peptides used in this study is presented in Table I. These peptides were synthesized on an Applied Biosystems 9050 automated peptide synthesizer (Applied Biosystems, Foster City, CA) using conventional fluorenylmethyloxycarbonyl chemistry as described previously (23). The addition of a lipophilic group (palmitoyl-lysine) to the N terminus of the synthetic peptide was performed as a last step using semiautomated protocol. Peptides were characterized by mass spectrometry and purified by HPLC.
Table I.
List of peptides used in this study
| Peptide | Sequence |
|---|---|
| pJAK2(1001–1013) | 1001LPQDKEYYKVKEP |
| JAK2(1001–1013)2A alanine-substituted control | 1001LPQDKEAAKVKEP |
| MuIFN-γ(95–106) | 95AKFEVNNPQVQR |
| MuIFN-γ(95–125) | 95AKFEVNNPQVQRQAFNELIRVVHQLLPESSL |
| MuIFN-γ(95–132) | 95AKFEVNNPQVQRQAFNELIRVVHQLLPESSLRKRKRSR |
| SOCS1-KIR | 53DTHFRTFRSHSDYRRI |
| SOCS3-KIR | 20LRLKTFSSKSEYQLVV |
All of the peptides were synthesized with an attached lipophilic group, palmitic acid, for cell penetration. JAK2(1001–1013)2A, MuIFN-γ(95–125), and MuIFN-γ(95–106) were used as control peptides. These peptides do not show significant biological activity in the assays for which they have been used as control peptides.
Mu, murine.
Mice
All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Florida. Female C57BL/6 mice (6–8 wk old) were purchased from The Jackson Laboratory (Bar Harbor, ME). Peptides dissolved in PBS in a volume of 100 μl were administered i.p. For the oral administration of the peptides, indicated amounts of peptide in 0.5 ml PBS were given using a feeding needle. Intraperitoneal administration of EMCV was done in a volume of 100 μl. For intranasal administration, vaccinia virus was taken in a volume of 10 μl, and 5 μl was delivered in each of the nostrils of a lightly anesthetized mouse. Following infection, mice were observed daily for signs of disease, such as lethargy, ruffled hair, weight loss, and eye secretions. Moribund mice were euthanized and counted as dead.
FITC labeling and detection of cell penetration
FITC was conjugated with lipo-pJAK2(1001–1013), according to the manufacturer’s (Pierce, Rockford, IL) instruction. Mice were injected i.p. with 15 μg FITC-labeled lipo-pJAK2(1010–1013) or an equivalent amount of FITC alone. Two hours later, peritoneal cells were harvested and viewed in a fluorescent microscope. L929 cells were similarly treated with 5 μM FITC-labeled lipo-pJAK2(1010–1013) or FITC alone for 2 h, followed by visualization in a fluorescent microscope.
Reporter gene assays
The plasmid pGL3 promoter, which expresses the firefly luciferase, was obtained from Promega (Madison, WI). A sequence containing three copies of the GAS promoter element from human IRF1 gene, 5′-AGCCTGATTTCCCCGAAATGACGCG-3′, was inserted in the multiple cloning site of pGL3. A constitutively expressed thymidine kinase promoter-driven Renilla luciferase gene (pRL-TK) was used as an internal control in all of the reporter plasmid transfections. WISH or L929 cells were seeded in 12-well plates at 50% confluency. The next day, 500 ng GAS promoter-driven firefly luciferase and 50 ng pRL-TK were cotransfected using lipofectamine reagent (Invitrogen, Carlsbad, CA). The indicated amounts of pJAK2(1001–1013) and/or IFN-γ(95–132) peptide were added. One day later, the cell lysates were used to assay for firefly and Renilla luciferase using a dual luciferase assay kit from Promega according to the manufacturer’s instructions. Luciferase activity in relative luciferase units was standardized by dividing firefly by Renilla luciferase activity in each sample. Where indicated, murine SOCS-1 or SOCS-3 cDNA in CMV promoter-driven constructs was included in the transfection.
Measurement of intracellular and extracellular vaccinia virus formation
BSC-40 cells were seeded and grown overnight to confluency. Peptides at concentrations indicated were added to cells for 1 h followed by infection with vaccinia virus at a multiplicity of infection (moi) of 5 for 1 h. This was followed by addition of growth medium containing the same amount of peptides as before and incubation for times indicated. Supernatants were harvested, and the cells were scraped in 0.2 ml cell lysis buffer consisting of 50 mM Tris HCl (pH 7.5), 250 mM NaCl, 0.1% Nonidet P-40, 50 mM NaF, and 5 mM EDTA, followed by three cycles of freeze thawing and sonication. The virus titer in the supernatant (extracellular) and cell extracts (intracellular) was measured by plaque assay on BSC-40 cells.
Western blot analysis
Western blot analysis was carried out to determine whether pJAK2(1001–1013) had an effect on endogenous pSTAT1, STAT1, SOCS-1, IFN-α, or IFN-β levels. Cells were incubated with various concentrations of lipophilic pJAK2(1001–1013) or JAK2(1001–1013)2A for 30–60 min. The cells were washed in cold PBS and harvested in RIPA buffer containing protease and phosphatase inhibitor cocktails (Santa Cruz Biotechnology, Santa Cruz, CA). Protein concentration was measured using a BCA kit (Pierce), and lysates were resolved on 12% SDS-PAGE, transferred onto nitrocellulose membranes, and probed with various Abs. The membranes were then stripped and reprobed with the indicated anti-protein Abs. IFNAbs were from PBL Biomedical Laboratories (Piscataway, NJ), and pSTAT1 and SOCS Abs were custom made by GenScript (Pisactaway, NJ). STAT1 Abs were from Santa Cruz Biotechnology. Scanning of the band intensity was carried out using ImageJ software from the National Institutes of Health (Bethesda, MD).
IFN-β ELISA
L929 cells were treated with peptides for 30 or 60 min and then lysed with RIPA lysis buffer containing protease inhibitor cocktails (Sigma-Aldrich, St. Louis, MO). The cell lysates were analyzed with a murine IFN-β ELISA kit (PBL Biomedical Laboratories), following the manufacturer’s instructions. Briefly, cell lysates were plated onto plate strips for 1 h at room temperature. The strips were washed three times with wash buffer and then incubated with the Ab solution for 1 h at room temperature. After washing the strips three times, they were incubated with the HRP solution for 1 h at room temperature. The strips were washed three times and incubated with TMB substrate solution for 15 min at room temperature. The reaction was stopped by addition of stop solution. The absorbance was measured at 450 nm with a standard plate reader (Bio-Tek Instruments, Winooski, VT).
Measurement of vaccinia virus-specific cellular response by proliferation assay
Spleens from naive or recovered mice at times indicated were homogenized to single-cell suspension. Splenocytes (105 cells/well) were incubated with medium alone or medium containing UV-inactivated vaccinia virus at 37°C for 96 h. The cultures were then pulsed with [3H]thymidine (1 μCi/well; Amersham Biosciences, Piscataway, NJ) for 8 h before harvesting onto filter paper discs using a cell harvester. Cell-associated radioactivity was counted using a scintillation counter. Stimulation index refers to the incorporation in splenocytes cultured with test Ag divided by incorporation in splenocytes cultured with medium alone.
Measurement of vaccinia virus-specific cellular response by IFN-γ ELISPOT
CD4 depletion of splenocytes from naive or recovered mice was carried out by using the L3T4 Ab bound to Dynabeads (Invitrogen). ELISPOT assay was carried out by using a kit from Mabtech (Mariemont, OH). Briefly, CD4-depleted cells (105/well) were seeded in a microtiter plate, previously coated with an Ab to IFN-γ, and incubated in the absence or presence of purified vaccinia virus (moi 0.01) for 24 h at 37°C. After washing, diluted mAb was added and incubated for 2 h, followed by washing and addition of streptavidin-HRP. After 1 h at room temperature, the wells were washed and TMB substrate was added, washed, and the spots were counted.
Measurement of anti-vaccinia Ab response by ELISA
Microtiter plates were coated with 106 PFU purified UV-inactivated vaccinia virus (900,000 μJ/cm2 for 5 min in a DNA cross-linker) in 100 μl binding buffer (carbonate-bicarbonate [pH 9.6]) overnight at 4°C. Plates were blocked for 2 h at room temperature with PBS containing 5% FBS. Mouse sera was serially diluted in PBS containing 0.1% Tween 20 (wash buffer); 0.1 ml diluted serum was added to each well. The plate was incubated for 2 h at room temperature and washed three times with wash buffer. Peroxidase-conjugated goat anti-mouse IgA (α-chain specific) or IgG (γ-chain specific) (both from Santa Cruz Biotechnology), diluted in a volume of 0.1 ml, was added to each well, incubated for 1 h, and washed five times with wash buffer. o-phenylenediamine (OPD) in a volume of 0.1 ml was added and incubated for 15 min. The reaction was stopped by addition of 50 μl 3 N HCl. The OD at 490 nm was determined using a microtiter plate reader.
