

Summary
FoxO transcription factors and TORC1 are conserved downstream effectors of Akt. Here we unraveled regulatory circuits underlying interplay between Akt, FoxO, and mTOR. Activated FoxO1 inhibits mTORC1 by TSC2-dependent and TSC2-independent mechanisms. First, FoxO1 binds the promoter region of Sestrin3 (Sesn3) gene and directly elevates Sesn3 expression, which in turn inhibits mTORC1 activity in Tsc2-proficient cells. Second, FoxO1 elevates the expression of Rictor leading to increased mTORC2 activity that consequently activates Akt. In Tsc2-deficient cells, the elevation of Rictor by FoxO increases mTORC2 assembly and activity at the expense of mTORC1, thereby activating Akt, while inhibiting mTORC1. FoxO may act as rheostat that maintains homeostatic balance between Akt and mTOR complexes activities. Indeed, in response to physiological stresses, FoxO is required to maintain high Akt activity and low mTORC1 activity. Thus, under stress conditions, FoxO inhibits the anabolic activity of mTORC1-a major consumer of cellular energy, while activating Akt, which increases cellular energy metabolism, thereby maintaining cellular energy homeostasis.
Introduction
The serine/threonine kinase Akt delivers the effect of growth factors and other extracellular signals on intracellular metabolism in mammalian cells. The regulation of metabolism in general and energy metabolism in particular both at the cellular and organismal levels is the most evolutionarily conserved function of Akt. The two most evolutionarily conserved downstream effectors of Akt are the FoxO transcription factors and the target of rapamycin complex 1 (TORC1), which comprises Raptor and LST8. The mammalian FoxO transcription factors regulate cell proliferation and lifespan through the transcriptional activation of certain genes (Greer and Brunet, 2005), while the mammalian TORC1 (mTORC1) regulates cell growth and proliferation largely through the increase in protein synthesis. mTORC1 elevates mRNA translation by phosphorylating and activating S6K1 and by phosphorylating and inhibiting the eIF4E binding protein (4E-BP), a repressor of mRNA translation. (Mamane et al., 2006). Akt phosphorylates and inactivates the FoxO transcription factors, while it indirectly activates TORC1. mTORC1 and the Drosophila TORC1 are inhibited by the TSC1/TSC2 heterodimer acting as a GAP for the small GTPase Rheb, thereby inhibiting Rheb activity, which is required for TORC1 activation. Akt activates TORC1 by phosphorylating and inhibiting TSC2 activity (Inoki et al., 2002). The increase in energy metabolism by Akt also contributes to the activation of mTORC1. As consequence of energy stress, AMPK is activated, and in turn inactivates mTORC1 by phosphorylating and activating TSC2 (Inoki et al., 2003), and by phosphorylating Raptor (Gwinn et al., 2008). The increase in intracellular energy metabolism by Akt inhibits AMPK and thereby contributing to mTORC1 activation (Hahn-Windgassen et al., 2005).
Cells have evolved a feedback mechanism to maintain an optimal balance between the activities of Akt and mTORC1, and thus after mTORC1 is activated by Akt it institutes a negative feedback loop to inhibit Akt (Harrington et al., 2004). The interplay between Akt and mTOR is further complicated as mTOR is the catalytic component of another protein complex, mTORC2, which includes, in addition to mTOR, Rictor, Sin1 and mLST8. mTORC2 phosphorylates Akt at the Ser 473 in the hydrophobic motif and activates Akt (Sarbassov et al., 2005).
In C. elegans and Drosophila, interplays between TORC1 and FoxO have been reported. In C. elegans the FoxO orthologue, DAF-16, was shown to inhibit TORC1 activity by inhibiting the expression of Raptor (Jia et al., 2004), which is required for the activity of TORC1. In Drosophila, FoxO was shown to elevate the expression of 4E-BP, thereby negating the effect of TORC1 on mRNA translation (Puig et al., 2003). However, it is not known if such interplay operates in mammalian cells.
Results
Activated FoxO1 inhibits mTORC1 and activates Akt
To determine whether there is interplay between FoxO and mTORC1 in mammalian cells, we introduced an inducible FoxO1(AAA) mutant, in which the three Akt phosphorylation sites were mutated to alanines, fused in frame with the estrogen receptor (ER) ligand binding domain (FoxO1(AAA)-ER). The activity of FoxO1(AAA) is elevated upon addition of 4-hydroxy tamoxifen (4-OHT) that binds the ligand-binding domain and triggers a conformational change to activate the latent FoxO1(AAA)-ER.
Rat1a cells or Rat1a cells expressing constitutively active myristoylated Akt (Rat1a-mAkt) were infected with FoxO1(AAA)-ER retrovirus to establish polyclonal stable cell lines (Fig. 1A, Fig. S1A). Upon addition of 4-OHT to activate FoxO1(AAA)-ER, a reduction in mTORC1 activity was observed in Rat1a cells as measured by S6K1 phosphorylation and 4E-BP1 mobility shift (Fig. 1A, Fig. S1A). The induction of FoxO activity was confirmed by the increased in p27 protein level (Fig. S1A), a known downstream target of FoxO (Greer and Brunet, 2005). Interestingly, the reduction in mTORC1 activity, as assessed by S6K1 phosphorylation, occurred even in Rat1a (mAkt) cells that display higher S6K1 phosphorylation (Fig. S1A), suggesting that activated FoxO is able to reduce mTORC1 activity even when Akt is hyperactivated. To further confirm these results, cells were infected with adenovirus expressing FoxO1(AAA). After infection of NIH3T3 with adenovirus expressing FoxO1(AAA), we observed, like in Rat1a cells, a reduced p-S6K1 as well as S6 phosphorylation (Fig. 1B). The results were also substantiated in human cells. After infection of the human cell lines DOV13, MCF7, and U2OS, with adenovirus expressing FoxO1(AAA), we found that, the same as observed with rodent cells, mTORC1 activity, as assessed by S6K1 phosphorylation, was reduced (Fig. 1C). Notably, we also observed Akt activation, as assessed by Ser 473 phosphorylation of Akt (Fig 1B, C, and see below). Overall these results show that mTORC1 activity is decreased when cells possess high FoxO activity.
Figure 1. Activation of FoxO1 down-regulates mTORC1 but elevates Akt activity.
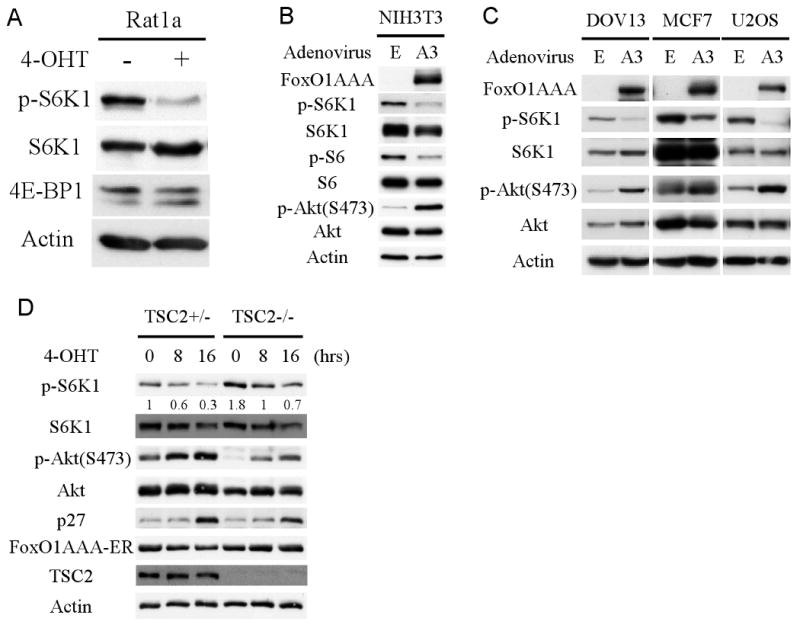
A. Activated FoxO1 represses mTORC1 activity. Rat1a cells stably expressing FoxO1(AAA)-ER were treated with 300 nM 4-hydroxy tamoxifen (4-OHT) to induce FoxO1(AAA) activity. Total proteins were extracted, adjusted for similar amounts of total S6K1, and subjected to immunoblotting with the indicated antibodies. B, C. FoxO1 down-regulates mTORC1 activity but elevates Akt activity. NIH3T3, DOV13, MCF7, and U2OS cells were infected with either FoxO1(AAA) adenovirus or control virus. Forty-eight hours after adenovirus infection, total protein was extracted and subjected to immunoblotting with the indicated antibodies. D. Tsc2+/- and Tsc2-/- MEFs stably expressing FoxO1(AAA)-ER were treated with 4-OHT to activate FoxO1. Sixteen hours after total proteins were extracted and subjected to immunoblotting.
mTORC1 activity is tightly regulated by multiple distinct signaling inputs such as growth factor stimulation, amino acid withdrawal, energy homeostasis, and hypoxia (reviewed in (Bhaskar and Hay, 2007)). Among all of the upstream regulators, tuberous sclerosis complex (TSC) is considered the most important regulator, which converges multiple upstream inputs to regulate mTORC1 activity. To further clarify the mechanism by which FoxO regulates mTORC1 activity, we stably expressed FoxO1(AAA)-ER in Tsc2+/- and Tsc2-/- MEFs (Fig. 1D). As expected, activation of FoxO1 led to a reduced mTORC1 activity in Tsc2-proficient cells (Fig. 1D). Surprisingly, however, we found that the activation of FoxO1 reduced mTORC1 activity even in Tsc2-deficient cells (Fig. 1D). Once again we observed a marked increase in Akt phosphorylation at Ser 473, both in Tsc2-proficient and Tsc2-deficient cells. Notably, activation of FoxO(AAA)-ER after addition of 4-OHT, could elevate Akt activity and reduce mTORC1 activity even when cells were cultured in medium with 0.1% serum (Fig. S1B). In the absence of FoxO(AAA)-ER, 4-OHT could not affect either mTORC1 or Akt activities (Fig. S1C).
