

Abstract
Human papillomavirus (HPV), particularly type 16, has been associated with a subset of head and neck cancers. The viral-encoded oncogenic proteins E6 and E7 represent ideal targets for immunotherapy against HPV-associated head and neck cancers. DNA vaccines have emerged as attractive approaches for immunotherapy due to its simplicity, safety, and ease of preparation. Intradermal administration of DNA vaccine via gene gun represents an efficient method to deliver DNA directly into dendritic cells for priming antigen-specific T cells. We have previously shown that a DNA vaccine encoding an invariant chain (Ii), in which the class II-associated Ii peptide (CLIP) region has been replaced by a Pan-DR-epitope (PADRE) sequence to form Ii-PADRE, is capable of generating PADRE-specific CD4+ T cells in vaccinated mice. In the current study, we hypothesize a DNA vaccine encoding Ii-PADRE linked to E6 (Ii-PADRE-E6) will further enhance E6-specific CD8+ T cell immune responses through PADRE-specific CD4+ T helper cells. We found that mice vaccinated with Ii-PADRE-E6 DNA generated comparable levels of PADRE-specific CD4+ T cell immune responses as well as significantly stronger E6-specific CD8+ T cell immune responses and antitumor effects against the lethal challenge of E6-expressing tumor compared to mice vaccinated with Ii-E6 DNA. Taken together, our data indicates that vaccination with Ii-E6 DNA with PADRE replacing the CLIP region is capable of enhancing the E6-specific CD8+ T cell immune response generated by the Ii-E6 DNA. Thus, Ii-PADRE-E6 represents a novel DNA vaccine for the treatment of HPV-associated head and neck cancer and other HPV-associated malignancies.
Keywords: Head and Neck Cancer, HPV, DNA vaccine, Invariant chain, PADRE, E6
Introduction
Head and neck cancer accounts for about 3% to 5% of all cancers in the United States based on a report from the American Cancer Society. In 2010, it is estimated that 49,260 people will develop head and neck cancer, and approx. 11,480 deaths will occur 1. Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer worldwide 2. With a 5-year survival rate of 50%, HNSCC has one of the lowest survival rates associated with the major cancers 2. Thus, head and neck cancer requires an alternative treatment for its control.
Recently it has been found that a subset of HNSCC is associated with human papillomavirus (HPV). The HPV-associated head and neck tumors are derived mainly from the oropharynx, including the tonsil and base of tongue (for review see 3). Thus, immunotherapy targeting HPV antigens has emerged as a promising approach for the treatment of head and neck cancers 4. More than 90% of HPV-associated HNSCC are caused by HPV type 16 5. The HPV antigens E6 and E7 represent ideal targets since they are constantly expressed, essential for tumor phenotype and not expressed on normal cells and contribute to tumor progression (for review see 6). Thus, they represent ideal targets for developing antigen-specific immunotherapy for the control of head and neck cancers.
DNA vaccines have emerged as an attractive antigen-specific immunotherapy due to their simplicity, safety, ability to be constantly delivered without being neutralized, ease of preparation, stability at room temperature, and relatively cheap cost compared to other biotherapy agents (for reviews see 7,8). Using gene gun, the vaccine will be delivered into the skin where many immature dendritic cells (DCs), also called Langerhans cells, are located. It is now clear that dendritic cells are key players in initiating an immune response. Employment of gene gun has been shown to allow direct delivery of DNA into the DCs 9,10. Thus, this system will allow us to employ various strategies to modify the multiple properties of dendritic cells to improve DNA vaccine potency.
We have used the gene gun delivery system to test several DNA vaccines encoding HPV type E6/E7 antigens (for review, see 11). We have previously used this system to show that a DNA vaccine encoding an invariant chain (Ii), in which the class II-associated Ii peptide (CLIP) region has been replaced by a Pan-DR-epitope (PADRE) sequence to form Ii-PADRE, is capable of generating PADRE-specific CD4+ T cells in vaccinated mice 12. The PADRE motif is a high-affinity and “promiscuous” CD4+ T cell epitope and has been shown to bind to different human as well as mouse MHC class II molecules 13. Thus, the PADRE peptide has been widely used by us as well as other groups for enhancing CD4+ T helper cells 12,14–16. In addition, it has been shown that DNA vaccines encoding Ii linked to a model antigen on the carboxyl end of Ii will enhance antigen-specific CD8+ T cell immune responses in vaccinated mice 17.
In the current study, we reason that a combination of these approaches by generating a DNA vaccine encoding Ii-PADRE linked to E6 (Ii-PADRE-E6) will further enhance E6-specific CD8+ T cell immune responses through PADRE-specific CD4+ T helper cells. Our data indicate that vaccination with Ii-PADRE-E6 generates potent E6-specific CD8+ T cell immune responses and antitumor effects against E6-expressing tumors, thus representing an innovative DNA vaccine for the treatment of HPV-associated head and neck cancer and other HPV-associated malignancies. The clinical implications of the current study are discussed.
