

Abstract
In recent years, cryo-electron microscopy (cryo-EM) has established itself as a key method in structural biology, permitting the structural characterization of large biomolecular complexes in various functional states. The data obtained through single-particle cryo-EM has recently seen a leap in resolution thanks to landmark advances in experimental and computational techniques, resulting in sub-nanometer resolution structures being obtained routinely. The remaining gap between these data and revealing the mechanisms of molecular function can be closed through hybrid modeling tools that incorporate known atomic structures into the cryo-EM data. One such tool, molecular dynamics flexible fitting (MDFF), uses molecular dynamics simulations to combine structures from X-ray crystallography with cryo-EM density maps to derive atomic models of large biomolecular complexes. The structures furnished by MDFF can be used subsequently in computational investigations aimed at revealing the dynamics of the complexes under study. In the present work, recent applications of MDFF are presented, including the interpretation of cryo-EM data of the ribosome at different stages of translation and the structure of a membrane-curvature-inducing photosynthetic complex.
Keywords: MDFF, flexible fitting, cryo-EM, X-ray crystallography, NAMD, VMD, docking, hybrid modeling
Introduction
Advances in cryo-electron microscopy (cryo-EM) single-particle reconstruction have transformed the field of structural biology. Macromolecular assemblies are now routinely investigated from a structural perspective at ever increasing resolution. In many cases cryo-EM provides the first glimpse into the structure of macromolecular complexes, long before X-ray crystal structures become available. However, it is only when combined with such structures that the full power of cryo-EM can be realized. The bacterial ribosome is a prototypical example. Although many insights into the function of the ribosome were obtained from cryo-EM before crystal structures were available, the landmark achievements of ribosome crystallography dramatically increased the amount of information that could be derived from the cryo-EM reconstructions. However, cryo-EM remains a unique tool to study key structural conformers of biomolecular complexes not accessible to crystallography as it has the ability to capture such complexes in different functional states in a native environment, albeit at lower resolution. Thus, several independent methods have been developed recently for flexibly fitting atomic structures into electron microscopy density maps. Here we review the first applications of the molecular dynamics flexible fitting (MDFF) method [1], which illustrate the power of hybrid methods for investigating the biological function of macromolecular assemblies.
In the MDFF method [1], a molecular dynamics (MD) simulation is performed using an initial atomic model, often just a crystal structure. In addition to the standard MD force field, forces proportional to the gradient of the density map are applied to each atom, locally driving the structure to occupy high-density regions. Secondary structure restraints can be applied to proteins and nucleic acids in order to prevent overfitting, i.e., a close fit of the map at the cost of unphysical distortions of the molecular structure. Furthermore, any feature available in regular MD simulations can be employed in MDFF; for instance, a system can be simulated in environments containing water, ions, and even a membrane bilayer, mimicking in vitro or in vivo conditions. MDFF simulations are performed with NAMD, a highly scalable MD simulation package [2], allowing for complexes of several megadaltons, i.e., systems comprising millions of atoms, to be studied with MDFF. The setup and analysis of MDFF simulations are performed with VMD, a molecular visualization package [3]. A practical guide for setting up, performing, and analyzing MDFF simulations has been provided elsewhere [4].
The challenge of morphing crystal structures of the ribosome into cryo-EM maps representing various functional states was the driving force for the development of MDFF [1]. Thus, the majority of the initial applications of the method involved study of the ribosome. MDFF was first applied to investigate ribosome-induced conformational changes in elongation factor Tu, a critical step in the decoding of genetic information [5]. MDFF also furnished the first atomic models of the ribosome bound to a protein-conducting channel, shedding light into how the ribosome modulates the channel's function [6, 7]. MDFF-derived atomic models of the ribosome stalled by the regulatory nascent chain TnaC revealed how it leads to translational arrest [8], as well as how it is recognized by the ribosomal exit tunnel [9]. In addition to studies of ribosome function, we review here an MDFF application addressing the process of protein-induced membrane curvature, which determines the morphology of bacterial photosynthetic membranes [10, 11, 12]. Other MDFF applications published after the submission of this review, and thus not covered here, include investigations of the interactions between ribosomal L1 stalk and tRNAs [13] and of the actin-myosin interface [14], as well as recovery of atomic detail from coarse-grained structural models [15].