Measurement of vaccinia virus-specific neutralizing Abs
Plaque reduction assay was carried out to test the ability of Abs to inhibit viral infection of target cells. BSC-40 cells were seeded to confluency in a six-well plate the day before the assay. Sera obtained from mice on days indicated were heated at 56°C for 30 min to inactivate the complement. Purified vaccinia virus (100 PFU) was incubated with a known dilution of serum at 37°C for 1 h, followed by addition to BSC-40 cells. One hour later, the virus containing media was replaced with fresh medium containing 0.5% agarose and 0.01% neutral red. Two days later, the number of plaques was counted. The number of plaques in wells with vaccinia alone was taken as 100%. Percent reduction in other treatments carried out in triplicates was measured and is presented as average with SD.
Binding assays
Binding assays were performed as previously described (19) with minor modifications. pJAK2(1001–1013) or JAK2(1001–1013)2A were bound to 96-well plates in binding buffer (in 0.1 M carbonate-bicarbonate [pH 9.6]), at a final concentration of 3 μg/well. Wells were then washed in wash buffer (PBS containing 0.9% NaCl and 0.05% Tween 20), blocked with 2% gelatin and 0.05% Tween 20 in PBS for 1 h at room temperature, washed three times with wash buffer, and incubated with various concentrations of SOCS1-KIR or SOCS3-KIR for 1 h at room temperature in blocking buffer. Following incubation, wells were washed five times to remove unbound peptide. Bound peptides were detected by incubation with 1:500 dilution of Abs to SOCS1-KIR or SOCS3-KIR. After washing, detection was carried out with a goat anti-rabbit IgG-HRP conjugate, followed by the addition of OPD substrate and 2 N H2SO4. Absorbance was measured using a 450-microplate reader (Bio-Rad, Hercules, CA) at 490 nm.
Macrophage stimulation
Murine macrophage cells, RAW 264.7, were seeded on 96-well plates at a concentration of 5 × 106 cells/well in 200 μl volume and allowed to adhere. Lipo peptides, pJAK2(1001–1013), or the control peptide JAK2 (1001–1013)2A at 25 μM were then added to the wells and the cells incubated for 4 h, after which 2 μg/ml LPS or 0.1 μg/ml polyinosinic-polycytidylic acid (poly I:C) was added, and the cells were incubated for 3 d. Supernatants were transferred to fresh tubes and assayed for NO production using Griess reagent according to the manufacturer’s instructions (Alexis Biochemicals, Plymouth Meeting, PA).
Antiviral assay for EMCV
Antiviral assays for EMCV were performed by using a cytopathic effect reduction assay. Murine L929 cells (6 × 104 cell/well) were seeded in a 96-well plate and grown overnight to confluence for optimal growth. Various concentrations of IFN-γ, IFN-γ(95–132), IFN-γ(95–125), pJAK2(1001–1013), and JAK2(1001–1013)2A were added and incubated for 2 h, after which 200 PFU/well of EMCV was added to the plate and incubated. After 1 h, virus was removed, fresh media was added and incubated for 24 h. Cells were stained with 0.1% crystal violet. Unbound crystal violet was aspirated, and the plates were thoroughly rinsed with deionized water, blotted, and allowed to air dry. Plates were then scanned and analyzed using ImageJ 1.29 software (National Institutes of Health) to assess cell survival. Percentages of cell survival were determined by comparing survival for the experimental treatment groups with that for the virus-only control group.
Statistical analysis
All experimental data were measured for statistical significance by Student t test for peptide binding assays, by Kaplan-Meier survival curve and log-rank test for the mice studies, and by nonparametric (Wilcoxon-Mann-Whitney) tests for relative intensities in Western blots, using the GraphPad Prism software from GraphPad Software (San Diego, CA).
Results
pJAK2(1001–1013) protects mice against lethal vaccinia virus infection
Key to SOCS-1 inhibition of JAK2 kinase activity is the binding to the activation loop comprising the residues 1001–1013 of JAK2, with phosphorylated tyrosine at 1007 (Table I) (19). We synthesized a peptide corresponding to the activation loop of JAK2 with a phosphotyrosine at 1007, pJAK2(1001–1013), with a lipophilic group, palmitate, attached for cell penetration (19). We reasoned that pJAK2(1001–1013) would enhance innate and possibly adaptive immunity by preventing SOCS-1 from inhibiting IFN activity. We therefore examined pJAK2(1001–1013) for its ability to protect mice challenged with a lethal dose of the Western Reserve strain of vaccinia virus. C57BL/6 mice were injected i.p. with 10, 50, and 200 μg pJAK2(1001–1013) on days −2, −1, and 0 and challenged with 2 × 106 PFU vaccinia virus given intranasally (Fig. 1). Complete protection was observed with 200 μg SOCS-1 antagonist, whereas 50 and 10 μg resulted in 80 and 20% protection from death, respectively. Recovered mice were completely free of any disease symptoms for the 30 d observed. A control peptide that consisted of alanine substitutions for tyrosines at residues 1007 and 1008, JAK2(1001–1013)2A, was not protective, as all of the mice died by day 9. Thus, pJAK2(1001–1013) was able to completely protect mice against a lethal dose of vaccinia virus.
FIGURE 1.
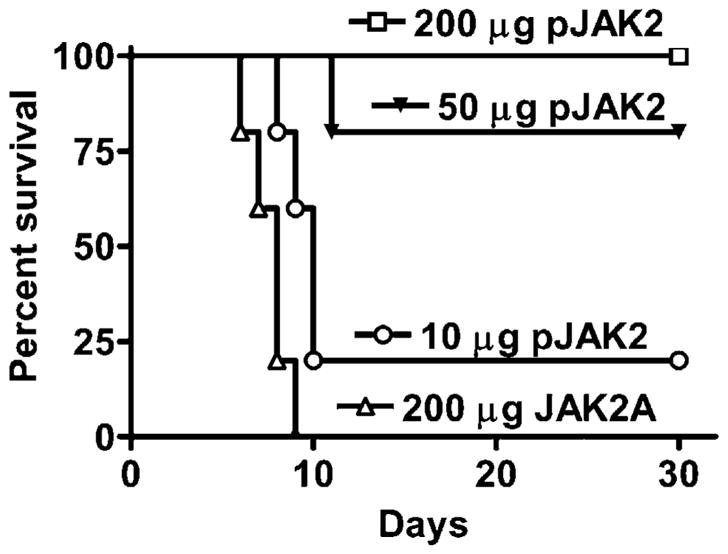
pJAK2(1001–1013) can rescue mice from an intranasal infection with vaccinia virus. Mice (C57BL/6, n = 5) were pretreated on days −2, −1, and 0 with 200 μg (□), 50 μg (▼), or 10 μg (○) lipo-pJAK2 (1001–1013) peptide or 200 μg (△) control peptide, lipo-JAK2(1001–1013) 2A. On day 0, vaccinia virus (2 ×106 PFU) was given intranasally. Survival of mice was followed over a period of 30 d. The significance of difference between different treatments was measured by log-rank survival method, which gave p values of 0.002, 0.002, and 0.02 for the administration of 200, 50, and 10 μg lipo-pJAK2(1001–1013) versus the control peptide, respectively.
To verify that palmitated (lipo)-pJAK2(1001–1013) was internalized by cells, we injected mice i.p. with 15 μg FITC–lipo-pJAK2 (1001–1013) or treated L929 cells in culture with 5 μM FITC–lipo-pJAK2(1001–1013). An equivalent amount of FITC alone was used as a control. After 2 h, peritoneal cells or L929 cells were examined by confocal fluorescence microscopy for FITC uptake. As shown in Fig. 2A, FITC–lipo-pJAK2(1001–1013), but not FITC alone was internalized by peritoneal cells. Similarly, FITC–lipo-pJAK2 (1001–1013) was specifically taken up by L929 cells as shown in Fig. 2B. Thus, the SOCS-1 antagonist is internalized where it has access to the SOCS-1 target.
FIGURE 2.