Results
The various E6 DNA constructs expressed comparable levels of E6 RNA
In the current study we planned to test whether a DNA vaccine encoding Ii-PADRE linked to E6 (Ii-PADRE-E6) will further enhance E6-specific CD8+ T cell immune responses through PADRE-specific CD4+ T helper cells. In order to test our hypothesis, we have come up with various DNA constructs, including pcDNA3-E6, pcDNA3-Ii, pcDNA3-Ii-E6, pcDNA3-Ii-PADRE, and pcDNA3-Ii-PADRE-E6 (see Figure 1). In order to characterize the E6 RNA expression of the various E6 DNA constructs, we performed quantitative RT-PCR to demonstrate the relative expression of E6 gene. DC cell line was transfected with pcDNA3-E6, Ii-E6, or PADRE-E6 plasmid. As shown in Supplementary Figure 1, we found that the various E6 DNA constructs expressed comparable levels of E6 RNA.
Figure 1. Schematic diagram depicting the various pcDNA3 constructs.

For the pcDNA3-Ii-E6 construct, the E6 gene was cloned at the carboxyl end of the invariant chain (Ii). For the pcDNA3-Ii-PADRE construct, the PADRE sequence was cloned replacing the CLIP region of the Ii between aa81 and 101 of the Ii chain. For the pcDNA3-Ii-PADRE-E6 construct, the E6 gene was cloned at the carboxyl end of the Ii and PADRE sequence was cloned replacing the CLIP region of the Ii.
Linkage of E6 to the carboxyl end of Ii or Ii-PADRE is able to improve E6 antigen processing and presentation through MHC class I molecules
We then wanted to determine whether the linkage of E6 to the carboxyl end of Ii or Ii-PADRE could enhance E6 antigen processing and presentation through MHC class I molecules. We did this by characterizing the activation of the E6-specific T cell line by 293-Kb cells transfected with the various DNA constructs encoding Ii, PADRE, E6, Ii-E6, and PADRE-E6 respectively. The cells were then analyzed using intracellular cytokine staining by staining with CD8 and IFN-γ. As shown in Figure 2, 293-Kb transfected with DNA encoding Ii-E6 and Ii-PADRE-E6 generated significantly higher frequency of activated IFN-gamma secreting E6-specific CD8+ T cells compared to 293-Kb transfected with Ii, Ii-PADRE, or E6 DNA. These results indicate that linking of E6 to the carboxyl end of Ii or Ii-PADRE is capable of improving E6 antigen processing and presentation through the MHC class I molecule to activate a greater frequency of E6-specific CD8+ T cells.
Figure 2. Characterization of the activation of the E6-specific T cell line by 293-Kb cells transfected with the various DNA constructs.

293 cells expressing murine H-2Kb (293-Kb) were transfected with pcDNA3 encoding Ii, PADRE, E6, Ii-E6, and PADRE-E6 respectively using Lipofectamine 2000. 24 hours later, transfected cells were used to stimulate a H-2Kb restricted, HPV-16 E6 aa48–57 specific T cell line for 20 hours at the presence of 1μl/ml of GolgiPlug. 293-Kb cells pulsed with HPV16 E6aa50–57 peptides were used as positive control. The cells were then analyzed using intracellular cytokine staining by staining with CD8 and IFN-γ. A. Representative flow cytometry data demonstrating the percentage of IFN-γ+ CD8+ T cells out of total CD8+ T cells. B. Bar graph depicting the percentage of IFN-γ+ CD8+ T cells out of total CD8+ T cells. The data shown here are from one representative experiment of two performed.
Vaccination with Ii-PADRE-E6 generated the highest frequency of E6-specific CD8+ T cells in vaccinated mice
To determine whether Ii-PADRE-E6 could generate the best E6-specific CD8+ T cell immune responses in vaccinated mice, we used C57BL/6 mice (5 per group) and vaccinated the mice intradermally via gene gun with 2 μg/mouse of Ii, Ii-E6, Ii-PADRE, Ii-PADRE-E6 DNA vaccines. The mice received a boost with the same dose and regimen of the DNA vaccines four days later. One week after the last vaccination, the splenocytes from vaccinated mice were harvested and characterized for E6-specific CD8+ T cells. The presence of E6-specific CD8+ T cells was determined by CD8-specific antibodies as well as intracellular cytokine staining for interferon gamma. As shown in Figure 3, mice vaccinated with Ii-PADRE-E6 DNA vaccine generated the highest frequency of E6-specific CD8+ T cells in mice among all the groups (p<0.05). Ii-E6 DNA vaccine also generated higher frequency of E6-specific CD8+ T cells compared to E6 DNA although not as high as Ii-PADRE-E6 DNA. Thus, the replacement of CLIP with PADRE in the Ii-E6 construct is able to further enhance E6-specific CD8+ T cells.
Figure 3. Characterization of the E6-specific CD8+ T cell immune responses in mice vaccinated with the various DNA constructs.

C57BL/6 mice (5 per group) were vaccinated with the various DNA constructs via gene gun delivery at a dose of 2μg/mouse. Four days later, mice were boosted with the same dose and regimen. One week after last vaccination, splenocytes from mice were harvested and characterized for E6-specific CD8+ T cells using intracellular IFN-γ staining followed by flow cytometry analysis. A. Representative data of intracellular cytokine staining followed by flow cytometry analysis showing the frequency of E6-specific IFNγ+ CD8+ T cells in after DNA vaccination. B. Bar graph depicting the number of E6-specific IFNγ+ CD8+ T cells per 2×105 splenocytes ±SEM following DNA vaccination. The data shown here are from one representative experiment of two performed.
