

Abstract
The Wnt/β-catenin signaling pathway is critical to embryonic development as well as adult tissue regeneration. Dysregulation of this pathway can lead to a variety of human diseases, in particular cancers. Chibby (Cby), a small and highly conserved protein, plays an antagonistic role in Wnt signaling by inhibiting the binding of β-catenin to Tcf/Lef family proteins, a protein interaction that is essential for the transcriptional activation of Wnt target genes. Cby is also involved in regulating intracellular distribution of β-catenin. Phosphorylated Cby forms a ternary complex with 14-3-3 protein and β-catenin, facilitating the export of β-catenin from the nucleus. On the other hand, the antagonistic function of Cby is inhibited upon binding to thyroid cancer-1 (TC-1). To dissect the structure−function relationship of Cby, we have used NMR spectroscopy, ESI-MS, CD, and DLS to extensively characterize the structure of human Cby. Our results show that the 126-residue Cby is partially disordered under nondenaturing conditions. While the N-terminal portion of the protein is predominantly unstructured in solution, the C-terminal half of Cby adopts a coiled-coil structure through self-association. Initial data for the binding studies of Cby to 14-3-3ζ (one of the isoforms in the 14-3-3 family) and TC-1 via these two distinct structural modules have also been obtained. It is noteworthy that in a recent large-scale analysis of the intrinsically disordered proteome of mouse, a substantial number of disordered proteins are predicted to have coiled-coil motif presence in their sequences. The combination of these two molecular recognition features could facilitate disordered Cby in assembling protein complexes via different modes of interaction.
The molecular mechanism of Wnt/β-catenin signaling has been intensively investigated in the past 2 decades because of the pivotal role this pathway plays in development processes and its association with human diseases, in particular cancers (1−4). Being a key protein in this signaling pathway, the level and intracellular distribution of β-catenin are tightly regulated by different mechanisms. In the absence of Wnt signal, β-catenin in cytosol is phosphorylated at multiple Ser/Thr sites located in its N-terminal domain by the destruction complex composed of axin, adenomatous polyposis coli (APC),1 glycogen synthase kinase-3β (GSK-3β), and casein kinase 1 (CK1) [reviewed in Kimelman and Wu (5)]. The phosphorylated β-catenin is subsequently ubiquitinated and targeted for proteasomal degradation (6). In the presence of the Wnt signal, however, the phosphorylation of β-catenin by the destruction complex is inhibited. The accumulation of hypophosphorylated β-catenin in the cytoplasm provokes the protein translocation to the nucleus, where it interacts with T cell factors (Tcf)/lymphoid enhancer factor (Lef) proteins to activate the transcription of Wnt target genes, with many of them being involved in cell proliferation (7). Because of its strong link to cancers, targeted inhibition of Wnt/β-catenin signaling becomes an active research area in therapeutic development (8).
One of the key binding partners of β-catenin is Chibby (Cby), a small (126 residues in human) and highly conserved protein, which was first identified as an antagonist of Wnt signaling in 2003 (9). It functions by competing with the Tcf/Lef family of proteins for binding to β-catenin (9). Besides functioning as an inhibitor of β-catenin-Tcf binding, in a recent study, Takemaru and co-workers demonstrated that Cby also plays an essential role in the intracellular distribution of β-catenin in conjunction with 14-3-3 (10), a family of proteins that function in a variety of signaling pathways by interacting with phosphorylated Ser/Thr targets (11−13). With a canonical mode II binding motif of 14-3-3 harbored in its N-terminal region between residues 16−22 (11−13), it has been shown that Cby can form a ternary complex with 14-3-3ζ and β-catenin in the nucleus upon phosphorylation at S20 by Akt1. This complex formation facilitates the export of β-catenin to the cytoplasm, which in turn diminishes Wnt signaling (10,14).
Cby has also been shown to bind to thyroid cancer-1 (TC-1), a tumorigenic and intrinsically disordered protein first found to be upregulated in thyroid cancer cells (15,16). Being a positive regulator in Wnt signaling, TC-1 interacts with Cby and inhibits its binding to β-catenin (17), which in turn leads to upregulation of many cancer-associated genes. Structural characterization of TC-1 by nuclear magnetic resonance (NMR) shows that the protein is partially disordered (16,18). While the N-terminal portion of the protein is largely unstructured, three segments of the protein in the C-terminal half are found to contain high helical propensity. Importantly, these preformed structural elements in TC-1 are involved in mediating its interaction with Cby (18).
Because of the crucial role Cby plays in the Wnt/β-catenin signaling pathway, it is not surprising that Cby is associated with different human diseases (19−22). For instance, Schuierer et al. discovered that the mRNA of Cby is downregulated in colon carcinoma cell lines (20). On the other hand, Singh et al. demonstrated that overexpression of Cby is related to the cardiac differentiation of embryonic stem cells (21). Further, Cby has also been found to be involved in polycystic kidney disease (23). By using the yeast two-hybrid screening technique, in 2004, Hidaka et al. identified Cby (the authors named the protein PIGEA-14; polycystin-2 interactor, Golgi- and endoplasmic reticulum-associated protein with a molecular weight of 14 kDa) as a binding target of polycystin-2, a protein that has been found to be mutated in some patients with polycystic kidney disease (23). The interaction between PIGEA-14/Cby and polycystin-2 is shown to be crucial in regulating the intracellular distribution of the latter (23).
Despite the fact that multiple binding partners of Cby have been identified, structural information of Cby is still limited. This hinders a detailed understanding of the molecular mechanism by which Cby interacts with its targets. For instance, the 781-residue human β-catenin consists of a core armadillo repeat region (residues 138−664) flanked by the highly unstructured N- and C-terminal domains (24,25). Consisting of 12 repeats and with each of these repeats composed of three α-helices (except for repeat 7, which has only two α-helices), the armadillo region adopts an elongated, superhelical structure with a positively charged groove, providing a large accessible surface area for protein−protein interactions (24,25). Crystal structures of the β-catenin/Tcf complex reveal that the disordered Tcf4 adopts an elongated structure upon binding to the positively charged groove of the armadillo region of β-catenin, spanning repeats 3−9 (26−28). On the other hand, by using a yeast Ras recruitment system and different truncated β-catenin constructs as baits for target screening, Takemaru et al. discovered that Cby binds specifically to the armadillo repeats 10−12 and the C-terminal region of β-catenin via its C-terminal half, which is predicted to contain a coiled-coil motif (9,14,23). Interestingly, even though Cby competes with Tcf4 to bind to β-catenin, the binding sites of these two proteins on β-catenin do not overlap.
In the present work, we have carried out an extensive structural characterization of Cby using NMR spectroscopy, CD, ESI-MS, and DLS. Our results demonstrate that Cby, the binding target of TC-1, β-catenin, and 14-3-3ζ in the Wnt/β-catenin pathway, is partially disordered in its free state. The NMR and CD data together suggest that the N-terminus of Cby, which has been shown to mediate the interaction with 14-3-3ζ upon phosphorylation (10), is largely unstructured in the absence of its target. On the other hand, the C-terminal portion of Cby, which harbors the binding sites of TC-1 and β-catenin, is likely to adopt a coiled-coil conformation that is highly stable even in the presence of 4 M urea. This potential coiled-coil domain is found to have a high tendency to self-associate, which renders the structural characterization of this region challenging (29). The biological implications of the coexistence of these two distinct structural modules in Cby are appraised in the Discussion.
Experimental Procedures
Protein Sample Preparation
The pDEST17 plasmid carrying the human Cby cDNA sequence with the N-terminal His6-tag was transformed into the Rosetta 2(DE3)pLysS strain of Escherichia coli (Novagen) for protein overexpression. For 15N,13C-labeled protein samples, cells were grown at 37 °C in 1 L of M9 media culture containing 1 g of 15NH4Cl (Cambridge Isotope Laboratories) and 3 g of 13C6-d-glucose (Isotec) as the only nitrogen and carbon sources, respectively, until the OD600 value reached 0.7−0.8. CaCl2 (0.1 mM), 1 mM MgSO4, 10 μg/mL of thiamin, and 10 μg/mL of biotin were also added to the media as supplements. The protein overexpression was then induced with 0.5 mM isopropyl β-d-thiogalactopyranoside (IPTG; BioShop), and the cells were allowed to grow overnight at 18 °C before harvesting. The cells were lysed by sonication (with lysozyme added) and followed by centrifugation at 16000g to sediment the inclusion bodies. The washed inclusion bodies were then solubilized in 8 M urea, and the resulting solution was mixed in a 1:4 volume ratio with an 8 M urea binding buffer (20 mM Tris, pH 7.5, 5 mM imidazole, 500 mM NaCl, and 8 M urea). Nickel Sepharose (GE Healthcare) beads pre-equilibrated with the binding buffer were then added, and the mixture was incubated at room temperature for 2 h on a rotator. The beads were loaded to an econocolumn and washed extensively with 8 M urea binding buffer. The protein was subsequently eluted with buffer containing 8 M urea and 1 M imidazole and then dialyzed overnight into a buffer with 8 M urea and 10 mM sodium acetate at pH 5. Finally, the urea was removed by dialysis, with the final buffer for the protein containing 10 mM sodium acetate at pH 5.0. The purity of the protein was confirmed both by SDS−PAGE gel electrophoresis and ESI-MS. The protein yield was ∼30 mg/L of growth. The 29-residue His6-tag sequence containing a tobacco etch virus cleavage site was retained in the experiments since the removal of the tag significantly reduced the solubility of the protein.