Taken together, these results suggest that activated FoxO1 is reducing mTORC1 activity while increasing Akt activity, thereby uncoupling Akt and mTORC1 activities. Interestingly, after a long-term FoxO activation, the protein level of S6K1 were also reduced (Figs. 1 B, D, and S1A) even though the mRNA levels of S6K1 remain constant (data not shown), suggesting that FoxO can also regulate S6K1 activity downstream of mTORC1 by reducing S6K1 protein level.
FoxO1 inhibits mTORC1 activity by elevating Sesn3 expression
Recent studies showed that p53 downregulates mTORC1 activity by elevating the expression of Sestrin1 (Sesn1) and Sestrin2 (Sesn2), which in turn activate TSC2 by activating AMPK (Budanov and Karin, 2008). Sestrin gene family comprises three members, Sesn1, Sesn2, and Sesn3. Since we had previously shown that FoxO exclusively elevates the expression of Sesn3 (Nogueira et al., 2008), we examined whether FoxO downregulates mTORC1 activity via the upregulation of Sesn3. We first substantiated our previous results and showed that activation of FoxO1 specifically elevates the expression of Sesn3 but not Sesn1 or Sesn2 (Fig. 2A). In Tsc2+/- cells FoxO1 elevates Sesn3 mRNA level up to seven fold after 24 hours of FoxO1 activation. However, Sesn1 or Sesn2 show no obvious induction at the same time point. Similar results were obtained in MCF7 or U2OS cells infected with adenovirus carrying FoxO1(AAA). FoxO1(AAA) elevates Sesn3 mRNA level up to five fold in MCF7 cells, though FoxO1(AAA) activated Sesn1 very modestly and did not affect Sesn2 in MCF7 cells.
Figure 2. FoxO1 binds the promoter region of Sestrin3 gene and directly induces its mRNA levels.
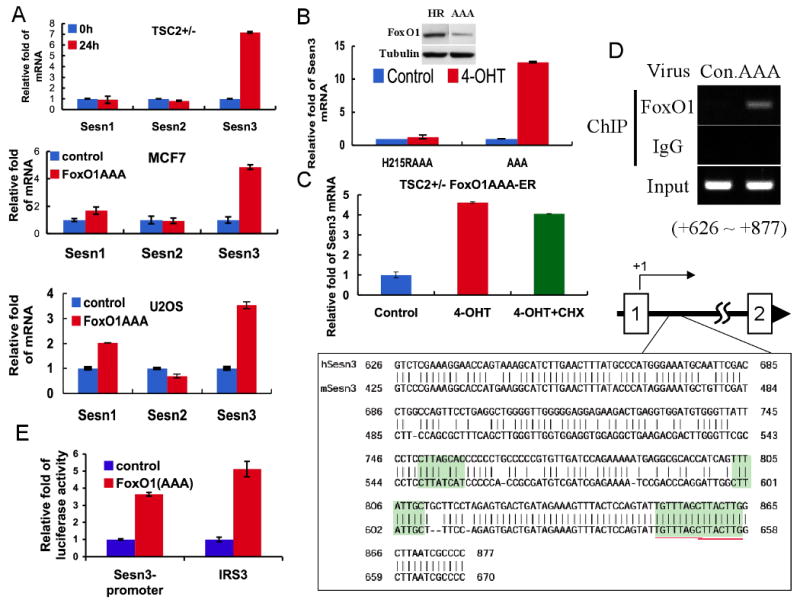
A. FoxO1 increases Sesn3 mRNA level. Total RNA was isolated from Tsc2+/- MEFs stably expressing FoxO1(AAA)-ER in the absence or presence of 4-OHT or from FoxO1(AAA) adenovirus or control adenovirus infected MCF7 and U2OS cells. mRNA levels were quantified by qRT-PCR as described in Experimental Procedures. Fold change in mRNA levels was calculated by normalizing to actin mRNA. B. DNA binding deficient FoxO1 cannot induce Sesn3 mRNA. Total RNAs were extracted from Tsc2+/-cells stably expressing FoxO1(AAA)-ER or FoxO1H215R(AAA)-ER in the absence or presence of 4-OHT. Relative mRNA levels were determined by qRT-PCR. Insert shows the levels of FoxO1AAA and FoxO1H215R(AAA). C. The induction of Sesn3 mRNA by FoxO1 does not require de novo protein synthesis. Tsc2+/- FoxO1(AAA)-ER cells were treated with cycloheximide (CHX) for one hour prior to the addition of 4-OHT to prevent de-novo protein synthesis. After addition of 4-OHT for four hours, relative levels of mRNA were determined by qRT-PCR. D. FoxO1 directly binds to the promoter region of the Sesn3 gene. U2OS cells were infected with FoxO1(AAA) adenovirus and then subjected to a ChIP assay using FoxO1 antibodies, as described in Experimental Procedures. The localization of the binding region within the first intron of Sesn3 gene is indicated schematically. Sequences show four predicted consensus FoxO binding sites highlighted within the binding region. The high homology of these sequences between the mouse and human Sesn3 genes is shown. E. FoxO1 regulates transcription from the Sesn3 promoter in a luciferase reporter assay. FoxO1(AAA) MEFs were transfected with pGL3-Sesn3 promoter or conventional pGL3-IRS3 (three copies of insulin response element of IGFBP1 promoter) construct as a positive control. Cells were treated with or without 4-OHT and subjected to Dual-Luciferase reporter assay (Promega). Luciferase activities were normalized to a co-expressed Renilla luminescent signal, and is shown as relative folds against control samples.
To identify the exact mechanism by which FoxO1 induces the expression of Sesn3 mRNA, we first determined whether the elevation of Sesn3 by FoxO1 is dependent on its DNA binding, and if Sesn3 is a direct target gene of FoxO. To verify whether the DNA binding activity of FoxO1 is required for the induction of Sesn3 mRNA, we used a mutant of activated FoxO1 in which histidine 215 was mutated to arginine, and that is impaired in DNA binding (Tang et al., 1999). The mutant H215R on the background of FoxO1 mutated in the three Akt phosphorylation sites was fused in frame to the ER ligand-binding domain to generate FoxO1H215R(AAA)-ER expressing retrovirus. The retrovirus was then used to generate a polyclonal cell line stably expressing the H215R mutant. As shown in Fig. 2B, upon addition of 4-OHT, the H215R mutant was unable elevate Sesn3 mRNA, suggesting that the elevation of Sesn3 mRNA by FoxO1 requires the DNA binding activity of FoxO1. To determine whether FoxO1 directly induces Sesn3 mRNA, we treated the cells with cycloheximide (CHX) prior to the induction of FoxO1(AAA)-ER activity by 4-OHT to prevent de-novo protein synthesis. As shown in Fig. 2C, CHX alone did not affect the level of Sesn3 mRNA, and did not inhibit the ability of FoxO1(AAA)-ER to induce Sesn3 mRNA, indicating that de novo protein synthesis is not required. These results imply that FoxO1 directly induces Sesn3 transcription. Indeed, we found, using chromatin immunoprecipitation (ChIP), that FoxO1 binds to a 250 bp region within the first intron of Sesn3 (Fig. 2D). This region possesses multiple predicted FoxO binding sites that are conserved in the human and the mouse genes (Fig. 2D). Cloning this region in a luciferase reporter plasmid show that it can activate transcription mediated by FoxO1 (Fig. 2D). The results indicate that FoxO1 binds directly to the promoter region of Sesn3 gene and transcriptionally upregulates Sesn3 mRNA expression.