We also characterized the E6-specific CD8+ T cell responses in mice vaccinated simultaneously with Ii-E6 DNA and Ii-PADRE DNA at the same site compared to mice vaccinated with Ii-PADRE-E6 DNA + Ii DNA (in order to match the amount of E6 and the total amount of DNA in the vaccination). We found that mice vaccinated with simultaneously with Ii-E6 DNA and Ii-PADRE DNA at the same site generated comparable E6-specific CD8+ T cell immune responses to mice vaccinated with Ii-PADRE-E6 + Ii DNA (Supplementary Figure 2). Taken together, our data suggests that the enhancement of the E6-specfic CD8+ T cell immune responses can be contributed by co-administration with Ii-PADRE DNA or linkage of Ii-PADRE to E6 DNA construct (Ii-PADRE-E6 DNA).
Both Ii-PADRE and Ii-PADRE-E6 generate significantly higher frequency of PADRE-specific CD4+ T cells in vaccinated mice
In order to determine whether the replacement of CLIP by PADRE in Ii-PADRE and Ii-PADRE-E6 can lead to the generation of PADRE-specific CD4+ T cell immune responses in vaccinated mice, we used C57BL/6 mice (5 per group) and vaccinated them as described in Figure 3. One week after the last vaccination, the splenocytes from vaccinated mice were harvested and characterized for PADRE-specific CD4+ T cells. The presence of PADRE-specific CD4+ T cells was determined by CD4-specific antibodies as well as intracellular cytokine staining for interferon gamma. As shown in Figure 4, mice vaccinated with Ii-PADRE-E6 or Ii-PADRE DNA vaccine both generated significantly higher frequency of PADRE-specific CD4+ T cells compared to mice vaccinated with E6, Ii, and Ii-E6, although Ii-PADRE-E6 generated significantly lower numbers of PADRE-specific CD4+ T cells than Ii-PADRE (p<0.05). Thus, the replacement of CLIP with PADRE in the Ii and Ii-E6 construct is able to generate a significant frequency of PADRE-specific CD4+ T cells in vaccinated mice.
Figure 4. Characterization of the PADRE-specific CD4+ T cell immune responses in mice vaccinated with the various DNA constructs.
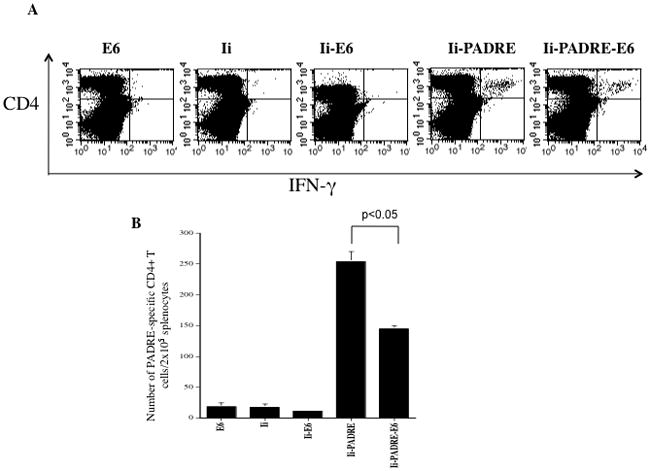
C57BL/6 mice (5 per group) were vaccinated with the various DNA constructs via gene gun delivery at a dose of 2μg/mouse. Four days later, mice were boosted with the same dose and regimen. One week after last vaccination, splenocytes from mice were harvested and characterized for PADRE-specific CD4+ T cells using intracellular IFN-γ staining followed by flow cytometry analysis. A. Representative data of intracellular cytokine staining followed by flow cytometry analysis showing the number of PADRE-specific IFNγ+ CD4+ T cells in after DNA vaccination. B. Bar graph depicting the number of PADRE-specific IFNγ+ CD4+ T cells per 2×105 splenocytes ± SEM following DNA vaccination. The data shown here are from one representative experiment of two performed.
Vaccination with Ii-PADRE-E6 generated a potent protective anti-tumor effect against E6-expressing tumors
In order to determine whether the observed enhanced E6-specific CD8+ T cell immune response generated by Ii-PADRE-E6 can be translated into enhanced protective anti-tumor effects, we first vaccinated C57BL/6 mice intradermally via gene gun with 2 μg/mouse of Ii, Ii-E6, Ii-PADRE, Ii-PADRE-E6 DNA vaccines. Four days later, the mice received a boost with the same dose and regimen of the DNA vaccines. Seven days after the last vaccination, the mice were challenged with a luciferase-expressing E6-positive murine tumor cell line, TC-1/luc in the buccal mucosa of the oral region. The tumor growth of the mice was followed by a non-invasive luminescence imaging system. As shown in Figure 5, both Ii-E6 and Ii-PADRE-E6 DNA vaccines generated a better protective anti-tumor effect against E6-expressing tumors in vaccinated mice among all the tested DNA vaccines (p<0.05). Thus, our data indicates that vaccination with Ii-PADRE-E6 generates a potent protective anti-tumor effect against E6-expressing tumors.
Figure 5. Characterization of protective antitumor effects by vaccination with various DNA constructs.