Site-Directed Mutagenesis of Wild-Type Human Cby
Wild-type human Cby in pDEST17 plasmid was subjected to point mutagenesis using the Stratagene QuikChange II site-directed mutagenesis kit. Overexpression and purification of mutational variants were carried out with the similar procedure as mentioned above for the wild-type human Cby.
Electrospray Ionization Mass Spectrometry
ESI-MS data were recorded on a Q-TOF spectrometer (Micromass) in positive ion nanospray mode. The cone voltage was 50 V, with a source temperature of 80 °C. Calibration of the instrument was performed with myoglobin. The experiment was performed under nondenaturing conditions with the buffer of 10 mM ammonium acetate at pH 5. Protein concentrations ranging from ∼20 to 50 μM were used. All data were acquired and analyzed using MassLynx software provided by the instrument manufacturer.
Circular Dichroism Spectropolarimetry
CD experiments were performed on a Jasco J-810 spectropolarimeter (Easton, MD), equipped with a PTC-423S Peltier temperature control unit, using a cuvette with 0.1 mm path length. The wavelength was calibrated using neodymium glass, and the CD signal was calibrated using ammonium d-10-camphorsulfonate. Both calibrations were carried out as described in the Jasco Model J-810 Spectropolarimeter Hardware/Function Manual. Spectra of 40 μM Cby (190−260 nm, in 10 mM sodium acetate at various pH values as well as various additives such as urea; see Results section) were recorded at 25 °C. For the melting curves of Cby, the temperature was scanned from 5 to 100 °C, and the ellipticity at 222 nm was monitored. All data sets were collected using Spectra Manager software, and the CD data (θ, in millidegrees) were converted to mean residue ellipticity ([θ]) using standard formula and the software supplied by the manufacturer. The programs SELCON3, CDSSTR, and CONTIN/LL included in the CDPro software package were used to deconvolute the CD spectra (30). A wavelength range of 190−240 nm and reference sets SMP56 and SDP48 were used in the calculations.
Dynamic Light Scattering
The Stokes radii of the wild-type Cby and its mutants were measured using the ALV/CGS-3 compact goniometer system at room temperature, with a laser source of 22 mW (ALV/LSE-S003). Protein samples (in ranges of 50 μM and above) were prepared in 10 mM sodium acetate buffer at pH 5.0, and all samples and buffers were filtered through a 0.2 μm filter prior to analysis to prevent interference from residual particulates. Lysozyme was used as the control protein. The refractive index of the 10 mM pH 5 acetate buffer was found to be close to water, and thus, the η value used was left as default. Each measurement was repeated two to three times (30 s acquisition intervals each), and all data were processed using ALV correlator software (version 3.0). The mass-averaged method, a built-in function of the ALV correlator software, was used to calculate the hydrodynamic radii of different protein samples. Since particles large in size scatter light more efficiently than small particles in DLS, even a trace amount of large particles (i.e., dust) can contribute significantly to the light scattering. The built-in mass-averaged function was used to correct this bias (31,32).
NMR Spectroscopy
Five heteronuclear triple resonance experiments, HNCACB, CBCA(CO)NH, C(CO)NH, HN(CA)CO, and HNCO (33−35), were recorded for backbone resonance assignments. 13C,15N-labeled protein samples (250−300 μM) in 10 mM sodium acetate at pH 5.0 in the presence of different concentrations of urea were used, and the experiments were performed on a Varian Inova 600 MHz spectrometer equipped with a xyz-gradient triple resonance probe (UWO Biomolecular NMR Facility) at 25 °C. 1H−15N TROSY-HSQC spectra of 13C,15N,2H-labeled Cby (∼200 μM) in 10 mM sodium acetate at pH 5.0 were also acquired at 25 °C on a Varian Inova 600 MHz spectrometer (UWO Biomolecular NMR Facility) and a Varian Inova 800 MHz spectrometer (Canadian National High Field NMR Centre), both equipped with a Z-gradient HCN cold probe. All chemical shifts were referenced to the internal DSS (2,2-dimethyl-2-silapentane-5-sulfonate) signal. Data were processed and analyzed using the programs NMRPipe (36) and NMRView (37).
Hydrodynamic radius (Rh) measurement by NMR was performed using the water-suppressed longitudinal encode−decode (water−sLED) pulse sequence on a sample with 360 μM N-Cby(S20D) and 9 mM 1,4-dioxane with a range of gradient strengths, G(38). The decay rates (D) of the protein and dioxane signals (I) were determined individually based on the linear plot of log(I) versus G2, as I(G) = A exp(−DG2). The Rh of the protein was then calculated based on the signal decaying rates of protein (Dprotein) and dioxane (Ddioxane) using the equation Rh = (Ddioxane/Dprotein) × 2.12 Å, proposed by Wilkins et al. (39). The experiment was recorded three times, and protein signals from backbone amide, side-chain amide, and aliphatic regions were used to calculate the averaged values of Dprotein.
Backbone 15N R1 and R1ρ experiments (40,41) were performed on 15N-labeled N-Cby (∼200 μM) and N-Cby(S20D) (∼360 μM) samples in 10 mM acetate buffer at pH 5.0, using sequences included in the Varian Biopack library. A series of relaxation delay times (t) between 10 and 640 ms were used in the R1 experiments, while delay times ranging from 10 to 150 ms were used in the R1ρ measurements. The R1 and R1ρ relaxation rates of assigned signals that are well-resolved were determined based on the attenuations of peak intensity versus relaxation times, which follow the equation I(t) = I(0) exp(−Rt), where I(t) is the peak intensity at relax time t. Errors in the relaxation rates were estimated based on the fits of the data. The transverse relaxation rate, R2, was then calculated on the basis of the observed R1 and R1ρ values, and the offset between the resonance and the carrier frequency (Δω) in hertz, using the equation
where tan θ = BSL/Δω. BSL (=1.5 kHz) is the spin-lock field used in the R1ρ experiments (42).
Steady-state 1H−15N NOE values were determined as the ratio of the peak intensities derived from the spectra acquired in the presence and the absence of 1H saturation (41). A 7 s delay between scans followed by 5 s of saturation period was used in recording the 1H−15N NOE with proton saturation, while a 12 s delay between scans was used for 1H−15N NOE without saturation. Errors were estimated on the basis of the signal-to-noise ratios of individual peaks.
To probe the interaction between 15N-labeled N-Cby(S20D) and unlabeled 14-3-3ζ, the following procedure was used for the NMR sample preparations. 15N-labeled N-Cby(S20D) (∼200 μM) in 10 mM sodium acetate buffer, 1 mM DTT, and 1 mM EDTA at pH 5.0 was mixed rapidly in a 1:1 (v/v) ratio with the buffer (250 mM NaCl, 50 mM Tris, 1 mM DTT, and 1 mM EDTA, at pH 7.0) containing either 0 (as the control) or ∼600 μM 14-3-3ζ. The pH of the final solutions were adjusted to 7.0 by adding small amounts of 50 mM NaOH (∼40 μL). Precipitated proteins were removed by centrifugation before the NMR experiments were performed.