Control of elongation factor Tu's GTPase activity
Elongation factor Tu (EF-Tu) delivers aminoacyl-tRNAs, i.e., transfer RNAs carrying an amino acid, to the elongating ribosome. EF-Tu is a GTPase, and it binds to aminoacyl-tRNAs with high affinity only in the GTP-bound state. When there is a cognate codon-anticodon interaction between the mRNA and the tRNA, GTP hydrolysis takes place and EF-Tu dissociates, allowing for the incoming aminoacyl-tRNA to be accommodated into the ribosome. The study of ribosome-induced conformational changes in EF-Tu was the first application of MDFF [5]. A 6.7-Å cryo-EM reconstruction of an E. coli 70S ribosome in complex with an aminoacyl-tRNA:EF-Tu:GDP ternary complex (TC), stalled by the antibiotic kirromycin, was obtained. The cryo-EM data were interpreted by employing MDFF, which furnished the first atomic model of a 70S:TC complex (Fig. 1A).
Fig. 1.
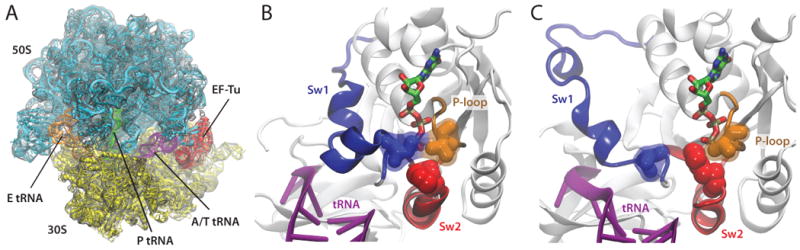
Ribosome-induced GTPase activation of EF-Tu. (A) 6.7-Å cryo-EM reconstruction of a 70S:EF-Tu:aminoacyl-tRNA:GDP complex stalled with kirromycin, together with an MDFF-derived model. (B) EF-Tu contains a hydrophobic gate formed by Val20 (P-loop, orange) and Ile60 (switch 1, blue), which prevents the catalytic residue His84 (switch 2, red) from accessing the GTP molecule. (C) When bound to the ribosome, switch 1 moves away from the P-loop, opening the hydrophobic gate; His84 can then reorient toward the nucleotide in a conformation conducive to GTP hydrolysis.
The GTPase activity of EF-Tu is very low when the factor is not bound to the ribosome [16]. Crystal structures of EF-Tu [17, 18] suggested that a hydrophobic gate, formed by switch I and the P-loop, blocks access of the catalytic residue His84 [19] to the GTP molecule (Fig. 1B; E. coli numbering is used throughout this paper), raising the question of how binding to the ribosome and recognition of the codon induces gate opening. This question was partially resolved by the MDFF-derived model of a 70S:TC complex, which revealed that EF-Tu's switch I moves away from the gate and interacts with the 16S rRNA (Fig. 1C). The other side of the gate, the P-loop, stays in place due to its interaction with the sarcin-ricin loop, a conserved region of the 23S rRNA. The model also displays a reorientation of the catalytic residue His84 toward the nucleotide, yielding a state consistent with GTP hydrolysis by EF-Tu. Thus, application of MDFF shed light onto ribosome-induced conformational changes in EF-Tu relevant for the control of its GTPase activity; however, the model of the 70S:TC complex by itself did not reveal yet how the codon-anticodon match signals to the hydrophobic gate.
More recently, a T. thermophilus 70S:TC structure was determined by X-ray crystallography at 3.6 Å resolution [20], confirming the mechanism of hydrophobic gate opening resolved via MDFF [5]. This X-ray structure can now be used to evaluate the accuracy of MDFF-derived models. The original E. coli 70S:TC model was obtained through MDFF performed in vacuo [5]. More accurate results, albeit at a much higher computational cost, can be expected from MDFF in explicit solvent [4]. A compromise can be achieved by fitting the entire complex in vacuo and refining in explicit solvent the region of interest, in this case the ternary complex and its surroundings. For comparison purposes, the region encompassing the ternary complex was refined with explicit-solvent MDFF with no secondary structure restraints using the same cryo-EM data as in [5]. Thus, the T. thermophilus 3.6-Å crystal structure can be compared with MDFF-derived models, one in vacuo and one in explicit solvent, obtained using the E. coli 6.7-Å cryo-EM reconstruction.
As shown in Table 1, using the ribosomal protein S12 as an example, many interactions between the ternary complex and the ribosome seen in the crystal structure are also captured by MDFF, both in vacuo and in explicit solvent. However, some of the contacts are only reproduced by MDFF when explicit solvent is used. This observation underlies the importance of solvent in the interpretation of cryo-EM data by flexible fitting methods. In MDFF, the force field contains not only intramolecular potentials, but also describes intermolecular interactions between water and solute as long as water is included in the simulation.