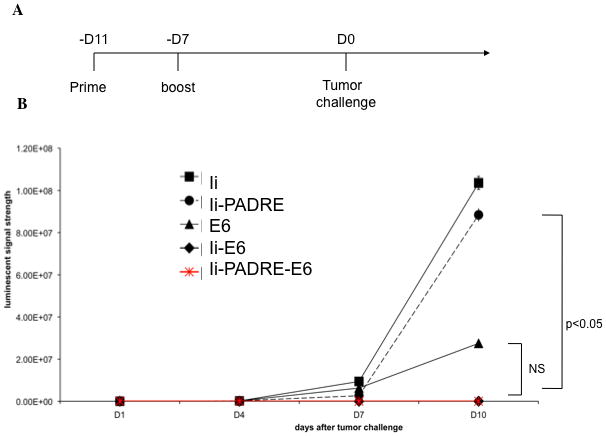
A. Schematic diagram demonstrating the time for prime boost and tumor challenge. C57BL/6 mice (5 per group) were primed with the various DNA vaccines at a dose of 2μg/mouse 11 days before tumor challenge. Mice were boosted with the same dose and regimen 4 days later. 7 days after the last vaccination, mice were orally inoculated at the buccal mucosa with 2 × 105 TC-1 cells/mouse. B. Graphical representation of the tumor growth in mice treated with the various DNA constructs (mean ± s.d.). The data shown here are from one representative experiment of two performed.
CD4+ and CD8+ T cells are essential for the antitumor effects generated by vaccination with Ii-PADRE-E6 DNA vaccine
In order to determine the subset of lymphocytes that are essential for the observed protective antitumor effects, mice were first primed and boosted with Ii-PADRE-E6 DNA vaccine. The vaccinated mice were depleted of CD4, CD8, or NK cells with various antibodies as described in the Materials and Methods. The antibody-depleted mice were then challenged with luciferase-expressing E6-positive murine tumor cell line, TC-1/luc in the buccal mucosa of the oral region. The tumor growth of the mice was followed by a non-invasive luminescence imaging system. As shown in Figure 6, the depletion of CD4+ or CD8+ T cells led to the significant loss of antitumor effects generated by the vaccination with Ii-PADRE-E6 DNA. In comparison, the depletion of NK cells did not lead to a loss of antitumor effects generated by the Ii-PADRE-E6 DNA vaccination. Taken together, these results indicate that both CD4 and CD8 T cells are essential for the antitumor effects mediated by Ii-PADRE-E6 DNA vaccine.
Figure 6. Characterization of the subset of lymphocytes contributing to the antitumor effects by in vivo antibody depletion.

A. Schematic diagram to depict the time course of vaccination depletion and tumor challenge. C57BL/6 mice (5 per group) were primed with the pcDNA3-Ii-PADRE-E6 vaccine at a dose of 2μg/mouse 11 days before tumor challenge. Mice were boosted with the same dose and regimen 4 days later. Mice underwent antibody depletion of CD4, CD8, or NK cells one day after the last vaccination. 7 days after the last vaccination, mice were orally inoculated at the buccal mucosa with 2 × 105 TC-1 cells/mouse. B. Graphical representation of the tumor growth in mice treated with pcDNA3-Ii-PADRE-E6 (mean± s.d.). C. Kaplan-Meier survival analysis of TC-1 tumor-bearing mice treated with pcDNA3-Ii-PADRE-E6. The data shown here are from one representative experiment of two performed.
Vaccination with Ii-PADRE-E6 generated the best therapeutic anti-tumor effects against E6-expressing tumors
In order to further determine whether the vaccination with Ii-PADRE-E6 can lead to enhanced therapeutic anti-tumor effects, we first challenged mice with luciferase-expressing E6-positive murine tumor cell line, TC-1/luc in the buccal mucosa of the oral region. Three days later, tumor-bearing-mice were vaccinated intradermally via gene gun with 2 μg/mouse of Ii, Ii-E6, Ii-PADRE, Ii-PADRE-E6 DNA vaccines. One week after tumor challenge, the mice received a boost with the same dose and regimen of the DNA vaccines. The tumor growth was followed by luminescence imaging twice a week following tumor challenge. As shown in Figure 7, Ii-PADRE-E6 DNA vaccine generated the best therapeutic anti-tumor effects against E6-expressing tumors in vaccinated mice among all the tested DNA vaccines. Furthermore, treatment with Ii-PADRE-E6 DNA generated the best over-all survival among all the DNA-treated groups. Taken together, Ii-PADRE-E6 is able to generate the best anti-tumor immune responses in tumor-challenged mice.
Figure 7. Characterization of antitumor effects against luciferase-expressing TC-1 tumors in mice treated with the various DNA constructs.


A. Schematic diagram to depict the time course for tumor challenge, prime and boost vaccination. C57BL/6 mice (5 per group) were orally inoculated at the buccal mucosa with 2 × 105 TC-1 cells/mouse on D0. Three days after tumor inoculation, all the tumor-bearing mice were treated with the various DNA constructs (Ii-E6, E6, Ii-PADRE or Ii-PADRE-E6) intradermally via gene gun at a dose of 2μg/mouse. Mice were boosted with the same dose and regimen 7 days later. Mice were imaged using the IVIS Imaging System Series 200. B. Representative bioluminescence images of TC-1 tumor-bearing mice treated with the various DNA constructs. Images were acquired on day 3, 7, 10, 13 and 16 after tumor inoculation. C. Graphical representation of the tumor growth in mice treated with the various DNA constructs (mean ± s.d.). D. Kaplan-Meier survival analysis of TC-1 tumor-bearing mice treated with the various DNA constructs. The data shown here are from one representative experiment of two performed.