Lipo-pJAK2(1001–1013) is internalized by mouse peritoneal cells in vivo and by L929 fibroblast cells in culture. Lipo-pJAK2(1001–1013) was coupled to FITC as per Materials and Methods. A, Peritoneal cell uptake. Mice were injected i.p. with 15 μg FITC–lipo-pJAK2(1001–1013) or an equivalent amount of FITC alone. Peritoneal cells were harvested after 2 h, and the cells were examined by confocal fluorescent and contrast microscopy for FITC labeling. B, L929 cells uptake. Cells were incubated with 5 μM FITC–lipo-pJAK2(1001–1013) or an equivalent amount of FITC alone for 2 h, after which they were examined as above for uptake of FITC. Magnification ×20.
pJAK2(1001–1013) synergizes with an IFN mimetic in protecting mice against lethal vaccinia virus infection and in activation of GAS promoter element
We have developed small peptide mimetics of IFN-γ, based not on the classical model of IFN-γ–initiated signaling by extracellular interaction, but rather on direct intracellular signaling by IFN-γ. IFN-γ, its receptor subunit IFNGR1, and transcription factor STAT1α are transported to the nucleus of cells as a complex, where IFN-γ provides a classical polycationic nuclear localization sequence (NLS) for such transport (23–26). The C terminus of IFN-γ, represented in this study by the mouse IFN-γ peptide IFN-γ(95–132), was capable of forming a complex with IFNGR1 and STAT1α when introduced intracellularly and provided the required NLS for nuclear transport (24–26). The IFN-γ mimetic is also a potent inhibitor of vaccinia virus infection in mice (11).
To determine if the SOCS-1 antagonist can synergize with the IFN-γ mimetic to protect mice against vaccinia virus, C57BL/6 mice were treated on days −2, −1, and 0 with suboptimal amounts of the two peptides either alone or in combination. Both peptides contain a lipophilic group (palmitate) for cell penetration. Combined treatment i.p. with 10 μg pJAK2(1001–1013) and 5 μg IFN-γ(95–132) resulted in complete protection of the mice against 1 × 106 PFU intranasally administered vaccinia virus (Fig. 3A). Ten micrograms SOCS-1 antagonist plus 2 μg IFN-γ mimetic resulted in 60% survival. Five micrograms and 2 μg IFN mimetic alone resulted in 40 and 20% protection, respectively. Ten micrograms SOCS-1 antagonist alone resulted in 20% protection. Thus, we have shown in this study that SOCS-1 antagonist and IFN mimetic can synergize to protect mice against vaccinia virus infection. The synergy or protection described in this paper is similar to that originally observed by us with regard to type I and type II IFNs in induction of antiviral activity (27).
FIGURE 3.
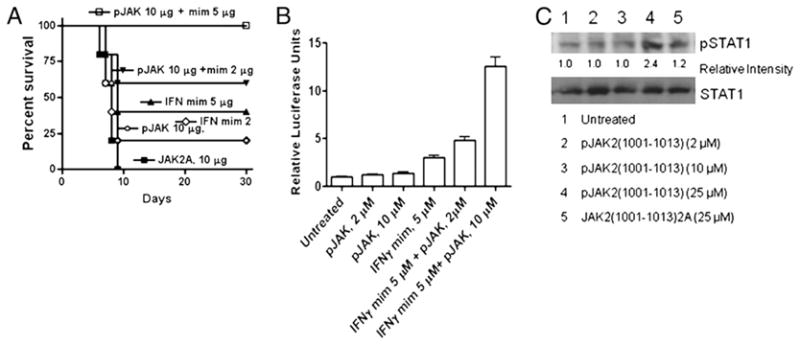
pJAK2(1001–1013) and IFN-γ(95–132) act synergistically. A, Mice were pretreated on days −2, −1, and 0 with 10 μg pJAK2 (○), 2 μg IFN-γ mimetic, indicated as mim (◇), 5 μg IFN-γ mimetic (▲), a control peptide JAK2(1001–1013)2A (■), or treated with a combination of 10 μg pJAK2 and 2 μg IFN-γ mimetic (▼) or 10 μg pJAK2 and 5 μg IFN-γ mimetic (□). On day 0, mice were infected intranasally with 1 × 106 PFU vaccinia virus. Survival of mice was followed over a period of 30 d. The significance of difference between different treatments was measured by log-rank survival method, which gave p values of 0.002, 0.04, 0.012, 0.017, and 0.017 for the administration of 10 μg pJAK2 and 5 μg IFN mimetic, 10 μg pJAK2 and 2 μg IFN mimetic, 5 μg of IFN mimetic, 2 μg IFN mimetic, and 10 μg pJAK2 versus the control peptide, respectively. B, IFN-γ mimetic and pJAK2(1001–1013) peptides activate GAS promoter element synergistically. WISH cells were cotransfected with plasmids expressing a GAS promoter element linked to a firefly luciferase and another plasmid constitutively expressing Renilla luciferase as an internal control, followed by addition of the peptides indicated. After overnight incubation, relative luciferase activity was measured and is expressed as average ± SD. C, pJAK2(1001–1013)-treated cells had increased levels of phosphorylated STAT1α. L929 cells were seeded onto six-well plates at 1 × 106 cells/well, grown overnight, and treated with pJAK2(1001–1013) (2, 10, 25 μM) or JAK2(1001–1013)2A (25 μM) for 1 h at 37°C. The cells were washed and lysed, and whole-cell extracts were resolved on 12% SDS-PAGE, transferred onto a nitrocellulose membrane, and probed with Abs to pSTAT1α or STAT1α. Similar results were obtained in three different experiments. Relative intensities of the pSTAT1α band are shown under the blot. As determined by the unpaired t test on nonnormalized readings, the intensity in the pJAK2-treated band versus the untreated had a p value of 0.0002.
We have shown in Fig. 3A that pJAK2(1001–1013) and the IFN-γ mimetic IFN-γ(95–132) synergized in protecting mice against vaccinia virus infection. To examine this synergy at the level of gene activation, we fused the GAS promoter with firefly luciferase and used this construct to transfect WISH cells. Cotransfection with a Renilla luciferase expression plasmid was carried out to normalize the expression of firefly luciferase, and activation is expressed as relative luciferase units. The transfected cells were incubated with pJAK2(1001–1013), IFN-γ(95–132), or the combination of the two peptides for 24 h, after which relative luciferase activity was measured. As shown in Fig. 3B, 10 μM SOCS-1 antagonist had minimal effect on reporter activation, whereas 5 μM IFN-γ mimetic was slightly more effective. Notably, 10 μM antagonist plus 5 μM IFN-γ mimetic together had a >4-fold effect on gene activation than did IFN-γ mimetic alone. This multiplicative effect is consistent with the synergy of protection against vaccinia virus seen in Fig. 3A. Thus, SOCS-1 antagonist and IFN-γ mimetic show synergy at the level of IFN-γ gene activation.
To determine if the SOCS-1 antagonist had any effect on STAT1α activation, we treated cells with pJAK2(1001–1013), or JAK2(1001–1013)2A and probed for phosphorylated STAT1α (Fig. 3C). Treatment with the SOCS antagonist at 25 μM increased pSTAT1α levels by >2-fold over untreated cells. Thus, pJAK2(1001–1013) enhances activation of STAT1α.
pJAK2(1001–1013) inhibits vaccinia virus replication as determined by a one-step growth curve
To determine if pJAK2(1001–1013) inhibits vaccinia virus replication as opposed to just inhibiting spread, we carried out a one-step growth curve experiment. BSC-40 cells were treated with lipophilic pJAK2(1001–1013) at 50 μM for 1 h, followed by infection with 5 moi of vaccinia virus to ensure simultaneous infection of all cells for a one-step growth curve. pJAK2(1001–1013) inhibited virus replication by ~92% as determined by intracellular virus yield (Fig. 4A) when compared with a variant, JAK2(1001–1013)2A, with alanine substituted for tyrosine at positions 1007 and 1008 (Table I). The alanine variant does not bind to KIR of SOCS-1 (L.D. Jager, R. Dabelic, and H.M. Johnson, unpublished observations). Inhibition was ~83% as determined by extracellular virus yield (Fig. 4B). Thus, the SOCS-1 antagonist pJAK2(1001–1013) inhibited vaccinia virus replication and not simply its release from cells. The fact that the alanine-substituted variant of the antagonist did not inhibit virus replication suggests that the specificity of the antiviral effects of the antagonist is directed at SOCS-1 in the cells.
FIGURE 4.

Time course of inhibition of vaccinia virus replication by pJAK2(1001–1013) by one-step growth curve. BSC-40 cells grown to confluency were left untreated, treated with lipo-pJAK2(1001–1013), or its alanine-substituted mutant JAK2(1001–1013)2A at 50 μM for 1 h. Cells were then infected with vaccinia virus at an moi of 5 for 1 h. After 1 h, the cells were washed and incubated in the presence of the same concentrations of peptides for the indicated times. Cell extracts (A) and supernatants (B) obtained from these were titrated for the amount of intracellular and extracellular virus, respectively. Note the difference of the scale on y-axis, indicating that there is less of extracellular virus than intracellular.