Results
Protein Sequence Analysis
Protein sequence analysis shows that Cby is highly conserved across a range of organisms spanning from fly to zebrafish, mouse, and human (9). Although limited structural information of human Cby is available to date, bioinformatic analysis predicts that a coiled-coil domain with four heptad repeats is located at the C-terminal part of the protein (residues 73−100; Figure 1a and Supporting Information Figure S1) (9,23,29,43). Meanwhile, amino acid composition analysis shows that overall Cby is richer in charged and polar residues and is mildly deficient in hydrophobic amino acids compared to globular proteins, despite a high number of leucine residues present in the predicted coiled-coil region. This unique composition places Cby on the borderline between ordered and disordered proteins in the charge−hydropathy plots (Supporting Information Figure S2b−d) based on the analysis proposed by Uversky et al. (44). The protein is also predicted to be largely disordered by PONDR (predictor of naturally disordered region; Supporting Information Figure S2a), one of the most commonly used predictors for disordered proteins (44−47), as a significant number of residues in the protein have PONDR scores above the threshold of 0.5.
Figure 1.

(a) Schematic diagram of the Cby primary structure. The canonical mode II binding motif of 14-3-3 is underlined. Negatively and positively charged residues are colored in red and blue, respectively. The arrows indicate the location of mutation sites. (b) Far-UV CD spectra of Cby recorded at pH 3 (blue), 4 (magenta), 5 (green), and 6 (black). The data were collected at 25 °C.
Cby Is Partially Disordered
In order to identify the residue-specific structure of Cby, extensive structural characterization of the protein was carried out by NMR spectroscopy. Due to the high tendency of sample precipitation at pH >5 or at high salt concentration (more discussion below), sample buffer with 10 mM sodium acetate at pH 5 was used for the NMR studies unless otherwise specified. Figure 2a shows the 1H−15N HSQC spectrum of Cby. The lack of chemical shift dispersion (crowded in a range of 7.6−8.8 ppm in 1H dimension) of peaks suggests that at least a significant portion of the protein is disordered. Intriguingly, the number of peaks in the spectrum is less than 50% of what is expected to be observed for the full-length Cby. The secondary structures of Cby were also characterized by CD spectropolarimetry (Figure 1b). To quantitatively determine the secondary structure contents of Cby, measured CD spectra were deconvoluted using the CDPro software (30). The result indicates that Cby contains 43 ± 2% of helical, 10 ± 1% of β-strand, and 47 ± 3% of random coil/turn structures in 10 mM acetate buffer, at pH 5. If we assume that the His-tag residues are in random coil conformations, the deconvolution result reveals that ∼67 residues of Cby are in helical conformation while ∼44 residues adopt random coil/turn structures. This clearly demonstrates that Cby is partially disordered yet with a high helical content present in the protein.
Figure 2.

(a) Assigned 1H−15N HSQC spectra of full-length Cby in 10 mM sodium acetate, pH 5. (b) Overlay of the HSQC spectra of full-length Cby (blue) and N-Cby (red), in 10 mM sodium acetate, pH 5.
To obtain residue-specific structural information of Cby, heteronuclear three-dimensional NMR experiments were performed for the backbone resonance assignment of the protein. The result indicates that most of the peaks that are observed in the 1H−15N HSQC spectrum of full-length Cby originate from the N-terminal portion of the protein (Figure 2a). This was further confirmed by the NMR analysis of the truncated Cby construct, which contains only the N-terminal half of the protein (N-Cby; residues 1−63). The fact that the majority of peaks observed in the 1H−15N HSQC of N-Cby can essentially overlay with the peaks observed in the spectrum of full-length Cby (except for residues 50−63) suggests that at least a large portion of the N-terminal part of the protein is disordered and may not interact significantly with the C-terminal segment (Figure 2b). The disordered nature of N-Cby is also confirmed by CD spectropolarimetry. The CD spectrum of N-Cby (Figure 7b) displays a large and negative signal at 190 nm and negligible ellipticities at 208 and 222 nm, indicating that N-Cby is largely unstructured (48). Interestingly, deconvoluted CD data reveal that N-Cby contains only 5 ± 2% of helical, 27 ± 7% of β-strand, and 68 ± 7% of random coil/turn structures. The fact that N-Cby contains significantly less helical content compared to the full-length Cby (5% vs 43%) strongly suggests that the helical structures of Cby are located at the C-terminal portion of the protein, which is predicted to adopt a coiled-coil conformation.
The residue-specific secondary structure propensities of Cby were then determined by using the SSP program. SSP (secondary structure propensity) is a method developed by Forman-Kay and co-workers to determine the residue-specific secondary structure propensity of IDPs based on their measured chemical shifts (49). In brief, the SSP score of a residue can be interpreted as the expected fraction of α-helical or β-strand/extended structure (a residue with fully formed α- or β-structure will have an SSP score of 1 or −1, respectively). For instance, a SSP score of 0.3 can be interpreted as 30% of the conformers in the disordered state ensemble adopt helical dihedral angles at that particular position (49). Secondary structure chemical shifts for different amino acid types in well-folded proteins are taken into consideration in the calculation, and the relative secondary chemical shifts of 13Cα and 13Cβ atoms are used to correct for potential chemical shift referencing error (49−52). Since most of the signals from the C-terminal half of Cby are unobservable, the SSP analysis was focused on the N-terminal portion of the protein. The result reveals that even though no stable secondary structure exists in the N-terminal region of the full-length Cby, a notable helical propensity is present between residues 20 and 50, even though it is less significant compared to residues 73 and 74, which are located at the beginning of the predicted coiled-coil region of the protein (Figure 3a). This helical propensity is shown to decrease as the concentration of urea increases (Figure 3b). Based on the SSP analysis, the rest of the N-terminal portion of Cby adopts β-strand/extended structure (with SSP score <0), which is more pronounced in the presence of denaturant (i.e., 4 M urea).
Figure 3.

Secondary structure propensity (SSP) scores for Cby in 0 M urea (a) and 4 M urea (b) calculated on the basis of the assigned 13Cα and 13Cβ chemical shifts using the SSP program.
Stable Helical Content Exists in the C-Terminal Part of Cby
Despite the NMR signals originating from the C-terminal region of Cby being unobservable, the CD data of full-length Cby (Figure 1b) and N-Cby (Figure 7b) together provide strong evidence that helical structure is present in the C-terminal portion of the protein. Further, this helical structure is stable over a wide pH range (Figure 1b) or in the presence of high concentrations of denaturant (up to 4 M urea; Supporting Information Figure S3). Figure 1b also shows that while the CD spectra of Cby at pH 3−6 display no dramatic difference, the ratio of the ellipticities at 222 and 208 nm ([θ]222/[θ]208) becomes closer to 1 as the pH increases, which suggests that the coiled-coil interaction may exist in the C-terminal portion of Cby and it becomes more stable at higher pH (53).
Self-Association of Cby via the C-Terminal Coiled Coil
The coiled-coil interaction may also explain the strong self-association tendency of Cby at high pH and salt concentration. For instance, the 18 kDa Cby elutes as a broad peak at ∼79 kDa from a size-exclusion column when 10 mM acetate (pH 5) with 100 mM NaCl was used as the buffer. Results of sedimentation velocity (data not shown) also indicate that the apparent molecular weight of Cby increases as the salt concentration of the buffer increases. The self-association of Cby has also been investigated using nondenaturing ESI-MS. The ESI-MS result shown in Figure 4 indicates that in a buffer with only 10 mM ammonium acetate at pH 5 (similar to the buffer conditions used in the NMR studies) Cby exists as a mixture of monomer, dimer, and tetramer. However, we should point out that the ESI-MS data could only be interpreted qualitatively. They cannot be used to quantify the populations of monomer and dimer since the ionization efficiency of the latter can be significantly lower due to the reduction in exposed hydrophobic surface area (54,55). We have also measured the averaged hydrodynamic radius (Rs) of Cby using dynamic light scattering. The result shows that the Rs of Cby is ∼30 Å, which is significantly larger than a globular protein with similar molecular weight. Even though the large value of Rs can be due to dimerization, partial unfolding, or formation of elongated coiled-coil structure of Cby, the ESI-MS and DLS results together strongly suggest the existence of oligomers in the ensemble.
Figure 4.

Oligomerization of Cby studied by ESI-MS. The ESI-MS spectrum was acquired under nondenaturing conditions with 35 μM Cby in 10 mM ammonium acetate, pH 5.
Unlike the full-length Cby, N-Cby is found to be more soluble at higher ionic strengths. This observation suggests that Cby may undergo self-association under the NMR conditions used via its putative C-terminal coiled-coil region. The elongated coiled-coil structure formed by self-association may render Cby to tumble significantly slower than a globular protein with similar molecular weight, resulting in severe broadening of the NMR peaks from this portion of the protein. Moreover, the association/dissociation of the coiled-coil structure on the intermediate NMR time scale may also contribute to the signal broadening. It is noteworthy that even though the self-association also increases the overall tumbling of the N-terminal part of Cby, this segment of the protein is highly flexible, and a large part of it does not interact with the C-terminal region. Therefore, the NMR signals from the N-terminus of Cby remain relatively sharp and observable despite the self-association.