Discussion
Our study demonstrated that vaccination with the Ii-PADRE-E6 DNA generated significantly higher frequency of E6-specific CD8+ T cells compared to vaccination with Ii-E6 DNA. We believe the PADRE-specific CD4+ T cells generated by the vaccination with the Ii-PADRE-E6 DNA likely contributed to the improved frequency of E6-specific CD8+ T cells. Our results are consistent with our previous studies using DNA vaccines co-administered with DNA encoding Ii-PADRE. This result may be due to the important roles of CD4+ T cells in facilitating the activation and proliferation of antigen-specific CD8+ T cells.
We observed that mice vaccinated with Ii-PADRE-E6 DNA vaccine had slightly lower PADRE-specific CD4+ T cells compared to mice vaccinated with Ii-PADRE DNA. This differs from our original expectations that mice vaccinated with Ii-PADRE-E6 DNA would generate comparable levels of PADRE-specific CD4+ T cell immune responses compared to mice vaccinated with Ii-PADRE DNA. The reduction of PADRE-specific CD4+ T cells may be related to the enhanced CD8+ T cells generated by Ii-PADRE-E6 16. Dendritic cells transfected with Ii-PADRE-E6 not only present PADRE-peptide through MHC II pathway to activate PADRE-specific CD4+ T cells, but also present E6-peptide through MHC I molecules to activate E6-specific CD8+ T cells. As a result, 293-Kb cells transfected with Ii-PADRE-E6 can serve as targets for specific killing by the generated E6-specific CD8+ T cells. In comparison, dendritic cells transfected with Ii-PADRE will not present E6-peptide through MHC I molecules and thus will not become targets for E6-specific T cell killing. This may explain why there is a reduction in the frequency of PADRE-specific CD4+ T cells in mice vaccinated with Ii-PADRE-E6 compared to mice vaccinated with Ii-PADRE.
We observed that the linkage of E6 antigen directly to the carboxyl end of Ii or Ii-PADRE can lead to enhanced E6 antigen processing and presentation through MHC class I molecules in cells transfected with Ii-E6 or Ii-PADRE-E6 DNA compared to cells transfected with E6 DNA (see Figure 2). Our data is consisting with previous studies using other antigenic systems 17,18. Targeting of an antigen to the endoplasmic reticulum (ER) can lead to enhanced E6 antigen processing on MHC I.
In our study, we observed that vaccination with Ii-PADRE-E6 generated the best therapeutic anti-tumor effects against E6-expressing tumors (See Figure 7). Although the vaccination with DNA encoding Ii-E6 also generated significantly higher level of E6-specific CD8+ T cells than DNA encoding E6 (Figure 3), it did not translate into a better therapeutic antitumor effect against E6-expressing tumors (See Figure 7). This may be related to the design of the experiment in our study. For example, if the dose of the tumor challenge is too high, it may be difficult to appreciate the difference in antitumor effects generated by the different suboptimal E6-containing DNA vaccines except the most potent DNA construct, Ii-PADRE-E6. Thus, in order to illustrate the difference in the therapeutic antitumor effects generated by the suboptimal E6-containing constructs, it may be necessary to reduce the dose or the interval between the tumor challenge and initiation of DNA vaccination.
The Ii-PADRE-E6 DNA vaccine represents a novel immunotherapeutic strategy that can enhance E6-specific CD8+ T cells through PADRE-specific CD4+ T helper cells. The success of Ii-PADRE-E6 DNA vaccine in preclinical mouse models has served as an important foundation for further clinical translation for the control of HPV-associated head and neck cancers in humans. Because the majority of HPV-associated head and neck cancers are related to HPV type-16, the current DNA vaccine that targets HPV-16 E6 can thus be used. However, for clinical translation, several issues need to be addressed. First, E6 is an oncogenic protein 6. Thus, it is important to modify E6 to alleviate the oncogenicity of E6 before it can be considered for vaccine. Second, the DNA vector used for the current study contains the ampicillin resistant gene, which is not suitable for clinical applications. Therefore, it is important to identify suitable vectors for administration to humans. Finally, it is important to determine the correct dose, appropriate route of administration, and suitable regimen (such as frequency and interval of vaccination) for optimal DNA vaccine effect (for reviews see 19,20).
The immunotherapeutic strategy used in the current study combining a strategy to enhance both CD4+ T helper cells as well as a strategy to augment antigen-specific cytotoxic T cells represents a platform for vaccine development. This strategy can potentially be used in different antigenic systems to improve vaccine potency. Not only can the current vaccine be used to treat HPV-16 associated head and neck cancers, but it also has the potential to treat other HPV-associated cancers, such as anogenital cancers (for review see 11). Furthermore, the current study should encourage the application of this strategy to control non-HPV-associated cancers and other infectious diseases.
Materials and Methods
Mice
C57BL/6 mice (6–8 weeks old) were purchased from the National Cancer Institute Frederick, MD). All animals were maintained under specific pathogen-free conditions at the Johns Hopkins Hospital (Baltimore, MD). All procedures were performed according to approved protocols and in accordance with recommendations for the proper use and care of laboratory animals.