There was a dose-response effect in the inhibition of vaccinia virus replication by pJAK2(1001–1013) in which 1–25 μM of antagonist resulted in a dose-response reduction in yield of both intracellular (Fig. 5A) and extracellular (Fig. 5B) virus, with ~87% inhibition of yield at 25 μM. Thus, SOCS-1 antagonist pJAK2 (1001–1013) specifically inhibited vaccinia virus replication.
FIGURE 5.

pJAK2(1001–1013) inhibits vaccinia virus replication in a dose-dependent manner as determined by a one-step growth curve. BSC-40 cells were grown to confluency and left untreated or treated with the indicated amounts of lipo-pJAK2(1001–1013) or the alanine-substituted control peptide for 1 h. Cells were next infected with vaccinia virus at an moi of 5. After 1 h, the cells were washed and incubated in the presence of the same concentrations of peptides for 1 d. Supernatant and cell extracts obtained were titrated for the amount of intracellular (A) and extracellular (B) virus, respectively.
pJAK2(1001–1013)-treated cells had increased levels of endogenous IFN-β
For efficient induction of an antiviral state, cells contain low levels of spontaneously or constitutively produced IFN-β (20). A subtle increase in this low level of IFN-β plays an important role in a positive-feedback loop to increase type I IFN production and induction of a potent antiviral state in cells (21). To determine if pJAK2(1001–1013) affected the level of spontaneous IFN-β, we treated L929 fibroblasts with the SOCS-1 antagonist as well as the alanine-substituted variant, JAK2(1001–1013)2A. As seen in Fig. 6A, cells treated with 12 or 24 μM pJAK2(1001–1013) for 30 or 60 min showed a significant increase in IFN-β as determined by Western blot, whereas the alanine-substituted variant had little or no effect on IFN-β levels. To confirm these findings, we determined IFN-β protein levels by ELISA (Fig. 6B) and found that the IFN-β protein levels correspond to the Western blot data in Fig. 6A. Western blots for IFN-α showed that IFN-α levels were not altered (Fig. 6C). Western blots for SOCS-1 in the cells showed significant declines in pJAK2(1001–1013)-treated cells, whereas the variant, JAK2(1001–1013)2A, had no significant effect on the SOCS-1 protein levels (Fig. 6D). Thus, the increase in IFN-β in the cells corresponded to a decrease in SOCS-1 and would suggest that the SOCS-1 antagonist played a role in SOCS-1 degradation, probably via proteasomal degradation mediated through the SOCS box of SOCS-1 (14).
FIGURE 6.

pJAK2(1001–1013)-treated cells had increased levels of endogenous IFN-β. pJAK2(1001–1013) increases levels of endogenous IFN-β. L929 cells were seeded onto six-well plates at 1 × 106 cells/well, grown to confluency, and treated with peptides at varying concentrations for 30 or 60 min at 37°C. The cells were washed and lysed, and whole-cell extracts were resolved on 12% SDS-PAGE, transferred onto a nitrocellulose membrane, and probed with Abs to IFN-β (A), IFN-α (C), or SOCS1-KIR (D). The relative intensities were tested by a nonparametric test (Wilcoxon-Mann-Whitney) and found to be statistically significant with a p value of <0.05 for antagonist versus control for A and C. B, Intracellular IFN-β levels were determined for the cell lysates with an IFN-β ELISA kit, following manufacturer’s instructions. Values represent the means (± SEM) of triplicate wells from three independent experiments. Asterisks (*) indicate statistically significant differences when compared with untreated cells as determined by two-way ANOVA with Bonferroni posttests.
Potent adaptive immunity develops in mice that are protected against lethal vaccinia virus infection by pJAK2(1001–1013) treatment
It has recently been shown that suppression of SOCS-1 in dendritic cells (DCs) by small interfering RNA (siRNA) enhances the immune response (28, 29). The question arises therefore as to whether mice protected by the SOCS-1 antagonist developed protective adaptive immunity to subsequent infection with a lethal dose of vaccinia virus. Mice protected by pJAK2(1001–1013) were rechallenged 10 wk later with a second dose of 2 × 106 PFU vaccinia virus administered intranasally without additional pJAK2(1001–1013) treatment. All five mice in the rechallenged group were protected against the lethality of the virus without showing symptoms of distress (Supplemental Fig. 1A). Naive control mice all died by day 9. Thus, the pJAK2(1001–1013), in addition to providing protection against the first exposure to vaccinia virus, also allowed the mice to develop protective adaptive immunity to subsequent challenge with vaccinia virus.
Various immunological parameters were examined in the combined vaccinia virus and pJAK2(1001–1013)-treated mice. Splenocytes obtained 3 wk after virus challenge from protected mice as well as from naive mice were incubated with purified UV-inactivated vaccinia virus in a proliferation assay. The protected mice had a stimulation index of 8 at 3 wk postchallenge with the virus (Supplemental Fig. 1B). Splenocytes from the naive mice did not respond to vaccinia virus. The proliferation results suggest the induction of virus-specific CD4+ T cells. Splenocytes were also tested for the production of the cytokine IFN-γ by ELISPOT assay. Similar to the proliferative response, CD4-depleted splenocytes (105/well) exposed to vaccinia virus showed increased secretion of IFN-γ by ELISPOT at 3 wk after virus challenge (Supplemental Fig. 1C), suggesting the induction of vaccinia virus-specific cytotoxic CD8+ T cells. Control cells did not respond to the virus.
Sera from protected mice were examined for Abs over 4 wk. As shown in Supplemental Fig. 1D, IgA Abs peaked at 2–4 wk, but were significant out to 4 wk postinfection, whereas the IgG Ab response peaked over 2–4 wk postchallenge (Supplemental Fig. 1E). The IgA Abs are particularly relevant to the intranasal route of virus challenge. The Ab response also resulted in the production of neutralizing Abs that peaked at 2–4 wk and probably involved both IgA and IgG Abs to vaccinia virus, as shown in Supplemental Fig. 1F. Thus, mice protected by pJAK2(1001–1013) mounted both a strong cellular and humoral immune response to vaccinia virus.
pJAK2(1001–1013) exerts an adjuvant effect on the immune system
In addition to its inhibitory effects on virus replication in cells and related to the potent antivaccinia response, we were interested in determining possible adjuvant effects of SOCS-1 antagonist on the immune response. Accordingly, C57BL/6 mice were immunized i.p. with 50 μg BSA, treated i.p. with 200 μg pJAK2(1001–1013) on days −2, −1, and 0, and then assessed for enhancement of cellular and humoral immune responses. BSA is a relatively weak Ag in mice. Four weeks postimmunization, splenocytes from the mice were stimulated in cell culture with 0.5 μg BSA. As shown in Fig. 7A, untreated mice or mice given PBS mounted a weak proliferation response. By comparison, mice treated with pJAK2 (1001–1013) had an ~8-fold greater proliferative response to BSA. The humoral immune response as assessed by the serum IgG Ab response to BSA in the mice was also significantly enhanced in the pJAK2(1001–1013)-treated mice at 3 and 4 wk postimmunization (Fig. 7B). The SOCS antagonist can also enhance the Ab response to the T cell-independent Ag LPS. This is shown in Fig. 7C, in which the Ab response of mice immunized with LPS was significantly enhanced by i.p. injection of pJAK2(1001–1013). We previously showed that staphylococcal enterotoxin superantigens staphylococcal enterotoxin A (SEA) and staphylococcal enterotoxin B (SEB) enhanced T cell-dependent Ab production (30), but SEA/SEB did not enhance the anti-LPS response. SEA/SEB did enhance the Ab response to BSA, a T cell-dependent Ag. Thus, the SOCS antagonist has a direct effect on B cell function independent of Th cells.
FIGURE 7.