To test whether the signal broadening is due to the slow molecular tumbling, a 1H−15N TROSY-HSQC experiment (56), which is designed for high molecular weight protein studies, was performed on an 15N,13C,2H-labeled Cby sample. The spectrum was compared to the one acquired with the regular 1H−15N HSQC experiment (Supporting Information Figure S4). Even though the NMR signals originating from the N-terminal part of Cby are sharper in the TROSY-HSQC spectrum, most of the signals from the C-terminal part of Cby are still unobservable. The data suggest that the severe peak-broadening problem may not be due to the slow molecular tumbling caused by dimerization but more likely be the result of conformational exchange on intermediate NMR time scale.
Multiple mutational variants of Cby have been created in an attempt to circumvent the signal-broadening problem due to conformational exchange and self-association occurring in the C-terminal region of the protein. The coiled-coil model (Supporting Information Figure S1) illustrates that there are two Asn residues (N81 and N88) in the a positions of the second and third heptad repeats, positions that are usually occupied by hydrophobic residues such as Leu, Ile, or Val. The presence of Asn residues in the dimer interface is also found in some other coiled-coil structures. It has been suggested that this confers specificity for dimerization/oligomerization (57). Even though it is not energetically favorable to bury a polar Asn in a hydrophobic interface, the buried Asn may still form hydrogen bonding to promote parallel assembly of the coiled coil (58).
The N88 residue in the third heptad repeat was mutated to a leucine, with the aim of strengthening the dimerization interface of the C-terminal coiled coil, which can in turn lead to the reduction of monomer−dimer exchange. An N88L/L93R double mutant has also been prepared, with the L93R mutation specifically designed to disrupt the possible hydrophobic patch formed by L90, L93, and I97 on the surface of the coiled coil. Finally, a L77A/L91A double mutant has also been included in this study. In a recent publication, Mofunanya et al. demonstrate that the mutations of these two leucine residues that are predicted to be critical for the coiled-coil formation to alanine can disrupt the dimerization of Cby (29).
Figure 5 shows the CD spectra of these three mutational variants collected at 25 °C, and their Tm values have been determined by thermal denaturation. Compared to the wild-type Cby, the N88L and N88L/L93R mutants display some minor changes in the CD spectra, and no significant change in the Tm of the protein was observed. On the other hand, the mutations of both L77 and L91 to alanines lead to an ∼60% decrease in the ellipticity at 222 nm and a dramatic decrease in Tm (from 70 to 18 °C) compared to the wild-type. In addition, the Rs of N88L and N88L/L93R mutants measured by DLS are very similar to the value obtained for the wild-type Cby (∼30 Å), while the Rs of L77A/L91A is significantly smaller (∼17 Å). The results together suggest that mutation of these two key leucine residues (L77 and L91) weakens the helical propensity and probably the dimerization tendency of the coiled-coil region (29).
Figure 5.
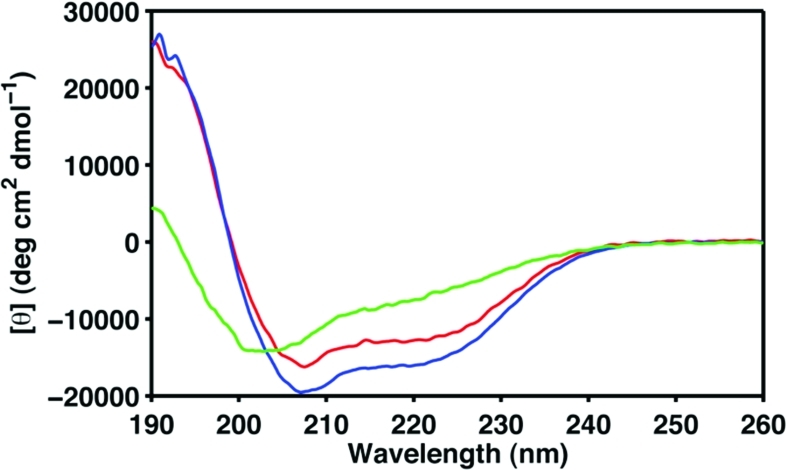
Secondary structure content and stability of Cby mutants. Far-UV CD spectra of N88L (red), N88L/L93R (blue), and L77A/L91A (green) mutational variants. Data were recorded in 10 mM sodium acetate (pH 5) at 25 °C with protein concentrations ∼35 μM.
Despite the fact that these three mutational variants show different extent of changes in the CD spectra, most of the NMR signals from the C-terminal portion of these mutants are still not observable in their 1H−15N HSQC spectra (data not shown). The results suggest that the conformational exchange that causes the severe line broadening of NMR signals may be due to a more complicated process than simply a monomer/dimer equilibrium. For instance, a number of hydrophobic residues near the C-terminus (residues 101−126) of Cby may very well also contribute to the self-association of this protein. In fact, a synthetic peptide with the sequence resembling only residues 70−100 of Cby and with sextuple mutations (N81L/N88L/L90R/L93R/I97R/D100R) incorporated to strengthen the dimer interface and to remove the hydrophobic patch on the coiled-coil surface is found to be highly soluble even at high pH and salt concentration. CD analysis shows that this peptide adopts a highly stable coiled-coil conformation with [θ]222/[θ]208 ≈ 1 and Tm ∼ 95 °C (data not shown).
Binding of Cby to TC-1
Besides the structural characterization of Cby in its free state, in this work, initial data on the binding of Cby to its protein targets have been obtained. In a previous study, we have shown that disordered TC-1 binds to Cby via its highly helical C-terminal region (18). Here we have carried out an NMR binding experiment by which unlabeled TC-1 was added to the 15N-labeled Cby. Panels a and b of Figure 6 show the 1H−15N HSQC spectra of full-length Cby (60 μM) in the absence and the presence of an excess amount (400 μM) of TC-1, respectively. No remarkable change in the chemical shift of the signals from the N-terminal part of Cby is observed. The result agrees with the previous finding that the N-terminal region of Cby is not directly involved in the binding to TC-1 (17). Intriguingly, even though no dramatic peak shifts are observed, the signals originating from the N-terminal portion of Cby sharpened up remarkably upon addition of TC-1. Figure 6c shows the relative intensity (Iwithout TC-1/Iwith TC-1) of the assigned Cby signals in the absence and the presence of unlabeled TC-1. The result quantitatively demonstrates the sharpening of N-terminal peaks of Cby upon binding to TC-1. This effect is more pronounced for residues 20−50, which are distant from the TC-1 binding site on Cby (17). Interestingly, the attenuations in peak intensity become more dramatic at higher ionic strength. In the presence of 100 mM ammonium acetate, a severe peak broadening occurs in the 1H−15N HSQC of Cby, and only a few peaks were observed, probably due to the significant self-association of the protein under this condition. The 1H−15N HSQC spectrum, however, changed dramatically when an excess amount of TC-1 was added to the sample. In the presence of TC-1, most of the peaks from the N-terminal part of Cby show significant increases in intensity without outstanding changes in chemical shifts (Supporting Information Figure S5a−c).
Figure 6.

Titration of unlabeled TC-1 to 15N-labeled Cby (in 10 mM ammonium acetate, pH 5, 25 °C). (a) 1H−15N HSQC spectrum of 60 μM 15N-labeled Cby. (b) 1H−15N HSQC spectrum of 60 μM 15N-labeled Cby + 400 μM unlabeled TC-1. (c) Relative peak intensities of (a) compared to (b), with peak intensities in (b) normalized to 1.
We have also performed 1H−15N HSQC experiments on the 15N-labeled L77A/L91A double mutant samples (60 μM) with different amounts of unlabeled TC-1 added (0, 200, and 400 μM). The results show that the L77A/L91A does not interact with TC-1 significantly as no considerable chemical shift changes have been observed (data not shown). Further, there is no increase in peak intensities upon addition of TC-1 as observed for the wild-type. It is noteworthy that even though the L77A/L91A mutations may weaken the dimerization, the double mutant may not necessarily bind stronger to TC-1. This is because while L77A/L91A is more monomeric, the mutations may also change the helical content of the C-terminus (as indicated by the CD data). Further, the two leucines in question may be directly involved in binding to TC-1. These together may diminish the binding of L77A/L91A to TC-1.