Cells, Antibodies, and Reagents
The HPV-16 E7-expressing murine tumor model, TC-1, has been described previously 21. In brief, HPV-16 E6 and E7 and the ras oncogene were used to transform primary C57BL/6 mouse lung epithelial cells to generate TC-1. The production and maintenance of E6-specific CD8+ T cells has been described in a previous paper 22. Firefly luciferase-expressing TC-1 cells (TC-1/luc) were generated as described previously 23. All cells were maintained in RPMI medium (Invitrogen, Carlsbad, CA) supplemented with 2 mM glutamine, 1 mM sodium pyruvate, 20 mM HEPES, 100 IU/ml penicillin, 100μg/ml streptomycin, and 10% fetal bovine serum (Gemini Bio-Products, Woodland, CA).
DNA Constructs
The pcDNA3-E6 and pcDNA3-Ii and pcDNA3-Ii-PADRE have been previously described 12,22. To generate pcDNA3-Ii-E6, the Ii DNA fragment was amplified using PCR with primers (AAATCTAGAATGGATGACCAACGCGACC and TTTGAATTCCAGGGTGACTTGACCCAGT) and pcDNA3-Ii as a template. The Ii DNA fragment was further cloned into XbaI/EcoRI sites of pcDNA3-E6 to form pcDNA3-Ii-E6. To generate pcDNA3-Ii-PADRE-E6, The Ii-PADRE DNA fragment was amplified by PCR using the same primers and pcDNA3-Ii-PADRE as a template. The Ii-PADRE DNA fragment was further cloned into XbaI/EcoRI sites of pcDNA3-E6 to form pcDNA3-Ii-PADRE-E6.
RT-PCR
For the real-time PCR, DC-1 cell line 24 was transfected with pcDNA3-E6, Ii-E6, or Ii-PADRE-E6 plasmid. From transfected cells, total RNA was purified using a Trizol reagent (Invitrogen, Carlsbad, CA) according to protocol. The immortalized DC line was kindly provided by Dr. Kenneth Rock (University of Massachusetts, Worcester, MA;25). With continued passage, we have generated subclones of DCs (DC-1) 24 that are easily transfected using Lipofectamine 2000 (Life Technologies, Inc., Rockville, MD). The cDNA was prepared using QIAGEN LongRange RT-PCT Kits and diluted for real time PCR. Relative transcript expression levels of E6 were measured by quantitative real-time PCR using method previously described 26. The primer sequences were HPV16 E6 foward: 5-TTACCACAGTTATGCACAGA-3 and reverse: 5-ACAGTGGCTTTTGACAGTTA-3 or GAPDH foward: 5-GGTGAAGGTCGGTGTGAACG-3 and reverse: 5-CTCGCTCCTGGAAGATGGTG-3. PCRs were performed in triplicates using an iCycler (Bio-Rad). The amplified products were quantified by fluorescence intensity of SYBR Green I (Molecular Probes). Average fold changes were calculated by differences in threshold cycles(Ct) between pairs of samples to be compared. GAPDH gene from the transfected DC-1 cells was used for normalization of the amount of sample loaded.
DNA vaccination by gene gun
DNA-coated gold particles were prepared, and gene gun particle-mediated DNA vaccination was performed, according to a protocol described previously 27. Gold particles coated with DNA vaccines were delivered to the shaved abdominal regions of mice by using a helium-driven gene gun (Bio-Rad Laboratories Inc., Hercules, Calif.) with a discharge pressure of 400 lb/in2. Mice were immunized with 2μg of the DNA vaccine and received one boost with the same dose after a 4 day interval. Splenocytes were harvested 1 week after the last vaccination.
Intracellular cytokine staining and flow cytometry analysis
Pooled splenocytes from the vaccinated mice were harvested 1 week after the last vaccination and incubated overnight with 1 μg/ml E6 peptide (aa50–57) or PADRE peptide (AKFVAAWTLKAAA) in the presence of GolgiPlug (BD Pharmingen, San Diego, CA, USA) (1 μl/ml). The stimulated splenocytes were then washed once with FACScan buffer and stained with phycoerythrin-conjugated monoclonal rat anti-mouse CD8a (clone 53.6.7) or CD4. Cells were subjected to intracellular cytokine staining using the Cytofix/Cytoperm kit according to the manufacturer’s instruction (BD Pharmingen, San Diego, CA, USA) 28,29. Intracellular IFN-γ was stained with FITC-conjugated rat anti-mouse IFN-γ. All antibodies were purchased from BD Pharmingen. Flow cytometry analysis was performed using FACSCalibur with CELLQuest software (BD Biosciences, Mountain View, CA, USA).
In vivo tumor protection experiment
C57BL/6 mice (5 per group) were primed with the various DNA vaccines at a dose of 2μg/mouse 11 days before tumor challenge. Mice were boosted with the same dose and regimen 4 days later. 7 days after the last vaccination, mice were orally inoculated at the buccal mucosa with 2 × 105 TC-1 cells/mouse and monitored twice a week for tumor growth using the non-invasive luminescence imaging system Xenogen 200 using methods similar to what we have described previously 30,31.