We next determined whether Sesn3 could regulate mTORC1 activity. Human Sesn3 gene gives rise to two alternatively spliced variants, which encode for two proteins, a 53kDa long from (Sesn3L) and a 44 kDa short form (Sesn3S), which is missing the N-terminus 79 amino acids. Overexpressing of Sesn3L inhibited the phosphorylation of S6K1 in HEK293 cells. Sesn3S overexpression also inhibited S6K1 phosphorylation but to a less extent (Fig. 3A). We next overexpressed Sesn3 in Tsc2+/- and Tsc2-/-MEFs (Fig. 3B), and found that Sesn3L inhibited S6K1 phosphorylation only in Tsc2+/- but not Tsc2-/- cells. Thus, like what had been shown for Sesn1 and Sesn2 (Budanov and Karin, 2008), Sesn3 also downregulates mTORC1 activity through TSC2.
Figure 3. FoxO1 inhibits mTORC1 activity by elevating Sesn3 expression.
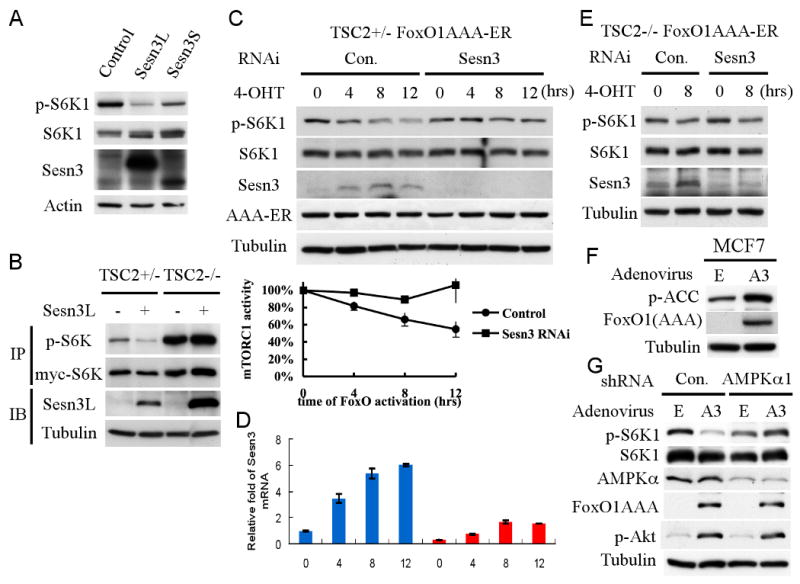
A. Overexpressing of Sesn3 downregulates mTORC1 activity. HEK293 cells were transiently transfected with Sesn3L, Sesn3S, or control plasmid. Forty-eight hour after transfection, total proteins were extracted and subjected to immunobloting with the specified antibodies. B. Sesn3 represses mTORC1 activity in a TSC2-dependent manner. Tsc2+/- and Tsc2-/- cells were transiently co-transfected with Sesn3 and myc-S6K1 plasmids. Twenty-four hour after transfection, total protein extracts were subjected to immunoprecipitation using 9E-10 myc-tag antibody followed by immunobloting with anti-p-S6K1, 9B-11 myc-tag, and anti-Sesn3 antibodies. C, D. Knockdown of Sesn3 attenuates the inhibition of mTORC1 activity by FoxO. Tsc2+/- FoxO1(AAA)-ER cells were transfected with Sesn3 or control RNAi. Forty-eight hour after transfection, cells were treated with 4-OHT and harvested at the indicated time points for protein (C) and RNA analyses (D). mTORC1 activity was deduced from three independent experiments and was quantified by the ratio of pS6K1/S6K1. E. The knockdown of Sesn3 has no effect in Tsc2-/- cells. Tsc2-/- FoxO1(AAA)-ER cells were transfected with Sesn3 or control RNAi. Forty-eight hour after transfection, cells were treated with 4-OHT and harvested at the indicated time points for protein and RNA analyses. F. Activated FoxO1 elevates AMPK activity as measured by pACC. MCF7 cells were infected with either FoxO1(AAA) (A3) adenovirus or control virus (E) for twenty-four hours. Cell lysates were subjected to immunobloting with indicated antibodies. G. The knockdown of AMPKα hindered FoxO1-mediated inhibition of mTORC1 activity without an effect on the increased Akt activity by FoxO1. MCF7 cells stably expressing AMPKα or control shRNA were infected with either FoxO1(AAA) (A3) adenovirus or control virus (E) for twenty-four hours. Whole cell lysates were subjected to immunoblotting with the indicated antibodies.
To verify if the elevation of Sesn3 by FoxO determines, at least in part, the ability of FoxO to attenuate mTORC1 activity, we knocked-down Sesn3, when FoxO was activated, and examined mTORC1 activity. Sesn3 RNA interference (RNAi) or control RNAi were transiently transfected into Tsc2+/-MEFs with FoxO1(AAA)-ER. Using Sesn3 RNAi, we reduced about 85% of Sesn3 mRNA as quantified by quantitative real-time PCR (qRT-PCR) at 0 hour time point (Fig. 3D). However, because FoxO forcefully elevates Sesn3 mRNA, the knockdown of Sesn3 was less effective at later time points. Nevertheless, p-S6K1 was monitored for up to 12 hours and the results revealed that the ability of FoxO1 to repress mTORC1 activity in the absence of Sesn3 is impaired (Fig. 3C). In Tsc2-/- cells, however, the knockdown of Sesn3 did not affect the reduced phosphorylation of S6K1 by the activated FoxO1 (Fig. 3E). We therefore concluded that the elevation of Sesn3 is one potential mechanism by which FoxO regulates mTORC1 activity in Tsc2-proficient cells.
The mechanism by which Sesn1 and Sesn2 inhibit mTORC1 was shown to be via the activation of AMPK preferentially towards TSC2 (Budanov and Karin, 2008). Consistently, activated FoxO1 elevated AMPK activity as measured by p-ACC (Fig. 3F), and the knockdown of AMPKα in MCF7 cells inhibited the effect of FoxO on mTORC1 activity (Fig. 3G). Notably, Akt activity was elevated even though mTORC1 activity was not significantly reduced, suggesting that the increase in Akt activity is not only due to the inhibition of the negative feedback loop normally operating through mTORC1 and S6K1 activation (see below). We also found that Sesn3 elevates AMPK activity, and the knockdown of AMPK or the inhibition of AMPK activity attenuated the effect of Sesn3 on mTORC1 activity (Fig. S2).
FoxO transcription factors upregulate mTORC1 activity by the elevation of Rictor
The results presented above clearly show that FoxO downregulates mTORC1 activity by the elevation of Sesn3 in a TSC2-dependent manner. However, as shown in Figures 1D 3E, and 3G, activation of FoxO could also downregulate mTORC1 activity, and elevate Akt phosphorylation in Tsc2-deficient cells, independently of Sesn3, suggesting additional TSC2-independent mechanism by which FoxO inhibits mTORC1, while elevating Akt activity.
To understand the mechanism by which FoxO exerts its effect independently of TSC2, we first examined the expression of mTORC1 and mTORC2 components by quantitative real time PCR (qRT-PCR) following FoxO1 activation. The elevation of p27 and the reduction of cyclin D1 mRNAs levels served as control for FoxO1(AAA) activity. As shown in Fig. 4A, none of the mRNAs encoding the various proteins comprising mTORC1 and mTORC2 were significantly affected by FoxO1 activation, except Rictor. Analysis of protein levels also did not show any significant change except for Rictor (Fig. 4B). Similar results were observed in Rat1a cells (Fig. 4C, D) and in Tsc2+/- MEFs (Fig. S3A). The increase in Rictor mRNA and protein levels by FoxO1 was also observed in MCF7 and U2OS cells (Fig. S3B). This increase is associated with an increase in mTORC2 levels (Fig. S3C).
Figure 4. FoxO1 upregulates Rictor mRNA and protein levels, which coincide with mTORC1 inhibition and Akt activation in Tsc2-/- cells.
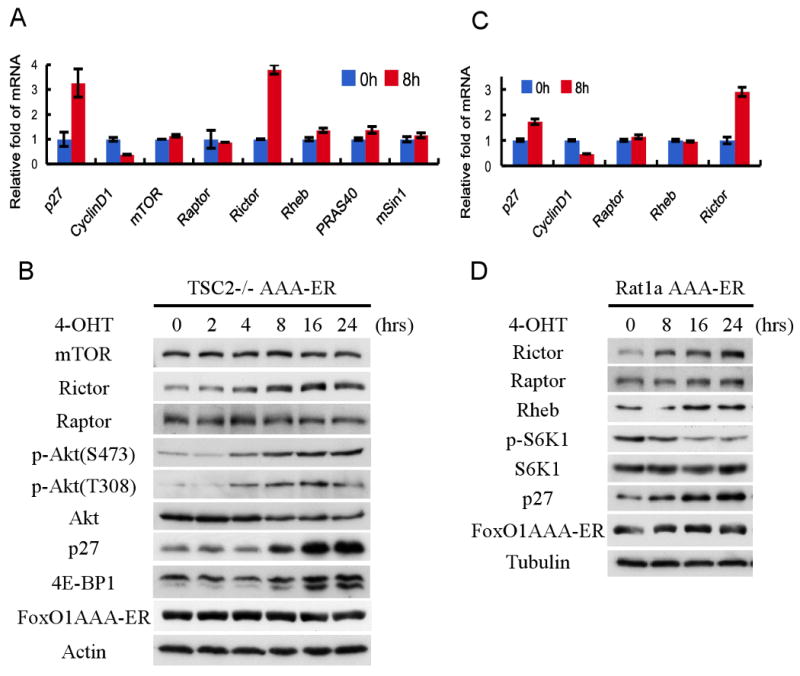
A, B. FoxO1 elevates Rictor mRNA and protein levels, but not other components of mTOR complexes in Tsc2-/- MEFs. Tsc2-/- FoxO1(AAA)-ER cells were untreated or treated with 4-OHT, and were harvested at the indicated time points for RNA and protein analyses. Total RNA was analyzed by qRT-PCR as described in Experimental Procedures. Total protein was subjected to immunoblotting with the specified antibodies. C, D. FoxO1 elevates Rictor mRNA and protein levels, but not other components of mTOR complexes in Rat1a cells. Experiments were performed as described above for Tsc2-/- MEFs.