Disordered N-Cby Interacts with 14-3-3ζ
We have also probed the interaction between the N-Cby and 14-3-3ζ using NMR spectroscopy. According to Li et al. (10), upon phosphorylation at serine 20 by Akt1, Cby can bind to the protein 14-3-3ζ in the nucleus. In the present work, the phosphorylation of Cby was mimicked via S20D site-directed mutagenesis. Compared to the wild-type of N-Cby, the single point mutation does not lead to a dramatic change in the overall structure of the protein, judging from the overlay of the 1H−15N HSQC and CD spectra of the wild-type and S20D mutant of N-Cby (Figure 7). The narrow dispersion of 1H−15N HSQC peaks indicates that N-Cby(S20D) remains disordered, and the peaks that show observable chemical shift changes upon mutation correspond to residues nearby the mutation site (Figure 7a).
Figure 7.

Comparison of the wild-type N-Cby and the S20D mutant. (a) 1H−15N HSQC spectra of WT N-Cby (black) and S20D mutant (red). Data were recorded in 10 mM sodium acetate (pH 5) at 25 °C with protein concentrations ∼300 μM. (b) Far-UV CD spectra of WT N-Cby (black) and S20D mutant (red). Data were recorded in 10 mM sodium acetate (pH 5) at 25 °C with protein concentrations ∼60 μM.
Backbone 15N spin-relaxation experiments have been performed to probe the dynamics of the N-Cby(S20D). The results of R1, R2, and steady-state 1H−15N NOE measurements are shown in Figure 8a−c. The relatively small R2 values for most of the residues indicate that the N-Cby(S20D) is highly flexible (Figure 8b). On the other hand, the observed positive NOE values for residues 28−62 suggest that even though the protein is largely unstructured, this particular segment of the protein is relatively restricted in motion (Figure 8c). The result agrees with the SSP analysis showing that residues 20−47 display mild helical propensity, while residues 48−62 may adopt β-strand structures in a significant fraction of conformers in the ensemble (Figure 8d). Compared to the N-Cby(WT), the S20D mutation does not cause significant changes in the flexibility of the protein (Supporting Information Figure S6a−c).
Figure 8.

Structural and dynamical characterization of N-Cby(S20D). Backbone 15N relaxation measurements of N-Cby(S20D) in 10 mM sodium acetate, pH 5, at 25 °C. (a) Longitudinal relaxation rate (R1), (b) transverse relaxation rate (R2), and (c) steady-state 1H−15N NOE. (d) SSP scores for N-Cby(S20D) were calculated on the basis of the assigned 13Cα and 13Cβ chemical shifts.
When the SSP scores of residues in N-Cby (Figure 8d) are compared to the corresponding values in the full-length Cby (Figure 3a), it is clear that the β-strand propensities of residues 50−62 increase upon truncation of the C-terminal part of the protein. This explains our earlier observation that the majority of peaks observed in the 1H−15N HSQC of N-Cby can essentially overlay with the peaks observed in the spectrum of full-length Cby, except for residues 50−63. The increase in β-strand/extended propensity of N-Cby revealed by the SSP calculation also qualitatively agrees with the deconvoluted CD data showing that N-Cby has a higher percentage of β-structure compared to that of the full-length protein (27% vs 10%). Interestingly, residues 50−62 of full-length Cby also display higher β-strand propensities (negative SSP scores) in 4 M urea. The results suggest that the dimerization of Cby via its coiled-coil domain may affect the secondary structure propensity and dynamics of this region of the protein.
The compactness of the N-terminal Cby was also determined by the pulsed field gradient NMR (PFG-NMR) diffusion experiment via the hydrodynamic radius (Rh) measurement. By using dioxane as the reference molecule in the measurements, the Rh of N-Cby(S20D) was determined to be 21.3 ± 0.8 Å. This corresponds to a compaction factor (C) of 0.68 (C has a value between 0 and 1, with C equal to 1 indicating a well-folded globular protein), suggesting that even though the protein is disordered, a significant residual structure is present (39).
To probe the interaction between disordered N-Cby(S20D) and 14-3-3ζ, unlabeled 14-3-3ζ was titrated to 15N-labeled N-Cby(S20D). Upon addition of 14-3-3ζ, the peaks on the 1H−15N HSQC of N-Cby(S20D) are significantly broadened out, albeit in a differential manner with some peaks affected much more than the others (Figure 9a,b). The changes in the relative intensity (Iwith 14-3-3ζ /Iwithout 14-3-3ζ) of all assigned peaks are plotted versus the residue number in Figure 9c. The severe signal broadening is expected to occur when the flexible and lower molecular weight N-Cby(S20D) binds to the dimeric 14-3-3ζ with molecular weight of ∼60 kDa. The differential changes in peak intensity may suggest that some parts of N-Cby(S20D) retain flexibility upon binding to 14-3-3ζ. Moreover, some residues (T17, A19, S22, L27, T31, G37, G41, S42, G49, Q50, A61) also demonstrate subtle chemical shift changes upon addition of 14-3-3ζ, indicating that a weak interaction may exist between these two proteins. We are currently in the process of preparing phosphorylated N-Cby for more detailed NMR characterization of its interaction with 14-3-3ζ.
Figure 9.

Transient interaction between N-Cby(S20D) and 14-3-3ζ. (a) 1H−15N HSQC spectra of ∼100 μM 15N-labeled N-Cby(S20D) (25 mM Tris, pH 7.0, 125 mM NaCl, 1 mM DTT, 1 mM EDTA, at 25 °C) in the absence (black) and the presence (red) of ∼300 μM unlabeled 14-3-3ζ protein. (b) Zoom in of the crowded region in (a). (c) Changes in intensity of N-Cby(S20D) peaks upon addition of 14-3-3ζ with the intensities of the apo form normalized to 1.
Discussion
In this work, we have performed the first detailed structural characterization of the 126-residue human Cby protein, a novel antagonist of β-catenin in the Wnt signaling pathway (9), by using NMR, CD, ESI-MS, and DLS techniques. The experimental and bioinformatic analyses together clearly show that Cby is partially disordered. While the N-terminal portion (residues 1−63) of the protein is intrinsically unstructured, the C-terminal region is strongly α-helical. Within this helical region, a stable four-heptad repeat coiled coil is present between residues 73 and 100. Importantly, our experimental data also add further evidence that Cby undergoes self-association via this putative C-terminal coiled-coil domain. By performing gel filtration chromatography and glutaraldehyde cross-linking analysis on His-tagged Cby purified from HEK293T cells, Mofunanya et al. demonstrated that Cby exists as a homodimer predominantly. Further, their results of coimmunoprecipitation experiments also illustrated that Cby forms an oligomer, which is stable even in the presence of 2 M NaCl or 2 M urea (29). These results very well agree with the observations we reported here.
Besides the dimerization interface, the presence of hydrophobic residues on the solvent-exposed surface of the coiled coil, namely, L90, L93, and I97, may possibly form a “hydrophobic patch”, further enhancing the self-assocation of Cby. Indeed, when these surface hydrophobic residues of the coiled-coil region were replaced with arginines in a synthetic peptide with the sequence resembling residues 70−100 of Cby and with sextuple mutations (N81L/N88L/L90R/L93R/I97R/D100R), the precipitate-prone coiled coil becomes highly soluble even at the physiological conditions of pH and salt concentration, without compromising the coiled-coil structure and stability. The fact that Cby exists in equilibrium between monomeric and dimeric (or even tetrameric) states very well explains the invisibility of NMR peaks from the C-terminal half of the protein, since significant conformational exchange on the intermediate time scale may occur, resulting in severe broadening of the signals. This also poses a substantial challenge in the detailed structural characterization of Cby.
While the C-terminal region of Cby is essential in mediating the interactions with β-catenin (9), TC-1 (17), polycystin-2 (23), importin-α (59), and NBPF1 (60), the N-terminal portion of the protein is also important for target recognition (10,59,60). Despite the lack of stable structure, results of SSP analysis and relaxation measurements indicate that a mild helical propensity exists in residues 20−50, while the rest of the N-Cby adopts transient β-strand structure. Residual structures of IDPs have been shown to play crucial roles in their interactions with targets (61,62). Indeed, the weak helical-like region of N-Cby seems to mediate at least two currently known interrelated biological functions. First, by harboring the binding motif of 14-3-3 proteins (residues 16−22), this region of Cby is involved in the trimolecular complex formation with β-catenin and 14-3-3ζ (10). Second, with a nuclear export signal located between residues 21 and 29, this region of Cby is also crucial for the interaction with CRM1 (chromosomal region maintenance protein 1) nuclear export receptor (59). Overall, the target interactions carried out by the N- and C-terminal of Cby together facilitate the dynamic nuclear−cytoplasmic shuttling of the protein, which in turn controls β-catenin subcellular distribution and signaling (59).