pJAK2(1001–1013) exerts an adjuvant effect at both cellular and humoral levels. A, Splenocyte stimulation. Mice (n = 5) were pretreated i.p. on day −2, −1, and 0 with pJAK2(1001–1013), control peptide JAK2(1001–1013)2A, or PBS. On day 0, 50 μg BSA was injected in mice in all groups, except the naive group. Four weeks later, isolated splenocytes (5 × 106/well) were seeded in quadruplicate and incubated with 0.5 μg BSA for 3 d with the addition of 1 μCi/well [3H]thymidine for the last 6 h, and its incorporation was measured. Data are representative of three individual experiments. B, IgG production. Mice (n = 5) were treated as in A. Sera obtained in the weeks indicated were diluted (1:1000) and added to microtiter plates. IgG Abs were measured in an ELISA assay. C, SOCS antagonist enhances T cell-independent Ab production. Mice (C57BL6, n = 3) were injected i.p. with T cell-independent Ag, LPS (50 μg each), or the T cell-dependent Ag BSA (50 μg). Some of the mice received SOCS antagonist (200 μg), the control peptide (JAK2A) (200 μg), or a combination of SEA/SEB (SAg, 25 μg each). A set of mice was also injected with BSA (50 μg) and SAg. Two weeks later, mice were bled. Sera were tested for IgG to LPS or BSA by ELISA. The secondary Ab used was anti-mouse IgG conjugated to HRP. After washing, substrate was added, and color was allowed to develop before reading absorbance at 490 nm. Comparison of LPS versus LPS and SOCS antagonist by Student t test resulted in p < 0.01 at 1/100 dilution. D, LPS stimulation. RAW264.7 cells (5 × 106/well) were seeded in triplicate and incubated overnight. The indicated amounts of pJAK2(1001–1013) or control peptide were added to the cells and incubated for 4 h, after which 2 μg/ml LPS was added, and the cells were incubated for 3 d. NO was measured by Griess reagent, and absorbance was read. E, Poly I:C stimulation. Murine macrophages (RAW264.7) were incubated with lipophilic pJAK2(1001–1013), or control peptide for 2 h, followed by stimulation with poly I:C at 0.1 μg/ml for 72 h. Culture supernatants were collected and nitrite concentration determined using Griess reagent. *p < 0.001.
At the level of macrophage TLR function, RAW264.7 cells treated with pJAK2(1001–1013) produced an ~5-fold increase in NO production upon LPS stimulation (via TLR4) compared with a control peptide (Fig. 7D). We also examined the effect of pJAK2 (1001–1013) on TLR3 activation. Poly I:C is a synthetic dsRNA that activates macrophages and DCs via TLR3 (31). TLR3 thus plays an important role in the antiviral responses to HSV-1, influenza virus, CMV, and respiratory syncytial virus, all of which have a dsRNA stage in their replication (32, 33). SOCS-1 negatively regulates TLR signaling at several stages including signaling by type I IFNs and by NF-κB transcription factor (14–17). Given the importance of TLR3 in the innate immune response against viruses, we treated the macrophage cell line RAW264.7 with poly I:C and determined the enhancing effect of the SOCS-1 antagonist pJAK2(1001–1013) on NO production. As shown in Fig. 7E, poly I:C at 0.1 μg/ml had a negligible effect on NO production, which was increased >20-fold by 25 μM pJAK2 (1001–1013). Alanine-substituted JAK2(1001–1013)2A had a negligible effect on NO production. Thus, the SOCS-1 antagonist enhanced the poly I:C activation of TLR3. The SOCS-1 effect would suggest that SOCS-1 induction has a regulatory effect on TLR3 activation and that the SOCS antagonist blocks this effect. These results demonstrate that pJAK2(1001–1013) has an adjuvant effect in terms of the cellular and humoral immune responses as well as in macrophage activation. Thus, in addition to direct inhibition of virus replication, the antagonist also has an adjuvant effect on the immune response.
SOCS-1 antagonist possesses antiviral activity against a picornavirus
EMCV is a rodent picornavirus, but can infect other species, including humans. In contrast to the complex, large double-stranded vaccinia virus, EMCV is a relatively simple, small plus-stranded RNA virus (6). It thus provides considerable contrast for assessing the antiviral effects of pJAK2(1001–1013). We treated L929 fibroblasts with the SOCS-1 antagonist prior to infection with EMCV, similar to the cell-culture treatments above with vaccinia virus. As shown in Fig. 8A, pJAK2(1001–1013) along with IFN-γ and the IFN-γ mimetic IFN-γ(95–132) significantly inhibited EMCV growth in cells treated with 200 PFU/well virus. Specifically, the antagonist and IFN-γ reduced CPE by ~50% at 24 μM and 100 U/ml, respectively, whereas the IFN-γ mimetic at 24 μM was completely protective. Of note, alanine-substituted SOCS-1 antagonist was only ~7% protective at 24 μM. This is consistent with the inability of the alanine-substituted antagonist to increase endogenous IFN-β protein in L929 cells. Synergy between pJAK2 (1001–1013) and IFN-γ(95–132) was observed at treatments with suboptimal concentrations in which 2 μM SOCS-1 antagonist and 5 μM IFN-γ mimetic combined completely protected L929 cells against EMCV, whereas separately, the peptides at these concentrations showed 20% or less protection (Fig. 8B). Thus, the SOCS-1 antagonist pJAK2(1001–1013) inhibits EMCV replication, similar to the inhibition of vaccinia virus replication.
FIGURE 8.

pJAK2(1001–1013) possesses antiviral activity against EMCV. A, L929 cells were treated with IFN-γ or different peptides for 2 h, after which 200 PFU/well EMCV was added. After 1 h, virus was removed, and media was added, followed by incubation for 24 h. Cells were stained with crystal violet, and plates were scanned. National Institutes of Health ImageJ software was used for analysis. B, Synergy between SOCS-1 antagonist and IFN-γ mimetic in inhibition of EMCV. pJAK2(1001–1013) at 2 μM and IFN-γ(95–132) at 5 μM together were incubated with L929 cells, after which the cells were infected as in A. C, L929 cells were incubated with the peptides in the presence or absence of 500 U/ml neutralizing Ab to IFN-β for 2 h, after which cells were infected with EMCV and processed as in A. D, pJAK2(1001–1013) protected mice from EMCV infection. Mice were injected daily i.p. beginning at day −2 with pJAK2(1001–1013) at 50, 100, and 200 μg and control peptide at 200 μg. On day 0, 50 PFU/mouse was injected i.p. Survival data are presented as Kaplan-Meier plots. The significances of difference were p < 0.005, p < 0.005, and NS for 200, 100, and 50 μg antagonist versus the control, respectively. E, Synergy in protection of mice infected with EMCV as for D above using suboptimal levels of pJAK2(1001–1013) (10 μg) and IFN-γ(95–132) (2 μg).
The increase in IFN-β in cells treated with the SOCS-1 antagonist raises the possibility that this IFN exerts its effects intracellularly and thus does not need to be secreted for subsequent interaction with the extracellular domain of the type I IFNR. To address this, we treated L929 cells with 24 μM pJAK2(1001–1013) in the presence or absence of neutralizing Abs to IFN-β prior to infection with 200 PFU EMCV. Complete protection by the antagonist was reduced to ~60% in the presence of a saturating level of anti–IFN-β Ab (Fig. 8C). Alanine-substituted antagonist was not protective. The data suggest that some of the increased intracellular IFN-β exerted its effects intracellularly. The ability of type I and type II IFNs to exert their effects intracellularly has recently been well established (24, 34).
Based on the antiviral effects of pJAK2(1001–1013) in tissue culture, we tested the therapeutic effects of the antagonist in a mouse model of lethal EMCV infection. C57BL/6 mice were treated i.p. with 50, 100, or 200 μg pJAK2(1001–1013) or 200 μg alanine-substituted antagonist, JAK2(1001–1013)2A, every day beginning at day −2. On day 0, the mice were challenged with 50 PFU EMCV per mouse and monitored daily for survival. As shown in Fig. 8D, mice treated with the alanine-substituted antagonist all died by day 5 after EMCV challenge. In contrast, mice treated with 100 and 200 μg pJAK2(1001–1013) showed 80% and 60% survival, respectively. Treatment with 50 μg pJAK2(1001–1013) resulted in 20% of mice surviving EMCV challenge. Thus, administration of pJAK2(1001–1013) at 100 and 200 μg daily resulted in significant protection of the mice.
Mice were infected with EMCV and treated with suboptimal doses of pJAK2(1001–1013) and IFN-γ(95–132) to assess synergy. As shown in Fig. 8E, treatment with pJAK2(1001–1013) at 10 μg and IFN-γ(95–132) at 2 μg resulted in 80% survival of infected mice, whereas pJAK2(1001–1013) treatment alone resulted in 40% survival, the same as that of PBS-treated mice. IFN-γ(95–132) alone resulted in 60% survival of mice. The synergy in this study with suboptimal doses of SOCS-1 antagonist and IFN-γ mimetic is similar to that shown for vaccinia virus, although the differential between protected and nonprotected mice was less dramatic. In general, we have found EMCV-infected mice to be more resistant to SOCS-1 antagonist and IFN mimetic therapy than those infected with vaccinia virus, probably because of the greater virulence of EMCV in these mice (9, 11).