Rictor is upregulated by activated FoxO1 at the mRNA level. At 8h after FoxO1 activation, we detected about 3-4 fold induction of Rictor mRNA, which is comparable to- or higher than the elevation p27 mRNA (Fig. 4A, C). Thus, FoxO1 elevates Rictor mRNA and protein levels in mammalian cells. However, unlike the induction of Sesn3 mRNA by FoxO, the elevation of Rictor mRNA appears to be independent of DNA binding as the H215R mutant was still able to elevate Rictor mRNA repress mTORC1 activity, and elevate Akt activity in Tsc2-/- cells (Fig. S3D, E). Therefore, unlike the induction of Sesn3 the DNA binding activity of FoxO1 is not required to elevate Rictor mRNA, suggesting that FoxO1 elevates Rictor mRNA through the association with another transcription factor that possesses the DNA binding activity. The onset of the increase in Rictor protein level was observed 4 hours following FoxO1 activation in Tsc2-/- cells, and remains high at the 24 hours time point (Fig. 4B and 4D). The elevation of Rictor protein level parallels the induction in Rictor mRNA level (Fig. S3F). Interestingly, in Tsc2-/- cells the timing of Rictor activation coincides with the change of both mTORC1 activity, and Akt activity, as measured by Ser473 and Thr308 phosphorylations (Fig. 4B). This is consistent with our observation that overexpression of Rictor could elevate Thr308 phosphorylation in addition to Ser 473 phosphorylation (Fig. S4A). This is in accord with previous results suggesting that Ser473 phosphorylation could enhance or sustain Thr308 phosphorylation (Yang et al., 2002). This is in contrast to what observed with cells deleted for Rictor from birth, in which the phosphorylation of Thr308 is elevated, possibly as a compensation mechanism (Guertin et al., 2006). However, acute knockdown of Rictor was shown to affect both Thr 308 and Ser 473 phosphorylations (Hresko and Mueckler, 2005; Sarbassov et al., 2005), which is consistent with results showing that pharmacological inhibitors of mTORC2 also inhibit Thr 308 phosphorylation (Feldman et al., 2009). Collectively, the results suggest that Rictor could be a potential candidate that determines FoxO mediated regulation of both Akt activity, and mTORC1 activity, particularly in Tsc2-deficient cells. Indeed, it was previously shown that the knockdown of Rictor not only reduces Akt activity but also elevates mTORC1 activity as measured by S6K1 phosphorylation (Sarbassov et al., 2004; Sarbassov et al., 2005). One potential mechanism by which Rictor could affect both mTORC1 and Akt activities is by competing with Raptor on the assembly of mTORC1 versus mTORC2. By increasing the assembly of mTORC2, at the expense of mTORC1, the elevation of Rictor could inhibit mTORC1 activity. Consequently, Akt activity would be elevated by both increasing mTORC2 activity and by decreasing mTORC1 activity that otherwise inhibits Akt activity via the negative feedback loop. However, as shown in Fig. 3G, Akt activity was elevated by FoxO even when S6K1 activity was not reduced, suggesting that FoxO can elevate Akt activity independently of the negative feedback loop. Furthermore, Akt activity was elevated in Tsc2-/- cells by H215R mutant of FoxO1 (Fig. S3E) that could not activate Sesn3 (Fig. 2B). To demonstrate that FoxO1 could elevate Akt activity through the elevation of Rictor even if the negative feedback loop is inhibited, we treated the cells with rapamycin in the absence or presence of activated FoxO1. We found that the combination of rapamycin and FoxO1 activation elevated Akt activity above of that observed with rapamycin alone (Fig. S4B).
To directly verify the possibility that changing Rictor levels could affect both mTORC2 and mTORC1 activities, we first knocked-down Rictor using lentivirus expressing Rictor short hairpin RNA (shRNA) in Tsc2-/- cells, which also express FoxO1(AAA)-ER. Seventy-two hours post lentivirus infection we detected about 60% reduction in Rictor protein level with concomitant increase in S6K1 phosphorylation as compared with cells infected with control virus (Fig. 5A). To determine if overexpression of Rictor could reduce mTORC1 activity we transiently overexpressed FoxO or Rictor in HEK293 cell together with S6K1, and observed a marked decrease in S6K1 phosphorylation as compared with control cells (Fig. 5B lanes 1 and 3). The reduction of mTORC1 activity by Rictor overexpression was similar to that observed following FoxO1(AAA) overexpression in HEK293 cell (Fig. 5B lanes 1, 2). Moreover, the overexpression of Rictor in HEK293 cells while increased its association with mTOR, decreased the association of Raptor with mTOR (Fig. 5C). Taken together, these results provide evidence that altering Rictor levels, the defining component of mTORC2, is able to affect mTORC1 activity.
Figure 5. Rictor regulates mTORC1 and mTORC2 activities downstream of FoxO.
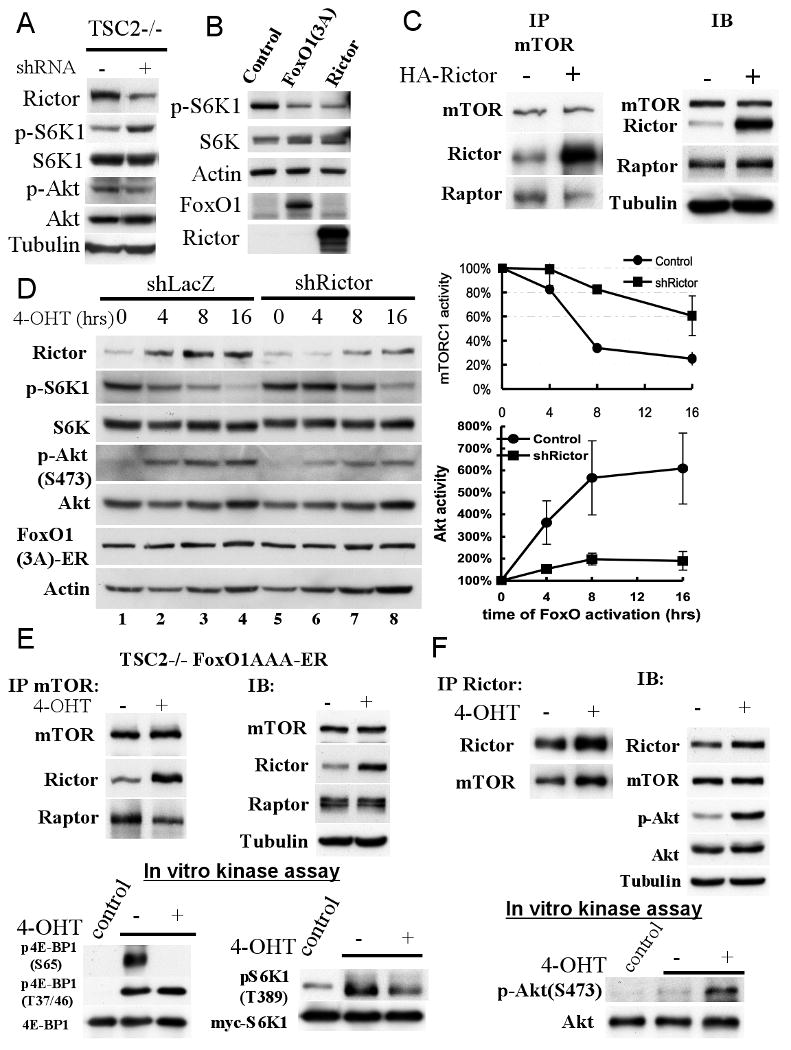
A. Knockdown of Rictor elevates mTORC1 activity. Tsc2-/- MEFs were infected with Lentivirus expressing shRNA targeting Rictor. Seventy-two hour after infection, cells were harvested for immunoblotting with the indicated antibodies. B. Overexpressing of Rictor represses mTORC1 activity to a similar extent as activated FoxO1. HEK293 cells were transiently transfected with FoxO1(AAA), Rictor, or control vectors. Twenty-four hour after transfection, total protein were extracted and subjected to immunoblotting. C. Overexpression of Rictor increases mTOR-Rictor interaction and decreases Raptor-mTOR interaction. HEK293 cells were transiently transfected with HA-Rictor or control DNA. Forty-eight hour after transfection, total protein were harvested and subjected to co-immunoprecipitation with mTOR antibody and immunoblotting with specific antibodies. D. The knockdown of Rictor in Tsc2-/- MEFs impairs the ability of FoxO1 to regulate mTORC1 and Akt activities. Tsc2-/- FoxO1(AAA)-ER cells were infected with lentivirus carrying shRNA targeting Rictor. Forty-eight hour after infection, cells were treated with 4-OHT. At the indicated time points cell lysates were prepared and subjected to immunobloting with the indicated antibodies. A representative immunoblot is shown. Right panels show quantification of mTORC1 and Akt activities after FoxO1 activation in the absence or presence of Rictor shRNA. Relative mTORC1 activity, as quantified by the ratio of pS6K1/S6K1, and relative Akt activity was quantified by the ratio pAkt/Akt in three independent experiments. F. Activated FoxO1 decreases Raptor-mTOR interaction, increases Rictor-mTOR interaction, and inhibits mTORC1 kinase activity in vitro. Tsc2-/- FoxO1(AAA)-ER cells were treated with or without 4-OHT and harvested for co-immunoprecipitation. Samples were immunoprecipitated by mTOR antibody and subjected to immunoblotting and in vitro kinase assays (bottom panels). In vitro kinase assays for mTORC1 activity was done using unphosphorylated S6K1 and recombinant 4E-BP1 (see Experimental Procedures). E. Activated FoxO1 increases mTORC2 kinase activity in vitro. Tsc2-/- FoxO1(AAA)-ER cells were treated with or without 4-OHT and harvested for co-immunoprecipitation. Samples were immunoprecipitated by Rictor antibody and subjected to immunoblotting and in vitro kinase assays (bottom panel). In vitro kinase assay for mTORC2 activity was done using unphosphorylated Akt1 (see Experimental Procedures).