In vivo antibody depletion experiments
C57BL/6 mice (5 per group) were primed with the pcDNA3-Ii-PADRE-E6 vaccine at a dose of 2μg/mouse 11 days before tumor challenge. Mice were boosted with the same dose and regimen 4 days later. Mice underwent antibody depletion of CD4, CD8, or NK cells one day after the last vaccination. 7 days after the last vaccination, mice were orally inoculated at the buccal mucosa with 2 ×105 TC-1 cells/mouse. Mice were depleted of CD8, CD4 T cells or NK cells using purified rat monoclonal antibodies mAb 2.43, 100 GK1.5 and mAb PK136, respectively, as described previously 32. The mice were injected with antibodies every other day for 3 times for the first week and then once every week using a protocol similar to one we have described previously 32. In addition, treated tumor-bearing mice without antibody depletion and untreated tumor-bearing mice were used as controls. Mice were monitored twice a week for tumor growth using the non-invasive luminescence imaging system Xenogen 200 using methods similar to what we have described previously 30,31.
In vivo tumor treatment experiment
C57BL/6 mice (5 per group) were orally inoculated at the buccal mucosa with 2 × 105 TC-1 cells/mouse on D0. Three days after tumor inoculation, all the tumor-bearing mice were treated with the various DNA constructs (Ii-E6, E6, Ii-PADRE or Ii-PADRE-E6) intradermally via gene gun at a dose of 2μg/mouse. Mice were boosted with the same dose and regimen 7 days later. The growth of tumor in mice was monitored using the IVIS Imaging System Series 200.
Statistical Analysis
All data expressed as means ±s.e. are representative of at least two different experiments. Data for intracellular cytokine staining with flow cytometry analysis were evaluated by ANOVA. Comparisons between individual data points were made using a Student’s t-test. For statistical analysis of the tumor protection experiment, we used Kaplan-Meier analysis.
Supplementary Material
Acknowledgments
We would like to thank Dr. T.-C. Wu for helpful discussion and critical review of the manuscript. This work was supported by the National Cancer Institute SPOREs (P50 CA098252 and P50 CA96784) and the 1 RO1 CA114425-01.
Footnotes
Conflict of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Cancer Facts & Figures. American Cancer Society. 2010. http://www.cancer.org/Research/CancerFactsFigures/CancerFactsFigures/cancer-facts-and-figures-2010.
- 2.Murdoch D. Standard, and novel cytotoxic and molecular-targeted, therapies for HNSCC: an evidence-based review. Curr Opin Oncol. 2007;19:216–221. doi: 10.1097/01.cco.0000264952.98166.99. [DOI] [PubMed] [Google Scholar]
- 3.Chung CH, Gillison ML. Human papillomavirus in head and neck cancer: its role in pathogenesis and clinical implications. Clin Cancer Res. 2009;15:6758–6762. doi: 10.1158/1078-0432.CCR-09-0784. [DOI] [PubMed] [Google Scholar]
- 4.Wu AA, Niparko KJ, Pai SI. Immunotherapy for head and neck cancer. J Biomed Sci. 2008;15:275–289. doi: 10.1007/s11373-008-9247-x. [DOI] [PubMed] [Google Scholar]
- 5.Capone RB, Pai SI, Koch WM, Gillison ML, Danish HN, Westra WH, et al. Detection and quantitation of human papillomavirus (HPV) DNA in the sera of patients with HPV-associated head and neck squamous cell carcinoma. Clin Cancer Res. 2000;6:4171–4175. [PubMed] [Google Scholar]
- 6.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 7.Donnelly JJ, Ulmer JB, Shiver JW, Liu MA. DNA vaccines. Annu Rev Immunol. 1997;15:617–648. doi: 10.1146/annurev.immunol.15.1.617. [DOI] [PubMed] [Google Scholar]
- 8.Gurunathan S, Klinman DM, Seder RA. DNA vaccines: immunology, application, and optimization. Annu Rev Immunol. 2000;18:927–974. doi: 10.1146/annurev.immunol.18.1.927. [DOI] [PubMed] [Google Scholar]
- 9.Condon C, Watkins SC, Celluzzi CM, Thompson K, Falo LD., Jr DNA-based immunization by in vivo transfection of dendritic cells. Nat Med. 1996;2:1122–1128. doi: 10.1038/nm1096-1122. [DOI] [PubMed] [Google Scholar]
- 10.Porgador A, Irvine KR, Iwasaki A, Barber BH, Restifo NP, Germain RN. Predominant role for directly transfected dendritic cells in antigen presentation to CD8+ T cells after gene gun immunization. J Exp Med. 1998;188:1075–1082. doi: 10.1084/jem.188.6.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hung CF, Ma B, Monie A, Tsen SW, Wu TC. Therapeutic human papillomavirus vaccines: current clinical trials and future directions. Expert Opin Biol Ther. 2008;8:421–439. doi: 10.1517/14712598.8.4.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hung CF, Tsai YC, He L, Wu TC. DNA Vaccines Encoding Ii-PADRE Generates Potent PADRE-specific CD4(+) T-Cell Immune Responses and Enhances Vaccine Potency. Mol Ther. 2007 doi: 10.1038/sj.mt.6300121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alexander J, Sidney J, Southwood S, Ruppert J, Oseroff C, Maewal A, et al. Development of high potency universal DR-restricted helper epitopes by modification of high affinity DR-blocking peptides. Immunity. 1994;1:751–761. doi: 10.1016/s1074-7613(94)80017-0. [DOI] [PubMed] [Google Scholar]