SOCS-1 antagonist inhibits both SOCS-1 and SOCS-3 function
As indicated, the SOCS-1 antagonist pJAK2(1001–1013) binds to SOCS-1 via the SOCS1-KIR region (14). SOCS-3 also regulates immune functions and has been suggested to play the central role in inhibition of STAT3 activation in Th17 cells (35). It is thus of interest with respect to possible regulation by the SOCS-1 antagonist. As shown in Fig. 9A and 9B, both SOCS1-KIR and SOCS3-KIR bound similarly to the SOCS-1 antagonist. Because pJAK2 (1001–1013) represents the activation loop of JAK2, this would suggest that the antagonist would inhibit the function of both SOCS-1 and SOCS-3. Accordingly, we cotransfected the mouse fibroblast L929 cell line with the IFN-γ GAS promoter coupled to the luciferase reporter gene and with SOCS-1 or SOCS-3 cDNA expression plasmids. Reporter gene activity was inhibited in cells transfected with either SOCS cDNA, which was reversed by treatment of cells with the SOCS-1 antagonist pJAK2(1001–1013) (Fig. 9C). These results show that pJAK2(1001–1013) inhibits both SOCS-1 and SOCS-3 function. Consistent with the binding results, JAK2 associated with SOCS-3 in L929 cells (Fig. 9D). This, along with the reporter gene results in L929 cells, suggests that pJAK2(1001–1013) interaction with SOCS3-KIR occurred at the level of SOCS-3 protein.
FIGURE 9.

pJAK2(1001–1013) binding to SOCS1-KIR and SOCS3-KIR as determined by an Ab ELISA and its reversal of SOCS-1 and SOCS-3 mediated inhibition of GAS promoter activity. SOCS1-KIR (A) and SOCS3-KIR (B) bind to pJAK2(1001–1013). pJAK2 or control peptide IFN-γ(95–106) was immobilized at 3 μg/well in a 96-well plate. Following blocking, various concentrations of SOCS1-KIR or SOCS3-KIR were added, and the plates were incubated for 1 h. Following washing, 1:500 dilution of SOCS1-KIR or SOCS3-KIR Ab was added for 1 h incubation. Bound SOCS peptide was detected with a goat anti-rabbit IgG-HRP conjugate, followed by the addition of OPD substrate and 2 N H2SO4. Absorbance was measured in a plate reader. The statistical significance of the binding was tested by a paired t test, and p values of <0.001 and 0.0166 for the binding in A and B, respectively, were observed. C, pJAK2(1001–1013) reverses SOCS-1 and SOCS-3 inhibition of GAS promoter activity. L929 cells were transfected with a GAS promoter linked to firefly luciferase reporter gene and a control plasmid with constitutively expressed Renilla luciferase. Where indicated, SOCS-1– or SOCS-3–expressing plasmids were included in the transfection. A plasmid without a promoter attached to firefly luciferase was used as a control in the first bar indicated as empty vector. After 24 h of transfection, cell extracts were assayed for relative luciferase activities. pJAK2 peptide was used at 30 μM. D, JAK2 associates with SOCS-3 in cells. Whole cell extracts of L929 cells were subjected to IP with Abs to JAK2. Western blot analysis of the immunoprecipitates showed association of SOCS-3. IP with nonspecific IgG was run as a control and showed no SOCS-3 association. IP, immunoprecipitation.
Discussion
The process of activation of cells by cytokines such as IFNs involves JAK/STAT signal transduction and gene activation, including SOCS genes involved in modulation of the IFN signaling (14–18). Thus, the signal to induce an antiviral or inflammatory response by IFNs initiates a signal to control the extent of the response. Knockout of SOCS-1 gene in mice results in death within 3 wk of birth as a result of T cell-dependent multiorgan inflammatory disease (36). Unregulated IFN-γ activity plays a major role in the resultant lethality, because SOCS-1−/− IFN-γ−/− double knockouts survive, even though they show evidence of inflammatory disease (37). SOCS-1 deficiency in the hematopoietic lineage of cellular development is sufficient to cause early death (38). Thus, linking the signal for SOCS-1 induction to that of cytokine signaling related to innate and adaptive immune responses is a prerequisite for survival of the individual.
Regulation or control of the SOCS-1 modulatory arm of the immune response provides an approach to enhancement of the response to infectious agents as well as to weak Ags that are the target of tumor vaccine studies. In this regard, the regulatory role of SOCS-1 extends to DCs and Ag presentation. DCs are probably the most efficient cells at capturing, processing, and presentation of Ags. Recently, it was shown that knockdown of DC SOCS-1 by siRNA led to more effective cancer vaccination (29). Specifically, presentation of murine melanocyte differentiation Ag tyrosine-related Ag 2 by DCs transfected with SOCS-1 siRNA protected C57BL/6 mice against the well-established B16 melanoma tumor. Protection was not observed in DC vaccination in which the siRNA was disrupted by GFP. The enhanced antitumor immunity was accompanied by enhanced tyrosine-related Ag 2-specific CTLs in protected mice as assessed by IFN-γ ELISPOT and CTL responses. The authors concluded that regulation of Ag presentation by suppression of DC SOCS-1 showed promise for more effective tumor vaccines. The SOCS-1 siRNA treatment also enhanced HIV-1 envelope-specific CD8+ CTL responses, which suggests that suppression of SOCS-1 in DCs is of potentially general value for immune enhancement against AIDS (28). Related to the SOCS-1 siRNA studies is the observation that SOCS-1−/− mice are more resistant to viral infection than their wild-type counterparts due to enhanced type I IFN activity involving the IFNAR1 receptor subunit (39).
The development of the small peptide SOCS-1 antagonist was based on our observation that the KIR region of SOCS-1 binds directly to the activation loop of JAK2, contained in peptide pJAK2 (1001–1013), thus raising the possibility that the peptide could function as an antagonist of SOCS-1 (19). We subsequently demonstrated that pJAK2(1001–1013) possessed SOCS-1 antagonist activity by four different approaches (19). First, pJAK2(1001–1013) enhanced suboptimal IFN-γ activity. Second, LNCaP prostate cancer cells transfected for constitutive production of SOCS-1 protein had reduced activation of STAT3 by IL-6 treatment. pJAK2 (1001–1013) reversed the SOCS-1 effect. Third, pJAK2(1001–1013) enhanced IFN-γ activation of the luciferase reporter gene via the GAS promoter element. Fourth, pJAK2(1001–1013) enhanced Ag-specific splenocyte proliferation.
The antiviral effects of the SOCS-1 antagonist appear to operate through direct effects on the cell as well as by indirect effects in mice by enhancement of both the cellular and humoral arms of the immune system. We have shown in this study one mechanism by which pJAK2(1001–1013) may exert direct antiviral effects on cells. A well-recognized but not fully understood aspect of IFN function in cells is that most cells constitutively produce low levels of intracellular IFN-β that are thought to play a role in induction of an antiviral state in cells treated with type I and type II IFNs (20). pJAK2(1001–1013) increased the level of intracellular IFN-β in cells in which it induced the antiviral effect. In addition, pJAK2 (1001–1013) enhanced STAT1α activation. Related to this, it exerted a direct effect and synergized with IFN-γ in the protection of mice against vaccinia and EMCV infection and, at the level of transcription, synergized with an IFN-γ mimetic to induce gene activation through the GAS promoter. pJAK2(1001–1013) similarly inhibited EMCV growth. These observations, along with that of inhibition of HSV-1 replication in keratinocytes (12), demonstrate the broad antiviral activity of the SOCS antagonist. The connection of the SOCS-1 antagonist with IFN appears to reduce the regulatory restraints imposed by SOCS-1 under normal physiological conditions as shown by SOCS-1 reduction in cells treated with pJAK2 (1001–1013). The mechanism of the reduction is currently not known, but may be related to proteasomal degradation via the SOCS box of SOCS-1 (40).
TLRs are key players in both the innate and adaptive arms of host defense. With respect to TLR3 and virus immunity, treatment of the macrophage cell line RAW264.7 with poly I:C in the presence of pJAK2(1001–1013) resulted in significant enhancement of NO production. Classically, poly I:C induces type I IFNs, and these in turn activate myeloid cells to produce NO, reflecting the activation of these cells (31).
Related to the SOCS antagonist findings of this study, targeting of SOCS-1 expression in cardiac myocytes by expression of a dominant negative SOCS-1 increased myocyte resistance to acute cardiac injury as well as reduced mortality in coxsackievirus-infected mice (41). This transfection study with a genetic approach to inhibition of SOCS-1 function is analogous to the SOCS antagonist results presented in this paper. Our antagonist peptide results provide an approach to targeting SOCS-1 and SOCS-3 with a flexible drug therapeutic potential. Induction of SOCS-1 and/or SOCS-3 in other viral infections such as influenza (32) further suggest a role for the SOCS antagonist in modulation of these infections.