To directly assess the effect of FoxO-induced Rictor on the elevation of Akt activity and on the inhibition of mTORC1 activity in Tsc2-/- cells, we determined whether the knockdown of Rictor could impair the ability of FoxO to attenuate mTORC1 activity and to elevate Akt activity. For this purpose we knocked down Rictor to the level that does not markedly affect basal S6K1 phosphorylation (Fig. 5D lanes 1 and 5). We followed the kinetics of Rictor elevation, S6K1 phosphorylation, and Akt phosphorylation in Tsc2-/- cells after activation of FoxO1 and the knockdown of Rictor. In cells expressing control shRNA, we observed a reduction of p-S6K1 following FoxO activation (Fig. 5D). The decline in S6K1 phosphorylation reached more than 60% at 8 hours after addition of 4-OHT (Fig. 5D). However, cells expressing Rictor shRNA displayed significantly slower rate in the decline of pS6K1 with only about 15% decline at 8 hours after addition of 4-OHT (Fig. 5D). The reduced ability of Rictor knockdown to overcome the effect of FoxO on S6K1 phosphorylation, at later time points, is probably due to the continuous elevation of Rictor mRNA by FoxO (Fig. 5D lane 8). Thus, knocking down Rictor hinders the ability of FoxO to downregulate mTORC1 activity. The results suggest that Rictor elevation is required for the effect of FoxO on mTORC1 in Tsc2-deficient cells.
Rictor is the primary determinant of mTORC2 activity as a kinase that phosphorylates Akt at Ser 473 (Sarbassov et al., 2005). To evaluate whether Rictor plays a role in FoxO-mediated Akt activation, we also followed Akt phosphorylation after Rictor knockdown. As shown in Fig. 5D, after the activation of FoxO1, for six hours, Ser 473 phosphorylation of Akt was elevated about six fold. However, the knockdown of Rictor significantly attenuated the increase in Akt phosphorylation, indicating that Rictor elevation is required for the effect of FoxO on Akt activity.
To determine if activated FoxO1 could increase the assembly of mTORC2, while decreasing the assembly of mTORC1, we performed a co-immunoprecipitation to pull down both complexes by mTOR antibody. Upon activation of FoxO1 we observed, as expected, elevated Rictor protein level in total cell extract (Fig. 5E). The same cell lysates were incubated with mTOR antibody to pull down both mTORC1 and mTORC2 complexes. The immunoprecipitates were subjected to immunoblotting with Rictor antibody and showed increasing amounts of Rictor-bound mTOR after FoxO1(AAA) activation suggesting an increase in mTORC2 formation (Fig. 5E). At the same time we observed a reduction of Raptor-bound mTOR suggesting a decrease in mTORC1 assembly. Thus, our data suggest that, by elevating the expression of Rictor, FoxO increases mTORC2 activity at the expense of mTORC1 thereby elevating Akt activity and reducing mTORC1 activity. To further corroborate the results, the immunoprecipitates were subjected to mTORC1 kinase assays. We first used S6K1 as a substrate and found that the phosphorylation of S6K1 was reduced to a similar extent as was observed in vivo (Fig. 5E). Interestingly using recombinant 4E-BP1, as a substrate, we found that after 10 min of incubation the phosphorylation of Thr37/46 was only modestly reduced (data not shown). After 20 min of incubation there was no difference in the in vitro phosphorylation of Thr 37/46 before and after FoxO1 activation. However, we reproducibly observed a dramatic reduction in the in vitro phosphorylation of Ser 65 following FoxO1 activation (Fig. 5E). This reduction in the in vitro phosphorylation of Ser 65 is much more pronounced than the reduction in the amount of Raptor associated with mTOR. Although we do not understand this discrepancy, one possibility is that FoxO only affects the rapamycin sensitive activity of mTORC1, since Thr37/46 phosphorylation is not affected by rapamycin while Ser 65 phosphorylation is rapamycin sensitive (Feldman et al., 2009) (see also Discussion).
To determine that indeed FoxO activation leads to an increase in mTORC2 activity, we conducted in vitro mTORC2 kinase assay using Akt as substrate. Because the level of mTORC2 is very low in TSC2-/- cells, and because the conditions for the extraction and immunoprecipitation of mTORC1 are not optimized for mTORC2 kinase assay, we could not perform mTORC2 kinase assay using the mTOR immunoprecipitates used in Fig. 5E. Therefore, immunoprecipitation with anti-Rictor antibodies was performed before and after induction of FoxO1 (Fig. 5F). Clearly mTORC2 kinase activity was increased to a similar extent as the observed increase in Akt phosphorylation by immunobloting (Fig. 5F). Collectively these results support the notion that, by elevating the expression of Rictor, FoxO could inhibit mTORC1 activity while increasing mTORC2 and Akt activities.
FoxO-deficiency activates mTORC1 activity while reducing Akt activity
The results shown above employed gain of function approaches to determine FoxO regulation of mTORC1 and Akt activities. We therefore employed a loss of function approach to further substantiate our results. As we have shown previously, Sesn3 levels are reduced in FoxO3a-/- MEFs, as FoxO3a is the major FoxO isoform, expressed in MEFs (Nogueira et al., 2008). However, in standard culture conditions of 10% FBS and 25mM glucose, we observed only a modest increase of p-S6K1 and a modest decrease of p-Akt in FoxO3a-deficient MEFs, and only after serum stimulation, as compared FoxO3a-wild type MEFs (data not shown). We therefore knocked down FoxO1 in FoxO3a-/- MEFs (Fig. 6A, Fig. S5A). By transiently transfecting FoxO1 RNAi into FoxO3a-/- MEFs, we reduced about 80% of FoxO1 mRNA and protein levels (Fig. 6A). In response to the knockdown of FoxO1 we observed an elevated S6K1 phosphorylation, indicating an increase in mTORC1 activity and a decrease in Akt phosphorylation, indicating an increase in mTORC1 activity (Fig. 6A). When FoxO3a-/- cells were cultured in 5mM glucose we observed a marked increase in S6K1 phosphorylation and a decrease in Akt phosphorylation as compared with WT cells (Fig. 6B). These results suggest that in the presence of high serum concentration (10% FBS) and high glucose level (25mM), Sesn3 level should be reduced below a certain threshold level in order to have a significant effect on mTORC1 activity. We also knocked down FoxO1 and FoxO3a in Tsc2-/- MEFs (Fig. S5B). Although Tsc2-/- cells maintain high endogenous mTORC1 activity, knocking down FoxO1 or both FoxO1 and FoxO3a reduced Rictor levels and elevated S6K1 phosphorylation as compared with control RNAi transfected cells (Fig. 6C and Fig. S5C). The reduction in Rictor level after the knockdown of FoxO1 in Tsc2-/- cells is also associated with a decrease in Akt phosphorylation and inn the amounts of Rictor that interacts with mTOR (Fig. 6C). Notably, the decrease in Rictor level by the knockdown of FoxO1 was not further decreased by the additional knockdown of FoxO3a (Fig. S5C), reinforcing the possibility that Rictor expression is primarily regulated by FoxO1. Taken together these results further confirmed that FoxO1 regulates Rictor expression in mammalian cells.