The fact that the N- and C-terminal regions of Cby are somewhat independent may be crucial for the protein to bind to multiple targets via the distinct structural modules located in these two segments. Yet the cooperativity of these two domains allows them to work in unison in eliciting the protein functions. Besides the aforementioned ternary complex with 14-3-3ζ and β-catenin, Cby has also been suggested to form another trimolecular complex with NBPF1 and clusterin, two candidate tumor suppressors linked to neuroblastoma, via its C- and N-terminal regions, respectively (60). Furthermore, based on the structural information of Cby obtained here and the data from the literature, we speculate that the mechanism Cby employs to block the interaction between β-catenin and Tcf4 is through “steric masking”, which involves both the N- and C-terminal regions of the protein, instead of direct competition for an overlapping binding site. By performing the in vitro pull-down assays, Takemaru et al. demonstrated that the sequence of armadillo repeat 10 to the C-terminus of β-catenin is essential for the binding to Cby (9). Surprisingly, this binding site does not overlap with the Tcf4 binding site on β-catenin, and yet Cby can inhibit the Tcf4/β−catenin interaction. One possible explanation for this phenomenon is that, upon binding to Cby, β-catenin undergoes significant conformational change, rendering the protein incapable of binding Tcf4. However, the structures of the armadillo-repeat region of β-catenin and the complexes it forms with a number of targets have been determined (24,26−28,63−66). Judging from these crystal structures, it does not seem that β-catenin undergoes dramatic structural change upon binding to its targets. On the basis of the result of our work, we speculate that Cby binds to β-catenin via its C-terminal helical region and then masks the Tcf4 binding site on β-catenin using its flexible N-terminal portion. This mechanism is very similar to the “anchor-and-kick” mechanism proposed by Graham et al. (64), which ICAT (inhibitor of β-catenin and Tcf4) uses to inhibit the interaction between β-catenin and Tcf4. The crystal structure of the ICAT/β-catenin complex shows that ICAT binds to the third helix of the armadillo repeat 12 of β-catenin via its N-terminal three-helix bundle (63,64). The “anchoring” of the N-terminal ICAT onto β-catenin allows the extended C-terminal tail of ICAT to interact with K312 and K435 of β-catenin, two key residues that mediate the β-catenin/Tcf4 binding. Further experiments confirm that even though the C-terminal tail of ICAT is not crucial for the binding to β-catenin, it is essential for the inhibition of the β-catenin/Tcf4 interaction (64).
Further, the self-associating nature of Cby may also interplay with its binding to targets. The incorporation of a coiled-coil domain into the partially disordered Cby may increase its stability in the absence of targets by self-association. In this work, evidence for the binding of Cby to TC-1 is presented. The sharpening of 1H−15N HSQC signals from the N-terminus of Cby upon addition of TC-1 suggests that the binding of TC-1 to Cby may compete with the self-association process of Cby by itself, thereby leading to changes in the flexibility and time scale of conformational exchange, as well as the molecular tumbling of Cby. However, since Cby interacts with TC-1 also via its C-terminal portion and the binding may not be very tight, signals from that part of the protein are still broad, possibly due to the exchange between the free and bound states on the intermediate time scale of NMR. Further experiments are needed to dissect the detailed molecular mechanism of binding between these two proteins.
Finally, the coexistence of intrinsic disorder with coiled coils appears to be widespread. In a recent large-scale analysis of the mouse intrinsically disordered proteome, Kriwacki and co-workers discovered that a significant number of partially disordered proteins are predicted to also contain coiled-coil domains (67). Since both disordered region and coiled-coil domain are known to mediate protein−protein interactions, the result suggests that the combination of these two structural modules could facilitate disordered proteins in assembling protein complexes via different modes of interaction (67). In line with this proteomic/bioinformatic discovery, several proteins with diverse biological functions have been structurally characterized to contain long disordered regions and coiled-coil motifs recently (68−73). For instance, human C/EBP homologous protein (CHOP), which functions as a Wnt-signaling inhibitor by binding to Tcf (74), has a disordered N-terminal half while its C-terminal portion is α-helical and harbors a putative coiled-coil domain (73). According to Singh et al., the oligomerization of CHOP mediated by the N-terminal domain has a direct implication in its biological function (73). As well, prostate apoptosis response factor-4 (Par-4), a proapoptotic and tumor suppressor protein, also employs both disordered region and coiled-coil domain to interact with multiple targets (72). Structural characterization by CD, DLS, and NMR revealed that, similar to Cby, Par-4 is partially disordered with a C-terminal coiled-coil domain mediating self-association. Intriguingly, over 50% of the residues in the full-length rat Par-4 are also not observable in its 1H−15N HSQC spectrum (72). The authors suggested that this may be due to the conformational exchange between a partially ordered monomer and a predominantly coiled-coil dimer (72,75).
The unique structural and dynamical properties of intrinsically disordered proteins are the reasons why they are frequently found to be involved in signaling pathways (76−78). The structural plasticity allows them to interact with multiple targets with high specificity and yet low binding affinity, which is crucial for the rapid response to signaling events (79,80). It is conceivable that the inclusion of a coiled-coil domain with dimerization and target-binding capabilities may radically expand the overall interaction diversity of IDPs, allowing them to act as “hubs” in protein−protein interaction networks. The ever-expanding number of Cby partners to date may very well be a testament to this: polycystin-2, TC-1, β-catenin, 14-3-3ζ, Akt1, NBPF1, clusterin, CRM-1, and importin-α (9,10,17,23,59,60). Detailed structural studies of Cby and its interactions with targets will undoubtedly augment our current understanding of the role of dual-module disordered and coiled-coil proteins in signaling cascades implicated in health and disease.
Acknowledgments
The authors thank Lee-Ann Briere for help with CD experiments, Dr. John deBruyn and Ron Dauphinee for help in setting up the DLS experiments and data analysis, and Dr. Gary Shaw for critical reading of the manuscript. We also thank the Canadian National High Field NMR Centre (NANUC) for assistance and use of their facilities. Operation of NANUC is funded by the Natural Science and Engineering Research Council of Canada and the University of Alberta.
Supporting Information Available
The coiled-coil model of Cby, results of protein disorder predictions, CD spectra of Cby at different urea concentrations, TROSY-HSQC spectrum of Cby, NMR binding results of Cby and TC-1 at high salt concentration, and backbone relaxation data of N-Cby (WT). This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by grants from the Canadian Institutes of Health Research (CIHR; MOP no. 74679), the Canada Foundation for Innovation (CFI), and the Ontario Government to W.-Y.C. An Ontario Graduate Scholarship in Science and Technology (OGSST) was awarded to S.M., and W.-Y.C. is the recipient of a CIHR New Investigator Award and an Ontario Early Researcher Award.
Footnotes
Abbreviations: APC, adenomatous polyposis coli; Cby, Chibby; C-Cby, C-terminus (residues 64−126) of human Chibby; CD, circular dichroism; CHOP, C/EBP homologous protein; DLS, dynamic light scattering; ESI-MS, electrospray ionization mass spectrometry; GSK-3β, glycogen synthase kinase-3β; HSQC, heteronuclear single-quantum coherence; ICAT, inhibitor of β-catenin and Tcf; N-Cby, N-terminus (residues 1−63) of human Chibby; NMR, nuclear magnetic resonance; Par-4, prostate apoptosis response factor-4; PIGEA-14, polycystin-2 interactor, Golgi- and endoplasmic reticulum-associated protein with a molecular mass of 14 kDa; TC-1, thyroid cancer-1; Tcf/Lef, T cell factors/lymphoid enhancer factor.