We made the original observation that type I and type II IFNs synergized to exert a multiplicative effect on the antiviral response (27), which is analogous to the above synergy of this study. This discovery predated our current insight into the complexities of the intracellular aspects of IFN signaling. The complexity of this synergism has been addressed in part by the demonstration that the type I IFNR component, IFNAR1, facilitates the activation of IFN-γ activated transcription factor (21). In fact, it functions also to enhance the induced type I IFN response via IFN response factors 3 and 7 (20). The synergism between IFNs in induction of antiviral and other effects suggests that there is considerable complexity to the IFN response.
The IFN-γ mimetic used in the synergism observed in this study was developed not on the classical model of IFN-γ initiated signaling by extracellular receptor interaction, but rather on direct intracellular signaling by IFN-γ (42). IFN-γ, its receptor subunit IFNGR1, and the transcription factor STAT1α are transported to the nucleus of cells as a complex where IFN-γ provides a classical polycationic NLS for such transport (24). The C terminus of IFN-γ, represented by the IFN-γ mimetic IFN-γ(95–132), was capable of forming a complex with IFNGR1 and STAT1α when introduced intracellularly and provided the NLS signaling for nuclear import. We have shown that IFNGR1 transport to the nucleus involves direct contact with the promoter region of IFN-γ–activated genes with associated increased gene activity, thus suggesting a transcriptional/cotranscriptional role of IFN-γ/IFNGR1 as well as a possible role in determining the specificity of IFN-γ action (25). Similar results were shown by others for the epidermal growth factor receptor in which transactivational activity of epidermal growth factor receptor was demonstrated (43). These findings address important issues of the nature of the unique specificity of signaling by ligands that use the JAK/STAT signaling pathways. Significantly, the IFN-γ mimetic is a logical result of these findings, but does not fit into the framework of the classical JAK/STAT model in which activated STATs are presented as the sole players in specific gene activation (25).
Intracellular signaling by type I IFN has been reported by several independent studies (34, 44). This includes both IFN-α and IFN-β, in which the IFNs were expressed in a nonsecretable form and induced an antiviral state without first undergoing secretion. This means that the antiviral activity was not dependent on IFN interaction with the extracellular domain of the IFNR complex. Although the involvement of ligand and receptor has not been developed with the type I IFN system to the extent of that with IFN-γ, there is evidence of some similarity. First, we have shown that the receptor unit IFNAR1 possesses a classical NLS and undergoes nuclear import in cells stimulated with IFN-β (45). Also, immunoprecipitation by importin β, a nuclear transporter (46), of cell extracts after type I IFN treatment, showed both constitutive and induced complex formation with IFNAR1. Additionally, GFP-coupled type I IFN, IFNAR1, or IFNAR2 all showed nuclear translocation after IFN treatment (C.M. Ahmed, E.N. Noon-Song, and H.M. Johnson, unpublished observations). Thus, these preliminary findings suggest that a complex sequence of events involving both ligands and receptors play a role in type I IFN signaling. Our demonstration that at least some of the increase in constitutive IFN-β in cells treated with pJAK2(1001–1013) remains intracellular would suggest that it plays an intracellular role in the signaling that is associated with SOCS-1 antagonist enhancement of the broad antiviral activity reported in this study.
Acknowledgments
We thank Dr. D. Hilton at the Walter and Eliza Hall Institute, Victoria, Australia, for the gift of murine SOCS-1 and SOCS-3 cDNAs in CMV promoter-driven constructs and Dr. Richard Condit at the University of Florida, Gainesville, FL, for the Western Reserve vaccinia virus.
This work was supported by National Institutes of Health Grants R01 AI 056152 and R01 NS 051245 to H.M.J.
Abbreviations used in this paper
- DC
dendritic cell
- EMCV
encephalomyocarditis virus
- GAS
IFN-γ activation site
- IP
immunoprecipitation
- KIR
kinase inhibitory region
- moi
multiplicity of infection
- Mu
murine
- NLS
nuclear localization sequence
- OPD
o-phenylenediamine
- poly I
C, polyinosinic-polycytidylic acid
- SAg
combination of staphylococcal enterotoxin A and staphylococcal enterotoxin B
- SEA
staphylococcal enterotoxin A
- SEB
staphylococcal enterotoxin B
- SH2
Src homology 2
- siRNA
small interfering RNA
- SOCS
suppressor of cytokine signaling
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Moss B. Poxviridae. In: Knipe DM, Howley PM, editors. Fields Virology. 3. Lippincott, Williams, and Wilkins; Philadelphia, PA: 2007. pp. 2905–2945. [Google Scholar]
- 2.McFadden G. Poxvirus tropism. Nat Rev Microbiol. 2005;3:201–213. doi: 10.1038/nrmicro1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith GL, McFadden G. Smallpox: anything to declare? Nat Rev Immunol. 2002;2:521–527. doi: 10.1038/nri845. [DOI] [PubMed] [Google Scholar]
- 4.Moss B, Shisler JL. Immunology 101 at poxvirus U: immune evasion genes. Semin Immunol. 2001;13:59–66. doi: 10.1006/smim.2000.0296. [DOI] [PubMed] [Google Scholar]
- 5.Alcamí A, Smith GL. The vaccinia virus soluble interferon-gamma receptor is a homodimer. J Gen Virol. 2002;83:545–549. doi: 10.1099/0022-1317-83-3-545. [DOI] [PubMed] [Google Scholar]
- 6.Racaniello VR. Picornaviridae: The viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 3. Lippincott, Williams, and Wilkins; Philadelphia, PA: 2007. pp. 795–838. [Google Scholar]
- 7.Rose NR. Viral damage or ‘molecular mimicry’-placing the blame in myocarditis. Nat Med. 2000;6:631–632. doi: 10.1038/76199. [DOI] [PubMed] [Google Scholar]
- 8.Yajima T, Knowlton KU. Viral myocarditis: from the perspective of the virus. Circulation. 2009;119:2615–2624. doi: 10.1161/CIRCULATIONAHA.108.766022. [DOI] [PubMed] [Google Scholar]
- 9.Mujtaba MG, Patel CB, Patel RA, Flowers LO, Burkhart MA, Waiboci LW, Martin JP, Haider MI, Ahmed CM, Johnson HM. The gamma interferon (IFN-gamma) mimetic peptide IFN-gamma (95–133) prevents encephalomyocarditis virus infection both in tissue culture and in mice. Clin Vaccine Immunol. 2006;13:944–952. doi: 10.1128/CVI.00021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahmed CM, Burkhart MA, Subramaniam PS, Mujtaba MG, Johnson HM. Peptide mimetics of gamma interferon possess antiviral properties against vaccinia virus and other viruses in the presence of poxvirus B8R protein. J Virol. 2005;79:5632–5639. doi: 10.1128/JVI.79.9.5632-5639.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahmed CM, Martin JP, Johnson HM. IFN mimetic as a therapeutic for lethal vaccinia virus infection: possible effects on innate and adaptive immune responses. J Immunol. 2007;178:4576–4583. doi: 10.4049/jimmunol.178.7.4576. [DOI] [PubMed] [Google Scholar]
- 12.Frey KG, Ahmed CM, Dabelic R, Jager LD, Noon-Song EN, Haider SM, Johnson HM, Bigley NJ. HSV-1-induced SOCS-1 expression in keratinocytes: use of a SOCS-1 antagonist to block a novel mechanism of viral immune evasion. J Immunol. 2009;183:1253–1262. doi: 10.4049/jimmunol.0900570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roizman BD, Knipe M, Whitley RJ. Herpes Simplex Viruses. In: Knipe DM, Howley PM, editors. Fields Virology. 3. Lippincott, Williams, and Wilkins; Philadelphia, PA: 2007. pp. 2501–2602. [Google Scholar]
- 14.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 15.Mansell A, Smith R, Doyle SL, Gray P, Fenner JE, Crack PJ, Nicholson SE, Hilton DJ, O’Neill LA, Hertzog PJ. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol. 2006;7:148–155. doi: 10.1038/ni1299. [DOI] [PubMed] [Google Scholar]