Figure 6. FoxOs-deficiency affects mTORC1 and mTORC2 activities.
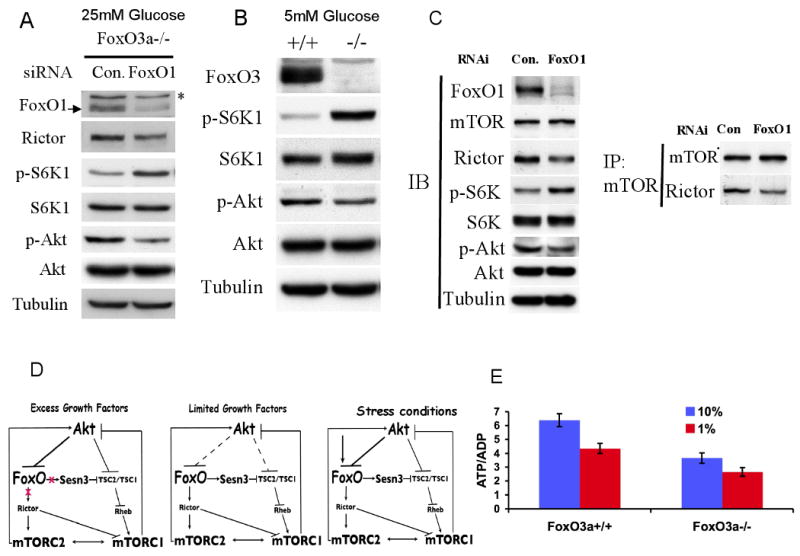
A. The knockdown of FoxO1 in FoxO3a-/- MEFs, under standard culture conditions, increases mTORC1 activity and decreases mTORC2 activity. FoxO3a-/- MEFs were transiently transfected with FoxO1 or control RNAi. Forty-eight hour after transfection, total protein lysates were subjected to immunobloting. mTORC1 and mTORC2 activities were deduced from the levels of Ser371 phosphorylation on S6K1 and of Ser473 on Akt respectively. B. mTORC1 activity is higher while Akt activity is lower in FoxOa3-/- cells than in control wild-type cells when cultured at low glucose levels. C. Knocking down of FoxO1 in Tsc2-/- cells decreases Rictor expression and Rictor-mTOR complex, decreasing Akt activity while increasing mTORC1 activity. TSC2-/- MEFs were transiently transfected with FoxO1 or control RNAi. Forty-eight hour after transfection, total proteins were harvested and subjected to co-immunoprecipitation with mTOR antibody and immunoblotting with specific antibodies. D. Schematic illustration depicting interplays between Akt, FoxO, and mTOR. In the excess of growth factors and nutrients, Akt inactivates FoxO and activates mTORC1. mTORC1 then induces a negative feedback loop to inhibit Akt. The inhibition of Akt by mTORC1 would activate FoxO to elevate Sesn3 and Rictor, which in turn inhibit mTORC1 and subsequently activate Akt, thereby maintaining a homeostatic balance between Akt and mTOR activities. In limited growth factors and nutrients or other stress conditions, the ability of Akt to inhibit FoxO is impaired or FoxO is activated regardless of Akt, thereby further elevating Sesn3, and Rictor to suppress mTORC1 activity and reduce energy consumption, while maintaining Akt activity to increase energy production. E. Intracellular ATP/ADP ratio in FoxO3a-proficient and FoxO3a-deficeint cells.
We propose that under growth factor limitations or other physiological stress conditions when FoxO transcription factors are activated, they inhibit mTORC1 activity while increasing Akt activity (Fig. 6D). Protein synthesis, ribosomal biogenesis, and lipid metabolism, which are mediated by mTORC1 (Porstmann et al., 2009), are major consumers of cellular energy metabolism, whereas Akt is a major positive regulator of cellular energy metabolism. Therefore, it is possible that the inhibition of mTORC1 and the subsequent activation of Akt by FoxO were evolved first, to maintain homeostatic balance between Akt and mTOR complexes activities, and second, to maintain cellular energy homeostasis under stress conditions. In support of this notion, we found that in conditions in which FoxO3a-/- cells, display higher activity of mTORC1 and lower activity of mTORC2 as compared to WT cells, they also have a significantly reduced ATP/ADP ratio than WT cells (Fig. 6E).
In certain stress conditions when FoxO is activated it could rescue cells from energy crisis prior to its full manifestation and the full activation of AMPK. By elevating Sesn3, FoxO reduces energy consumption, while by elevating Akt activity it increases energy production. These effects of FoxO are likely transient and occur in a temporal manner because once Akt is activated, it inhibits FoxO and activates mTORC1, and thus returning the cells to a normal status (Fig. 6D), unless FoxO is activated regardless of Akt status. This could occur under stress conditions that activate FoxO, overcoming its inhibition by Akt phosphorylation (Fig. 6D). For example, it was shown that the phosphorylation of FoxO by Jun-N-terminal kinase (JNK) (Essers et al., 2004) or by the mammalian Ste20-like kinase (MST1) (Lehtinen et al., 2006), after oxidative stress, induces its translocation to the nucleus.
To test this hypothesis experimentally we used isogenic pair of WT and FoxO3a-/- MEFs. We first examined the relative activities of mTORC1 and Akt in these cells under reduced serum concentrations, and 5mM instead of 25mM glucose. As shown in Fig. 7A, in all serum concentrations that were tested, the relative activity of Akt was higher than the relative activity of mTORC1 in WT cells. In contrast, in FoxO3a-/- cells, the relative activity of mTORC1 was higher than the relative activity of Akt in all serum concentrations. Importantly, at the lowest serum concentration (1% FBS), mTORC1 activity was markedly reduced relative to Akt activity in WT cells, while in FoxO3a-/- cells mTORC1 activity remains relatively higher and to a similar extent as in higher serum concentrations. The relative mTORC1/Akt activity in the two cell lines, in the different serum concentrations, is correlated with levels of Sesn3 expression (Fig. S6A). These results support the idea that FoxO serves as rheostat that coordinates energy consumption (mTORC1 activity) and energy production (Akt activity) in particular when growth factors are limited. To further verify this possibility we followed the kinetics of mTORC1 and Akt activities immediately after exposure to 1% FBS. Clearly, Akt and mTORC1 activities are reduced with a similar kinetics in WT cells, but not in FoxO3a-/- cells (Fig. 7B).
Figure 7. FoxO is required to maintain homeostatic balance between Akt and mTORC1 activities under stress conditions.
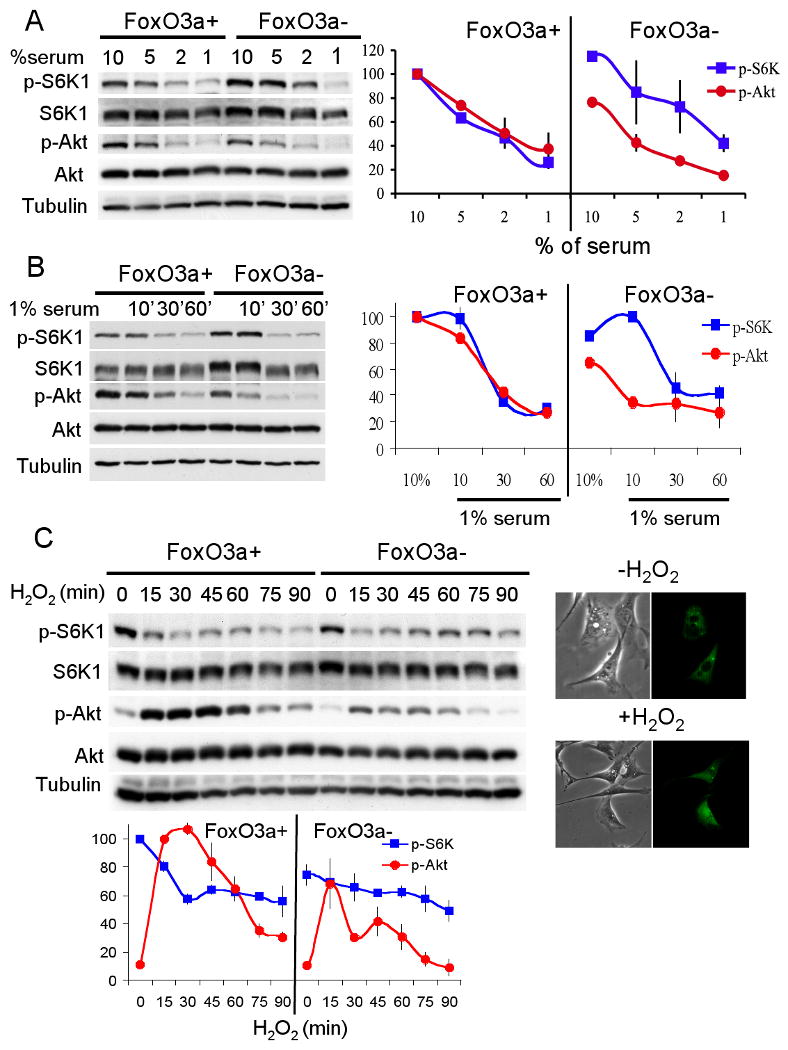
A. FoxO regulates the relative activities mTORC1 and Akt in response to growth factor limitations. Immortalized isogenic wild-type or FoxO3a-/- MEFs were cultured in DMEM medium contained different percentage of serum for twenty-four hours. Whole cell lysates and mRNA were extracted and subjected to immunoblotting. Representative immunoblot is shown (upper panel). Relative mTORC1 activity (p-S6K/S6K) and Akt activity (p-Akt/Akt) were quantified from two independent experiments (bottom panel). B. Immortalized FoxO3a wild-type and knockout MEFs were cultured in 10% serum containing medium for overnight. Cells were placed in 1% serum containing medium for the indicated time points. Total cell lysates were extracted and subjected to immunoblotting analyses. Representative immunoblot is shown (upper panel). Relative mTORC1 activity (p-S6K/S6K) and Akt activity (p-Akt/Akt) were quantified from two independent experiments (bottom panel). C. FoxO regulates the relative activities mTORC1 and Akt in response to oxidative stress. Immortalized isogenic wild-type or FoxO3a-/- MEFs were treated with 0.5 mM H2O2, and were harvested at the indicated time points at 26°C, and then subjected to immunoblotting analyses. Upper panel- shows the translocation of FoxO to the nucleus, in a response to H2O2 at 26°C. Wild-type MEFs were transiently transfected with FoxO1-GFP plasmid and treated with 0.5 mM H2O2. Subcellular localization of FoxO was observed by following GFP signal with fluorescence microscopy. FoxO's nuclear translocation was observed 15min after exposure to H2O2. Middle panel-representative immunoblot shows mTORC1 and Akt activities following exposure to H2O2. Bottom panel- relative mTORC1 activity (p-S6K/S6K) and Akt activity (p-Akt/Akt) were quantified from three independent experiments.