Supplementary Material
References
- Clevers H. (2006) Wnt/β-catenin signaling in development and disease. Cell 127, 469–480. [DOI] [PubMed] [Google Scholar]
- Logan C. Y.; Nusse R. (2004) The Wnt signaling pathway in development and disease. Annu. Rev. Cell. Dev. Biol. 20, 781–810. [DOI] [PubMed] [Google Scholar]
- Moon R. T.; Kohn A. D.; De Ferrari G. V.; Kaykas A. (2004) WNT and β-catenin signalling: diseases and therapies. Nat. Rev. Genet. 5, 691–701. [DOI] [PubMed] [Google Scholar]
- Polakis P. (2000) Wnt signaling and cancer. Genes Dev. 14, 1837–1851. [PubMed] [Google Scholar]
- Kimelman D.; Xu W. (2006) β-catenin destruction complex: insights and questions from a structural perspective. Oncogene 25, 7482–7491. [DOI] [PubMed] [Google Scholar]
- Aberle H.; Bauer A.; Stappert J.; Kispert A.; Kemler R. (1997) β-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 16, 3797–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J.; von Kries J. P.; Kuhl M.; Bruhn L.; Wedlich D.; Grosschedl R.; Birchmeier W. (1996) Functional interaction of β-catenin with the transcription factor LEF-1. Nature 382, 638–642. [DOI] [PubMed] [Google Scholar]
- Herbst A.; Kolligs F. T. (2007) Wnt signaling as a therapeutic target for cancer. Methods Mol. Biol. 361, 63–91. [DOI] [PubMed] [Google Scholar]
- Takemaru K.; Yamaguchi S.; Lee Y. S.; Zhang Y.; Carthew R. W.; Moon R. T. (2003) Chibby, a nuclear β-catenin-associated antagonist of the Wnt/Wingless pathway. Nature 422, 905–909. [DOI] [PubMed] [Google Scholar]
- Li F. Q.; Mofunanya A.; Harris K.; Takemaru K. (2008) Chibby cooperates with 14-3-3 to regulate β-catenin subcellular distribution and signaling activity. J. Cell Biol. 181, 1141–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muslin A. J.; Tanner J. W.; Allen P. M.; Shaw A. S. (1996) Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84, 889–897. [DOI] [PubMed] [Google Scholar]
- Yaffe M. B.; Rittinger K.; Volinia S.; Caron P. R.; Aitken A.; Leffers H.; Gamblin S. J.; Smerdon S. J.; Cantley L. C. (1997) The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 91, 961–971. [DOI] [PubMed] [Google Scholar]
- Yang X.; Lee W. H.; Sobott F.; Papagrigoriou E.; Robinson C. V.; Grossmann J. G.; Sundstrom M.; Doyle D. A.; Elkins J. M. (2006) Structural basis for protein-protein interactions in the 14-3-3 protein family. Proc. Natl. Acad. Sci. U.S.A. 103, 17237–17242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemaru K.; Fischer V.; Li F. Q. (2009) Fine-tuning of nuclear-catenin by Chibby and 14-3-3. Cell Cycle 8, 210–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua E. L.; Young L.; Wu W. M.; Turtle J. R.; Dong Q. (2000) Cloning of TC-1 (C8orf4), a novel gene found to be overexpressed in thyroid cancer. Genomics 69, 342–347. [DOI] [PubMed] [Google Scholar]
- Sunde M.; McGrath K. C.; Young L.; Matthews J. M.; Chua E. L.; Mackay J. P.; Death A. K. (2004) TC-1 is a novel tumorigenic and natively disordered protein associated with thyroid cancer. Cancer Res. 64, 2766–2773. [DOI] [PubMed] [Google Scholar]
- Jung Y.; Bang S.; Choi K.; Kim E.; Kim Y.; Kim J.; Park J.; Koo H.; Moon R. T.; Song K.; Lee I. (2006) TC1 (C8orf4) enhances the Wnt/β-catenin pathway by relieving antagonistic activity of Chibby. Cancer Res. 66, 723–728. [DOI] [PubMed] [Google Scholar]
- Gall C.; Xu H.; Brickenden A.; Ai X.; Choy W. Y. (2007) The intrinsically disordered TC-1 interacts with Chibby via regions with high helical propensity. Protein Sci. 16, 2510–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. Q.; Singh A. M.; Mofunanya A.; Love D.; Terada N.; Moon R. T.; Takemaru K. (2007) Chibby promotes adipocyte differentiation through inhibition of β-catenin signaling. Mol. Cell. Biol. 27, 4347–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuierer M. M.; Graf E.; Takemaru K.; Dietmaier W.; Bosserhoff A. K. (2006) Reduced expression of β-catenin inhibitor Chibby in colon carcinoma cell lines. World J. Gastroenterol. 12, 1529–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A. M.; Li F. Q.; Hamazaki T.; Kasahara H.; Takemaru K.; Terada N. (2007) Chibby, an antagonist of the Wnt/β-catenin pathway, facilitates cardiomyocyte differentiation of murine embryonic stem cells. Circulation 115, 617–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronina V. A.; Takemaru K.; Treuting P.; Love D.; Grubb B. R.; Hajjar A. M.; Adams A.; Li F. Q.; Moon R. T. (2009) Inactivation of Chibby affects function of motile airway cilia. J. Cell Biol. 185, 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidaka S.; Konecke V.; Osten L.; Witzgall R. (2004) PIGEA-14, a novel coiled-coil protein affecting the intracellular distribution of polycystin-2. J. Biol. Chem. 279, 35009–35016. [DOI] [PubMed] [Google Scholar]
- Huber A. H.; Nelson W. J.; Weis W. I. (1997) Three-dimensional structure of the armadillo repeat region of β-catenin. Cell 90, 871–882. [DOI] [PubMed] [Google Scholar]
- Xing Y.; Takemaru K.; Liu J.; Berndt J. D.; Zheng J. J.; Moon R. T.; Xu W. (2008) Crystal structure of a full-length β-catenin. Structure 16, 478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham T. A.; Ferkey D. M.; Mao F.; Kimelman D.; Xu W. (2001) Tcf4 can specifically recognize β-catenin using alternative conformations. Nat. Struct. Biol. 8, 1048–1052. [DOI] [PubMed] [Google Scholar]
- Graham T. A.; Weaver C.; Mao F.; Kimelman D.; Xu W. (2000) Crystal structure of a β-catenin/Tcf complex. Cell 103, 885–896. [DOI] [PubMed] [Google Scholar]
- Poy F.; Lepourcelet M.; Shivdasani R. A.; Eck M. J. (2001) Structure of a human Tcf4-β-catenin complex. Nat. Struct. Biol. 8, 1053–1057. [DOI] [PubMed] [Google Scholar]
- Mofunanya A.; Li F. Q.; Hsieh J. C.; Takemaru K. (2009) Chibby forms a homodimer through a heptad repeat of leucine residues in its C-terminal coiled-coil motif. BMC Mol. Biol. 10, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreerama N.; Woody R. W. (2000) Estimation of protein secondary structure from CD spectra: comparison of CONTIN, SELCON and CDSSTR methods with an expanded reference set. Anal. Biochem. 282, 252–260. [DOI] [PubMed] [Google Scholar]
- Blouwolff J.; Fraden S. (2007) Crystallization conditions of membrane protein CLC-ec1: an example outside the crystallization slot. J. Cryst. Growth 303, 546–553. [Google Scholar]
- Koppel D. E. (1972) Analysis of macromolecular polydispersity in intensity correlation spectroscopy: the method of cumulants. J. Chem. Phys. 57, 4814–4820. [Google Scholar]
- Clubb R. T.; Thanabal V.; Wagner G. (1992) A constant-time three-dimensional triple-resonance pulse scheme to correlate intraresidue 1HN, 15N, 13C′ chemical shifts in 15N-13C-labeled proteins. J. Magn. Reson. 97, 213–217. [Google Scholar]
- Grzesiek S.; Anglister J.; Bax A. (1993) Correlation of backbone amide and aliphatic side-chain resonances in 13C/15N-enriched proteins by isotropic mixing of 13C magnetization. J. Magn. Reson. B 101, 114–119. [Google Scholar]
- Muhandiram D. R.; Kay L. E. (1994) Gradient-enhanced triple-resonance three-dimensional NMR experiments with improved sensitivity. J. Magn. Reson. B 103, 203–216. [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Johnson B. A. (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 278, 313–352. [DOI] [PubMed] [Google Scholar]
- Altieri A. S.; Hinton D. P.; Byrd R. A. (1995) Association of biomolecular systems via pulsed field gradient NRM self-diffusion measurements. J. Am. Chem. Soc. 117, 7566–7567. [Google Scholar]
- Wilkins D. K.; Grimshaw S. B.; Receveur V.; Dobson C. M.; Jones J. A.; Smith L. J. (1999) Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 38, 16424–16431. [DOI] [PubMed] [Google Scholar]
- Akke M.; Palmer A. G. (1996) Monitoring macromolecular motions on microsecond-millisecond time scales by R1ρ and R1 constant relaxation time NMR spectroscopy. J. Am. Chem. Soc. 118, 911–912. [Google Scholar]
- Farrow N. A.; Muhandiram R.; Singer A. U.; Pascal S. M.; Kay C. M.; Gish G.; Shoelson S. E.; Pawson T.; Forman-Kay J. D.; Kay L. E. (1994) Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33, 5984–6003. [DOI] [PubMed] [Google Scholar]