- 16.Yasukawa H, Misawa H, Sakamoto H, Masuhara M, Sasaki A, Wakioka T, Ohtsuka S, Imaizumi T, Matsuda T, Ihle JN, Yoshimura A. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J. 1999;18:1309–1320. doi: 10.1093/emboj/18.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi T, Takaesu G, Yoshimura A. Mal-function of TLRs by SOCS. Nat Immunol. 2006;7:123–124. doi: 10.1038/ni0206-123. [DOI] [PubMed] [Google Scholar]
- 18.Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. 2008;19:414–422. doi: 10.1016/j.semcdb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waiboci LW, Ahmed CM, Mujtaba MG, Flowers LO, Martin JP, Haider MI, Johnson HM. Both the suppressor of cytokine signaling 1 (SOCS-1) kinase inhibitory region and SOCS-1 mimetic bind to JAK2 auto-phosphorylation site: implications for the development of a SOCS-1 antagonist. J Immunol. 2007;178:5058–5068. doi: 10.4049/jimmunol.178.8.5058. [DOI] [PubMed] [Google Scholar]
- 20.Taniguchi T, Takaoka A. A weak signal for strong responses: interferon-alpha/β revisited. Nat Rev Mol Cell Biol. 2001;2:378–386. doi: 10.1038/35073080. [DOI] [PubMed] [Google Scholar]
- 21.Takaoka A, Mitani Y, Suemori H, Sato M, Yokochi T, Noguchi S, Tanaka N, Taniguchi T. Cross talk between interferon-γ and -α/β signaling components in caveolar membrane domains. Science. 2000;288:2357–2360. doi: 10.1126/science.288.5475.2357. [DOI] [PubMed] [Google Scholar]
- 22.Chen HM, Tanaka N, Mitani Y, Oda E, Nozawa H, Chen JZ, Yanai H, Negishi H, Choi MK, Iwasaki T, et al. Critical role for constitutive type I interferon signaling in the prevention of cellular transformation. Cancer Sci. 2009;100:449–456. doi: 10.1111/j.1349-7006.2008.01051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szente BE, Soos JM, Johnson HW. The C-terminus of IFN gamma is sufficient for intracellular function. Biochem Biophys Res Commun. 1994;203:1645–1654. doi: 10.1006/bbrc.1994.2375. [DOI] [PubMed] [Google Scholar]
- 24.Ahmed CM, Burkhart MA, Mujtaba MG, Subramaniam PS, Johnson HM. The role of IFNgamma nuclear localization sequence in intracellular function. J Cell Sci. 2003;116:3089–3098. doi: 10.1242/jcs.00528. [DOI] [PubMed] [Google Scholar]
- 25.Ahmed CM, Johnson HM. IFN-gamma and its receptor subunit IFNGR1 are recruited to the IFN-gamma-activated sequence element at the promoter site of IFN-gamma-activated genes: evidence of transactivational activity in IFNGR1. J Immunol. 2006;177:315–321. doi: 10.4049/jimmunol.177.1.315. [DOI] [PubMed] [Google Scholar]
- 26.Fulcher AJ, Ahmed CM, Noon-Song EN, Kwan RYQ, Subramaniam PS, Johnson HM, Jans DA. Interferon γ is recognised by importin α/β: enhanced nuclear localising and transactivation activities of an interferon γ mimetic. FEBS Lett. 2008;582:1569–1574. doi: 10.1016/j.febslet.2008.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fleischmann WR, Jr, Georgiades JA, Osborne LC, Johnson HM. Potentiation of interferon activity by mixed preparations of fibroblast and immune interferon. Infect Immun. 1979;26:248–253. doi: 10.1128/iai.26.1.248-253.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song XT, Evel-Kabler K, Rollins L, Aldrich M, Gao F, Huang XF, Chen SY. An alternative and effective HIV vaccination approach based on inhibition of antigen presentation attenuators in dendritic cells. PLoS Med. 2006;3:e11. doi: 10.1371/journal.pmed.0030011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen L, Evel-Kabler K, Strube R, Chen SY. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity. Nat Biotechnol. 2004;22:1546–1553. doi: 10.1038/nbt1035. [DOI] [PubMed] [Google Scholar]
- 30.Torres BA, Perrin GQ, Mujtaba MG, Subramaniam PS, Anderson AK, Johnson HM. Superantigen enhancement of specific immunity: antibody production and signaling pathways. J Immunol. 2002;169:2907–2914. doi: 10.4049/jimmunol.169.6.2907. [DOI] [PubMed] [Google Scholar]
- 31.Matsumoto M, Seya T. TLR3: interferon induction by double-stranded RNA including poly(I:C) Adv Drug Deliv Rev. 2008;60:805–812. doi: 10.1016/j.addr.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 32.Vercammen E, Staal J, Beyaert R. Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clin Microbiol Rev. 2008;21:13–25. doi: 10.1128/CMR.00022-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pothlichet J, Chignard M, Si-Tahar M. Cutting edge: innate immune response triggered by influenza A virus is negatively regulated by SOCS1 and SOCS3 through a RIG-I/IFNAR1-dependent pathway. J Immunol. 2008;180:2034–2038. doi: 10.4049/jimmunol.180.4.2034. [DOI] [PubMed] [Google Scholar]
- 34.Ahmed CMI, Wills KN, Sugarman BJ, Johnson DE, Ramachandra M, Nagabhushan TL, Howe JA. Selective expression of nonsecreted interferon by an adenoviral vector confers antiproliferative and antiviral properties and causes reduction of tumor growth in nude mice. J Interferon Cytokine Res. 2001;21:399–408. doi: 10.1089/107999001750277871. [DOI] [PubMed] [Google Scholar]
- 35.Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu B, Tato C, Yoshimura A, Henninghausen L, O’Shea JJ. Selective regulatory function of Socs3 in the formation of IL-17-secreting cells. Proc Natl Acad Sci USA. 2006;103:8137–8142. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marine JC, Topham DJ, McKay C, Wang D, Parganas E, Stravopodis D, Yoshimura A, Ihle JN, Ihle JN. SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell. 1999;98:609–616. doi: 10.1016/s0092-8674(00)80048-3. [DOI] [PubMed] [Google Scholar]
- 37.Brysha M, Zhang JG, Bertolino P, Corbin JE, Alexander WS, Nicola NA, Hilton DJ, Starr R. Suppressor of cytokine signaling-1 attenuates the duration of interferon gamma signal transduction in vitro and in vivo. J Biol Chem. 2001;276:22086–22089. doi: 10.1074/jbc.M102737200. [DOI] [PubMed] [Google Scholar]
- 38.Metcalf D, Alexander WS, Elefanty AG, Nicola NA, Hilton DJ, Starr R, Mifsud S, Di Rago L. Aberrant hematopoiesis in mice with inactivation of the gene encoding SOCS-1. Leukemia. 1999;13:926–934. doi: 10.1038/sj.leu.2401440. [DOI] [PubMed] [Google Scholar]
- 39.Zimmerer JM, Lesinski GB, Kondadasula SV, Karpa VI, Lehman A, Raychaudhury A, Becknell B, Carson WE., 3rd IFN-alpha-induced signal transduction, gene expression, and antitumor activity of immune effector cells are negatively regulated by suppressor of cytokine signaling proteins. J Immunol. 2007;178:4832–4845. doi: 10.4049/jimmunol.178.8.4832. [DOI] [PubMed] [Google Scholar]
- 40.Zhang JG, Farley A, Nicholson SE, Wilson TA, Zugaro LM, Simpson RJ, Mortiz RL, Cary D, Richardson R, Hausman G, et al. The conserved SOCs box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci USA. 1999;96:2071–2076. doi: 10.1073/pnas.96.5.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yasukawa H, Yajima T, Duplain H, Iwatate M, Kido M, Hoshijima M, Weitzman MD, Nakamura T, Woodard S, Xiong D, et al. The suppressor of cytokine signaling-1 (SOCS1) is a novel therapeutic target for enterovirus-induced cardiac injury. J Clin Invest. 2003;111:469–478. doi: 10.1172/JCI16491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson HM, Ahmed CM. Gamma interferon signaling: insights to development of interferon mimetics. Cell Mol Biol (Noisy-le-grand) 2006;52:71–76. [PubMed] [Google Scholar]
- 43.Lo HW, Hsu SC, Hung MC. EGFR signaling pathway in breast cancers: from traditional signal transduction to direct nuclear translocalization. Breast Cancer Res Treat. 2006;95:211–218. doi: 10.1007/s10549-005-9011-0. [DOI] [PubMed] [Google Scholar]
- 44.Shin-Ya M, Hirai H, Satoh E, Kishida T, Asada H, Aoki F, Tsukamoto M, Imanishi J, Mazda O. Intracellular interferon triggers Jak/Stat signaling cascade and induces p53-dependent antiviral protection. Biochem Biophys Res Commun. 2005;329:1139–1146. doi: 10.1016/j.bbrc.2005.02.088. [DOI] [PubMed] [Google Scholar]
- 45.Subramaniam PS, Johnson HM. The IFNAR1 subunit of the type I IFN receptor complex contains a functional nuclear localization sequence. FEBS Lett. 2004;578:207–210. doi: 10.1016/j.febslet.2004.10.085. [DOI] [PubMed] [Google Scholar]
- 46.Wagstaff KM, Jans DA. Importins and beyond: non-conventional nuclear transport mechanisms. Traffic. 2009;10:1188–1198. doi: 10.1111/j.1600-0854.2009.00937.x. [DOI] [PubMed] [Google Scholar]