To determine if the same phenomenon occurs upon exposure to oxidative stress that activates FoxO regardless of the phosphorylation of FoxO by Akt (Brunet et al., 2004), we examined the kinetics of Akt and mTORC1 activities following exposure to H2O2. Because the induction of FoxO's nuclear translocation by H2O2 occurs very rapidly, this experiment was performed at 26°C to enable kinetics analyses. In these conditions the onset of FoxO's entry to the nucleus is about 15min after exposure to H2O2 (see insert Fig. 7C, upper panel). Subsequently FoxO transiently elevated the expression of Sesn3 and to a less extent Rictor (Fig. S6B, C). As expected, Akt activity was elevated immediately after exposure to H2O2 due to the inactivation of PTEN (Leslie et al., 2003) (Fig. 7C). However, at these time points, in WT cells, mTORC1 activity was reduced relative to Akt activity. At later time points, in WT cells, Akt and mTORC1 activities were coordinated (Fig. 7C). In contrast, mTORC1 activity in FoxO3a-/- cells had remained relatively higher than Akt activity at all time points and did not significantly go down even when Akt activity was markedly reduced (Fig. 7C).
Collectively these results support the model depicted in Fig. 6D, and provide strong evidence that FoxO is required to coordinate mTORC1 and Akt activities particularly under stress conditions.
Discussion
We have demonstrated that FoxO regulates mTORC1 and Akt activities in mammalian cells. Activation of FoxO inhibits mTORC1 but upregulates Akt activity, whereas FoxO-deficiency elevates mTORC1 but diminishes Akt activity. Mechanistically, we unraveled new regulatory circuits underlying interplay between Akt, FoxO, and mTOR.
Our results identified two mechanisms by which FoxO suppresses mTORC1 activity. The first mechanism is TSC2-dependent and is mediated by the elevation of Sesn3. Like Sesn1 and Sesn2, Sesn3 elevates the activity of AMPK. The exact mechanism by which Sestrins elevate AMPK activity and inhibit Tsc2 activity is not known, but it was shown that Sesn2 interacts with AMPK (Budanov and Karin, 2008). Interestingly, it was recently shown that the scaffolding proteins, kinase suppressors of Ras1 and 2 (KSR1 and KSR2), physically interact with AMPK and activate it (Costanzo-Garvey et al., 2009). Although the exact mechanism by which these interactions with AMPK result in its activation is not known, they set the stage for a new paradigm whereby interaction of AMPK with certain proteins that by themselves may not possess enzymatic activity, could increase its activity.
The second mechanism is TSC2-independent and is mediated, at least in part, through the elevation of Rictor by FoxO. The elevation of Rictor reduced mTORC1 activity, in part, through the decrease in Raptor-mTOR association. However, this may not fully explain the dramatic effect of FoxO1 activation on the rapamycin-sensitive phosphorylation site of 4E-BP1. One possibility to consider is that the increase in Rictor re-distributes mTOR within the cell. The redistributed mTOR may re-assemble with Raptor but in a non-active complex, as it may not have access to Rheb (Sancak et al., 2008). We also showed two mechanisms by which FoxO elevates Akt activity; first, the suppression of mTORC1 by FoxO contributes to the activation of Akt via the inhibition of the negative regulatory loop, and second, the elevation of Rictor and the subsequent increase in mTORC2 activity. Obviously the contribution of each mechanism to the inactivation of mTORC1 and to the activation of Akt would be dependent on the cell's milieu. It is likely that the ratio of mTORC1/mTORC2 in a given cell type could also determine the extent by which these mechanisms contribute to mTORC1 and Akt activities.
We have considered other potential mechanisms by which FoxO might elevate Akt activity and reduce mTORC1 activity. It was reported that the pseudokinase tribble 3 (Trb3) inhibits Akt activity (Du et al., 2003), and that FoxO1 may elevate Akt activity by suppressing Trb3 expression (Matsumoto et al., 2006). FoxO1 suppressesed Trb3 mRNA expression (Fig. S7A), but while the onset of Akt activation occurs 4 hr post FoxO activation (Fig. 4B), we observed down regulation of Trb3 mRNA only 8 hr after FoxO activation (Fig. S7A). Importantly, the knockdown of Trb3 did not have any significant effect on Akt phosphorylation either in the absence or the presence of activated FoxO1 (Fig. S7B).
It was reported that FoxO elevates the expression of Bnip3 (Mammucari et al., 2007), and it was independently reported that Bnip3 inhibits mTORC1 activity downstream of TSC2 by interfering with Rheb activity (Li et al., 2007). We therefore examined whether the elevation of Bnip3 by FoxO could contribute to the attenuation of mTORC1 activity by FoxO in Tsc2-deficient cells. However, although we confirmed that activated FoxO1 elevates the expression of Bnip3 mRNA, the onset of Bnip3 mRNA induction was at 16hr post FoxO1 activation, whereas the mTORC1 activity is fully suppressed after 8hr post FoxO1 activation (Fig. S7C). Furthermore, the knockdown of Bnip3 did not interfere with the effect of FoxO on mTORC1 activity.
Our results strongly support the idea that FoxO coordinates mTORC1 and Akt activities, to maintain a balance between a major cellular energy consumer- mTORC1 and a major energy producer-Akt. In normal physiological conditions FoxO may act as a rheostat that mediate a homeostatic balance between the activities of Akt and mTOR complexes (Fig. 6D). This could also explain why we have not observed a dramatic inhibition of mTORC1 when we express activated FoxO. While FoxO inhibits mTORC1, it is also activating Akt, which in turn activates mTORC1; therefore the net result is a significant mTORC1 inhibition but not a dramatic inhibition. In stress conditions FoxO, by modulating Akt and mTORC1 activities, may act as a gatekeeper to prevent energy crisis. The role of FoxO under stress conditions is manifested by the results in Fig. 7 showing that FoxO is required to maintain low mTORC1 activity, relative to Akt activity, under stress conditions. Thus, reducing energy consumption while maintaining energy production under stress conditions.
The inhibition of mTORC1 activity by FoxO transcription factors could also be evolved as a negative feedback mechanism that restrains hyperactivation of mTORC1. When mTORC1 is hyperactivated it elicits the inhibition of Akt, enabling the activation of FoxO transcription factors, which in turn inhibit the hyperactivated mTORC1.
Several of the cellular responses to the activation of FoxO phenocopy the cellular responses to the inhibition of mTORC1, and therefore it is possible that FoxO exerts some of its reported effects through the inhibition of mTORC1. For example, both the activation of FoxO and the inhibition of mTORC1 elicit cell cycle arrest or attenuation of cell proliferation. Likewise, both the activation of FoxO and the inhibition of mTORC1 elicit cellular atrophy (Ohanna et al., 2005; Sandri et al., 2004). The activation of mTORC1 is known to inhibit autophagy, and it was also shown that autophagy could be mediated by the activation of FoxO (Mammucari et al., 2007). Our results suggest that this could be explained, in part, by the ability of FoxO to inhibit mTORC1. Finally, the activation of FoxO could extend organismal and cellular lifespan (Greer and Brunet, 2005), while the activation of mTORC1 could promote cellular senescence (Zhang et al., 2003), and the inhibition of TORC1 extends lifespan in worms and flies (Kapahi and Zid, 2004). Based on our results it is possible that FoxO activation extends organismal and cellular lifespan, in part, via the inhibition of mTORC1.