- 14.Alexander J, del Guercio MF, Maewal A, Qiao L, Fikes J, Chesnut RW, et al. Linear PADRE T helper epitope and carbohydrate B cell epitope conjugates induce specific high titer IgG antibody responses. J Immunol. 2000;164:1625–1633. doi: 10.4049/jimmunol.164.3.1625. [DOI] [PubMed] [Google Scholar]
- 15.Franke ED, Hoffman SL, Sacci JB, Jr, Wang R, Charoenvit Y, Appella E, et al. Pan DR binding sequence provides T-cell help for induction of protective antibodies against Plasmodium yoelii sporozoites. Vaccine. 1999;17:1201–1205. doi: 10.1016/s0264-410x(98)00341-7. [DOI] [PubMed] [Google Scholar]
- 16.Kim D, Monie A, He L, Tsai YC, Hung CF, Wu TC. Role of IL-2 secreted by PADRE-specific CD4+ T cells in enhancing E7-specific CD8+ T-cell immune responses. Gene Ther. 2008;15:677–687. doi: 10.1038/sj.gt.3303102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grujic M, Holst PJ, Christensen JP, Thomsen AR. Fusion of a viral antigen to invariant chain leads to augmented T-cell immunity and improved protection in gene-gun DNA-vaccinated mice. J Gen Virol. 2009;90:414–422. doi: 10.1099/vir.0.002105-0. [DOI] [PubMed] [Google Scholar]
- 18.Holst PJ, Sorensen MR, Mandrup Jensen CM, Orskov C, Thomsen AR, Christensen JP. MHC class II-associated invariant chain linkage of antigen dramatically improves cell-mediated immunity induced by adenovirus vaccines. J Immunol. 2008;180:3339–3346. doi: 10.4049/jimmunol.180.5.3339. [DOI] [PubMed] [Google Scholar]
- 19.Tsen SW, Paik AH, Hung CF, Wu TC. Enhancing DNA vaccine potency by modifying the properties of antigen-presenting cells. Expert Rev Vaccines. 2007;6:227–239. doi: 10.1586/14760584.6.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hung CF, Wu TC. Improving DNA vaccine potency via modification of professional antigen presenting cells. Curr Opin Mol Ther. 2003;5:20–24. [PubMed] [Google Scholar]
- 21.Lin K-Y, Guarnieri FG, Staveley-O’Carroll KF, Levitsky HI, August T, Pardoll DM, et al. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Research. 1996;56:21–26. [PubMed] [Google Scholar]
- 22.Peng S, Hung C-F, Trimble C, He L, Yeatermeyer J, Boyd D, et al. Development of a DNA vaccine targeting HPV-16 oncogenic protein E6. J Virol. 2004;78:8468–8476. doi: 10.1128/JVI.78.16.8468-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang B, Mao CP, Peng S, He L, Hung CF, Wu TC. Intradermal administration of DNA vaccines combining a strategy to bypass antigen processing with a strategy to prolong dendritic cell survival enhances DNA vaccine potency. Vaccine. 2007;25:7824–7831. doi: 10.1016/j.vaccine.2007.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim TW, Hung CF, Boyd DA, He L, Lin CT, Kaiserman D, et al. Enhancement of DNA vaccine potency by coadministration of a tumor antigen gene and DNA encoding serine protease inhibitor-6. Cancer Res. 2004;64:400–405. doi: 10.1158/0008-5472.can-03-1475. [DOI] [PubMed] [Google Scholar]
- 25.Shen Z, Reznikoff G, Dranoff G, Rock KL. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol. 1997;158:2723–2730. [PubMed] [Google Scholar]
- 26.Lin CT, Tsai YC, He L, Yeh CN, Chang TC, Soong YK, et al. DNA vaccines encoding IL-2 linked to HPV-16 E7 antigen generate enhanced E7-specific CTL responses and antitumor activity. Immunol Lett. 2007;114:86–93. doi: 10.1016/j.imlet.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen C-H, Wang T-L, Hung C-F, Yang Y, Young RA, Pardoll DM, et al. Enhancement of DNA vaccine potency by linkage of antigen gene to an HSP70 gene. Cancer Research. 2000;60:1035–1042. [PubMed] [Google Scholar]
- 28.Kim JW, Hung CF, Juang J, He L, Kim TW, Armstrong DK, et al. Comparison of HPV DNA vaccines employing intracellular targeting strategies. Gene Ther. 2004;11:1011–1018. doi: 10.1038/sj.gt.3302252. [DOI] [PubMed] [Google Scholar]
- 29.Kim TW, Hung CF, Ling M, Juang J, He L, Hardwick JM, et al. Enhancing DNA vaccine potency by coadministration of DNA encoding antiapoptotic proteins. J Clin Invest. 2003;112:109–117. doi: 10.1172/JCI17293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hung CF, Tsai YC, He L, Wu TC. Control of mesothelin-expressing ovarian cancer using adoptive transfer of mesothelin peptide-specific CD8+ T cells. Gene Ther. 2007;14:921–929. doi: 10.1038/sj.gt.3302913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hung CF, Tsai YC, He L, Coukos G, Fodor I, Qin L, et al. Vaccinia virus preferentially infects and controls human and murine ovarian tumors in mice. Gene Ther. 2007;14:20–29. doi: 10.1038/sj.gt.3302840. [DOI] [PubMed] [Google Scholar]
- 32.Cheng WF, Hung CF, Chai CY, Hsu KF, He L, Ling M, et al. Tumor-specific immunity and antiangiogenesis generated by a DNA vaccine encoding calreticulin linked to a tumor antigen. J Clin Invest. 2001;108:669–678. doi: 10.1172/JCI12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.