- Davis D. G.; Perlman M. E.; London R. E. (1994) Direct measurements of the dissociation-rate constant for inhibitor-enzyme complexes via the T1 rho and T2 (CPMG) methods. J. Magn. Reson. B 104, 266–275. [DOI] [PubMed] [Google Scholar]
- Lupas A. (1996) Prediction and analysis of coiled-coil structures. Methods Enzymol. 266, 513–525. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Gillespie J. R.; Fink A. L. (2000) Why are “natively unfolded” proteins unstructured under physiologic conditions?. Proteins 41, 415–427. [DOI] [PubMed] [Google Scholar]
- Li X.; Romero P.; Rani M.; Dunker A. K.; Obradovic Z. (1999) Predicting protein disorder for N-, C-, and internal regions. Genome Inform. Ser. Workshop Genome Inform. 10, 30–40. [PubMed] [Google Scholar]
- Obradovic Z.; Peng K.; Vucetic S.; Radivojac P.; Dunker A. K. (2005) Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins 61, Suppl. 7176–182. [DOI] [PubMed] [Google Scholar]
- Romero P.; Obradovic Z.; Li X.; Garner E. C.; Brown C. J.; Dunker A. K. (2001) Sequence complexity of disordered protein. Proteins 42, 38–48. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. (2002) Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 11, 739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh J. A.; Singh V. K.; Jia Z.; Forman-Kay J. D. (2006) Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synuclein: implications for fibrillation. Protein Sci. 15, 2795–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Jardetzky O. (2002) Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 11, 852–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Sykes B. D. (1994) Chemical shifts as a tool for structure determination. Methods Enzymol. 239, 363–392. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Sykes B. D. (1994) The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 2, 171–180. [DOI] [PubMed] [Google Scholar]
- Lau S. Y.; Taneja A. K.; Hodges R. S. (1984) Synthesis of a model protein of defined secondary and quaternary structure. Effect of chain length on the stabilization and formation of two-stranded alpha-helical coiled-coils. J. Biol. Chem. 259, 13253–13261. [PubMed] [Google Scholar]
- Cech N. B.; Enke C. G. (2001) Practical implication of some recent studies in electrospray ionization fundamentals. Mass Spectrom. Rev. 20, 362–387. [DOI] [PubMed] [Google Scholar]
- Kuprowski M. C.; Konermann L. (2007) Signal response of co-existing protein conformers in electrospray mass spectrometry. Anal. Biochem. 79, 2499–2506. [DOI] [PubMed] [Google Scholar]
- Pervushin K.; Riek R.; Wider G.; Wuthrich K. (1997) Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anistropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. U.S.A. 94, 12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X.; Herndon A. M.; Hu J. C. (1997) Buried asparagines determine the dimerization specificities of leucine zipper mutants. Proc. Natl. Acad. Sci. U.S.A. 94, 3673–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfson D. N. (2005) The design of coiled-coil structure and assemblies. Adv. Protein Chem. 70, 79–112. [DOI] [PubMed] [Google Scholar]
- Li F. Q.; Mofunanya A.; Fischer V.; Hall J.; Takemaru K. I. (2010) Nuclear-cytoplasmic shuttling of Chibby controls beta-catenin signaling. Mol. Biol. Cell 21, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandepoele K.; Staes K.; Andries V.; van Roy F. (2010) Chibby interacts with NBPF1 and clusterin, two candidate tumor suppressors linked to neuroblastoma. Exp. Cell Res. 316, 1225–1233. [DOI] [PubMed] [Google Scholar]
- Fuxreiter M.; Simon I.; Friedrich P.; Tompa P. (2004) Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol. 338, 1015–1026. [DOI] [PubMed] [Google Scholar]
- Oldfield C. J.; Cheng Y.; Cortese M. S.; Romero P.; Uversky V. N.; Dunker A. K. (2005) Coupled folding and binding with α-helix-forming molecular recognition elements. Biochemistry 44, 12454–12470. [DOI] [PubMed] [Google Scholar]
- Daniels D. L.; Weis W. I. (2002) ICAT inhibits β-catenin binding to Tcf/Lef-family transcription factors and the general coactivator p300 using independent structural modules. Mol. Cell 10, 573–584. [DOI] [PubMed] [Google Scholar]
- Graham T. A.; Clements W. K.; Kimelman D.; Xu W. (2002) The crystal structure of the β-catenin/ICAT complex reveals the inhibitory mechanism of ICAT. Mol. Cell 10, 563–571. [DOI] [PubMed] [Google Scholar]
- Huber A. H.; Weis W. I. (2001) The structure of the β-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by β-catenin. Cell 105, 391–402. [DOI] [PubMed] [Google Scholar]
- Spink K. E.; Fridman S. G.; Weis W. I. (2001) Molecular mechanisms of β-catenin recognition by adenomatous polyposis coli revealed by the structure of an APC-β-catenin complex. EMBO J. 20, 6203–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea C. A.; High A. A.; Obenauer J. C.; Mishra A.; Park C. G.; Punta M.; Schlessinger A.; Ma J.; Rost B.; Slaughter C. A.; Kriwacki R. W. (2009) Large-scale analysis of thermostable, mammalian proteins provides insights into the intrinsically disordered proteome. J. Proteome Res. 8, 211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbar E. (2008) Dynein light chain LC8 is a dimerization hub essential in diverse protein networks. Biochemistry 47, 503–508. [DOI] [PubMed] [Google Scholar]
- Benison G.; Nyarko A.; Barbar E. (2006) Heteronuclear NMR identifies a nascent helix in intrinsically disordered dynein intermediate chain: implications for folding and dimerization. J. Mol. Biol. 362, 1082–1093. [DOI] [PubMed] [Google Scholar]
- Carrica M. C.; Craig P. O.; Alonso S. V.; Goldbaum F. A.; Cravero S. L. (2008) Brucella abortus MFP: a trimeric coiled-coil protein with membrane fusogenic activity. Biochemistry 47, 8165–8175. [DOI] [PubMed] [Google Scholar]
- Gazi A. D.; Bastaki M.; Charova S. N.; Gkougkoulia E. A.; Kapellios E. A.; Panopoulos N. J.; Kokkinidis M. (2008) Evidence for a coiled-coil interaction mode of disordered proteins from bacterial type III secretion systems. J. Biol. Chem. 283, 34062–34068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libich D. S.; Schwalbe M.; Kate S.; Venugopal H.; Claridge J. K.; Edwards P. J.; Dutta K.; Pascal S. M. (2009) Intrinsic disorder and coiled-coil formation in prostate apoptosis response factor 4. FEBS J. 276, 3710–3728. [DOI] [PubMed] [Google Scholar]
- Singh V. K.; Pacheco I.; Uversky V. N.; Smith S. P.; MacLeod R. J.; Jia Z. (2008) Intrinsically disordered human C/EBP homologous protein regulates biological activity of colon cancer cells during calcium stress. J. Mol. Biol. 380, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horndasch M.; Lienkamp S.; Springer E.; Schmitt A.; Pavenstadt H.; Walz G.; Gloy J. (2006) The C/EBP homologous protein CHOP (GADD153) is an inhibitor of Wnt/TCF signals. Oncogene 25, 3397–3407. [DOI] [PubMed] [Google Scholar]
- Schwalbe M.; Dutta K.; Libich D. S.; Venugopal H.; Claridge J. K.; Gell D. A.; MacKay J. P.; Edwards P. J. B.; Pascal S. M. (2010) Two-state conformational equilibrium in the Par-4 leucine zipper domain. Proteins 78, 2433–2449. [DOI] [PubMed] [Google Scholar]
- Mittag T.; Kay L. E.; Forman-Kay J. D. (2010) Protein dynamics and conformational disorder in molecular recognition. J. Mol. Recognit. 23, 105–116. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Oldfield C. J.; Midic U.; Xie H.; Xue B.; Vucetic S.; Iakoucheva L. M.; Obradovic Z.; Dunker A. K. (2009) Unfoldomics of human diseases: linking protein intrinsic disorder with diseases. BMC Genomics 10, Suppl. 1S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright P. E.; Dyson H. J. (2009) Linking folding and binding. Curr. Opin. Struct. Biol. 19, 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson H. J.; Wright P. E. (2002) Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 12, 54–60. [DOI] [PubMed] [Google Scholar]
- Dyson H. J.; Wright P. E. (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell. Biol. 6, 197–208. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.