In summary, our results provided an additional mechanism that maintains a delicate balance between Akt and mTORC1 activities, and an additional mechanism by which Akt regulates mTORC1 activity. Our results have also broader implications, suggesting that some of the cellular or the organismal consequences of FoxO activation could be explained by its ability to inhibit mTORC1.
Experimental Procedures
Details of reagents, antibodies, plasmids, retroviruses, lentiviruses, adenoviruses, and quantitative real-time polymerase chain reaction (qPCR) are provided in Supplemental Experimental Procedures.
Cell lines, cell culture, transfection, and protein extraction
Mouse embryonic fibroblast (MEFs) include Tsc2+/-p53-/-, Tsc2-/-p53-/-, and dominant-negative p53 immortalized FoxO3a+/+ and FoxO3a-/-, MCF7 (human breast adenocarcinoma), NIH3T3 (mouse fibroblast cell line), Rat1a (mAkt) (rat fibroblast cell line), and U2OS (human osteosarcoma) cell lines were cultured in DMEM containing 10% FBS. DOV13 (human ovarian carcinoma) cell line was cultured in MEM containing 10% FBS. For experiments comparing mTORC1 and Akt activities in wild-type and FoxO3a-/- MEFs (Figure 7), cells were culture in final 5mM Glucose with indicated percentage of dialyzed serum. DNA or RNAi transfections were performed using Lipofectamine 2000 (Invitrogen). For the transfection of RNAi, 80,000 cells were seeded on 3cm dish one day before transfection. 100nM siRNAs (Thermo Scientific Dharmacon) were diluted and mixed with 5μl Lipofectamine 2000 to transfect one sample. For other transient transfection, 6-10μg DNA of expression plasmids were used to transfect one 6cm dish with 60% confluent cells. For immunoblotting, protein extract was prepared as described previously (Hahn-Windgassen et al. 2005).
Immunoprecipitation and in vitro kinase assays
For co-immunoprecipitation, cells were incubated in 0.3%-CHAPS mTOR lysis buffer as described previously (Sarbassov et el. 2004). For each sample, 500μg protein was used to incubate with mTOR antibody (Cell Signaling) 1: 50 dilution and rotation for 3 hours. Thirty μl of 50% protein A/G agarose was then added and incubated for additional 1 hour. Immunoprecipitates were washed three times with mTOR lysis buffer and once with IP wash buffer (50 mM HEPES pH 7.5, 40 mM NaCl, 2 mM EDTA) and resuspended in sample buffer for immunoblotting analysis. Kinase assays were performed in 15μl at 30°C for 20min and contained washed mTOR or Rictor immunoprecipitates, myc-S6K1 (purified from HEK293 cells treated with rapamycin) or 100 ng recombinant 4E-BP protein, purified from E. coli, or inactive Akt (Upstate 14-279), in kinase assay buffer (100 μM ATP, 25 mM HEPES pH 7.4, 50 mM KCl, 20% glycerol, 10 mM MgCl2, 4 mM MnCl2, 1 mM DTT, and Roche phosphatase inhibitor cocktail) modified form previous publication (Kim et al., 2002). Reaction stopped by adding 5μl 4× sample buffer were analyzed by 12% SDS-PAGE and detected by phospho-4E-BP (S65) and 4E-BP antibodies.
To prepare S6K1 as a substrate for the kinase assay, HEK293 cells were transiently transfected with myc-S6K1 vector for 48 hours. Transfected HEK293 cells were serum starved for 24 hour and treated with 100 nM rapamycin for one hour before harvesting. Cells were lysed by CHAPS lysis buffer and subjected to myc antibody for one-hour incubation at 4°C Protein A/G agarose beads were added to pull down myc-S6K1 and then incubated with mTOR immunoprecipitates to bind mTOR complexes. Immunoprecipitates were washed three times with mTOR lysis buffer followed by the in-vitro kinase assay as described above.
To prepare Akt1 as substrate the inactive Akt purchased from Upstate was subjected to Lambda protein phosphatase (NEB) for one hour at 3°C. The phosphatase was heat inactivated for 30 min at 65°C and the reaction was subjected to pan-Akt antibody (Cell Signaling) for one hour incubation at 4°C. Protein A/G agarose beads were added to pull down Akt and then incubated with Rictor immunoprecipitates to bind mTORC2 complexes. Immunoprecipitates were washed three times with mTOR lysis buffer followed with in-vitro kinase assay as described above.
Supplementary Material
Acknowledgments
These studies were supported by NIH grants CA090764, AG016927, and AG025953 to N.H., and by ACS-IL 06-24 to Y.P.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bhaskar PT, Hay N. The Two TORCs and Akt. Dev Cell. 2007;12:487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Budanov AV, Karin M. p53 Target Genes Sestrin1 and Sestrin2 Connect Genotoxic Stress and mTOR Signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo-Garvey DL, Pfluger PT, Dougherty MK, Stock JL, Boehm M, Chaika O, Fernandez MR, Fisher K, Kortum RL, Hong EG, et al. KSR2 is an essential regulator of AMP kinase, energy expenditure, and insulin sensitivity. Cell Metab. 2009;10:366–378. doi: 10.1016/j.cmet.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300:1574–1577. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- Essers MA, Weijzen S, de Vries-Smits AM, Saarloos I, de Ruiter ND, Bos JL, Burgering BM. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. Embo J. 2004;23:4802–4812. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in Mice of the mTORC Components raptor, rictor, or mLST8 Reveals that mTORC2 Is Required for Signaling to Akt-FOXO and PKCalpha, but Not S6K1. Dev Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem. 2005;280:32081–32089. doi: 10.1074/jbc.M502876200. [DOI] [PubMed] [Google Scholar]
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem. 2005;280:40406–40416. doi: 10.1074/jbc.M508361200. [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131:3897–3906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- Kapahi P, Zid B. TOR pathway: linking nutrient sensing to life span. Sci Aging Knowledge Environ. 2004;2004:PE34. doi: 10.1126/sageke.2004.36.pe34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villen J, Becker EB, DiBacco S, de la Iglesia N, Gygi S, Blackwell TK, Bonni A. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. Embo J. 2003;22:5501–5510. doi: 10.1093/emboj/cdg513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang Y, Kim E, Beemiller P, Wang CY, Swanson J, You M, Guan KL. Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J Biol Chem. 2007;282:35803–35813. doi: 10.1074/jbc.M705231200. [DOI] [PubMed] [Google Scholar]
- Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N. mTOR, translation initiation and cancer. Oncogene. 2006;25:6416–6422. doi: 10.1038/sj.onc.1209888. [DOI] [PubMed] [Google Scholar]
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Han S, Kitamura T, Accili D. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest. 2006;116:2464–2472. doi: 10.1172/JCI27047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, Unterman T, Hay N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008;14:458–470. doi: 10.1016/j.ccr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohanna M, Sobering AK, Lapointe T, Lorenzo L, Praud C, Petroulakis E, Sonenberg N, Kelly PA, Sotiropoulos A, Pende M. Atrophy of S6K1(-/-) skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control. Nat Cell Biol. 2005;7:286–294. doi: 10.1038/ncb1231. [DOI] [PubMed] [Google Scholar]
- Porstmann T, Santos CR, Lewis C, Griffiths B, Schulze A. A new player in the orchestra of cell growth: SREBP activity is regulated by mTORC1 and contributes to the regulation of cell and organ size. Biochem Soc Trans. 2009;37:278–283. doi: 10.1042/BST0370278. [DOI] [PubMed] [Google Scholar]
- Puig O, Marr MT, Ruhf ML, Tjian R. Control of cell number by Drosophila FOXO: downstream and feedback regulation of the insulin receptor pathway. Genes Dev. 2003;17:2006–2020. doi: 10.1101/gad.1098703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggero D, Montanaro L, Ma L, Xu W, Londei P, Cordon-Cardo C, Pandolfi PP. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med. 2004;10:484–486. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Tang ED, Nunez G, Barr FG, Guan KL. Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem. 1999;274:16741–16746. doi: 10.1074/jbc.274.24.16741. [DOI] [PubMed] [Google Scholar]
- Yang J, Cron P, Thompson V, Good VM, Hess D, Hemmings BA, Barford D. Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol Cell. 2002;9:1227–1240. doi: 10.1016/s1097-2765(02)00550-6. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, Vazquez F, Carpenter CL, Kwiatkowski DJ. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest. 2003;112:1223–1233. doi: 10.1172/JCI17222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.