Table 1.
Comparison of contacts between ribosomal protein S12 and ternary complex in 70S:TC structures obtained from X-ray crystallography, MDFF in vacuo, and MDFF in explicit solvent.
| MDFF in vacuo [5] | MDFF in water (this work) | X-ray crystallography [20] |
|---|---|---|
| - | Lys119 (S12) - Glu249 (EF-Tu) | Lys123 (S12) - Glu261 (EF-Tu) |
| His76 (S12) - U69 (tRNA) | His76 (S12) - C68-G69 (tRNA) | His80 (S12) - U68 (tRNA) |
| Gln74 (S12) - U68 (tRNA) | Gln74 (S12) - C68 (tRNA) | Gln78 (S12) - A67 (tRNA) |
| - | Asp112 (S12) - C68 (tRNA) | Asp106 (S12) - U68 (tRNA) |
| Arg35 (S12) - U12 (tRNA) | Arg35 (S12) - U12 (tRNA) | - |
When compared to an isolated TC, the tRNA from the 70S:TC crystal structure shows a distinct conformational difference, namely a swing of the tRNA's D loop (Fig. 2A). Such a swing is not present in MDFF-derived models. We note, however, that the tRNA present in the 70S:TC crystal structure is tRNAThr, whereas the isolated TC and 70S:TC cryo-EM reconstruction both contain tRNAPhe. Thus, the question arises whether the swing of the D loop is not captured by MDFF due to a methodological limitation or if it is specific to tRNAThr. To address this question, a 6.7-Å simulated density map was generated from the 70S:TC crystal structure and used to fit the 70S:TC model containing tRNAPhe with MDFF in explicit solvent. As Fig. 2A shows, the swing of the tRNA's D loop is actually captured by MDFF in this case. To avoid any interpretation bias due to structural alignment, distances between residues in the D loop and a residue in the rigid acceptor stem were calculated (Fig. 2B). The correspondence in distances between structures confirms that MDFF refinement reproduces the D loop swing when a simulated density based on the 70S:TC crystal structure is used as a target. These results suggest that the swing of the D loop occurs for tRNAThr, as in the crystal structure, but not for tRNAPhe, as in the 70S:TC cryo-EM map and the corresponding MDFF-derived structure.
Fig. 2.
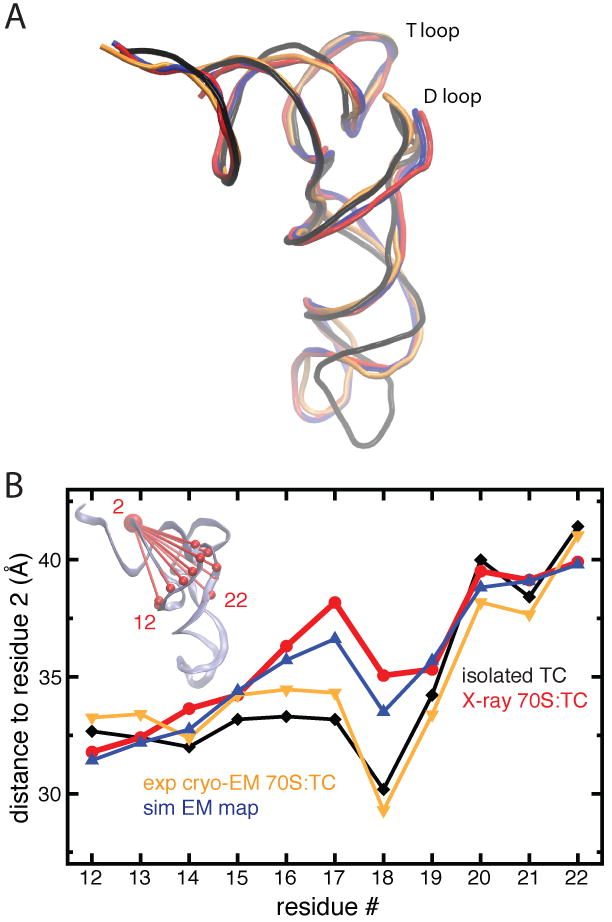
A/T tRNA structure as seen in cryo-EM and X-ray crystallography. (A) Swing of the D loop relative to the T loop in tRNA in ternary complexes. (B) Distances between the phosphorus atoms of residues in the D loop relative to residue 2 in the acceptor stem (see inset). The structure and distances of the tRNA in a free ternary complex (PDB 1OB2, black, diamonds) are compared to a tRNA in a ribosome-bound ternary complex obtained by MDFF interpretation of experimental cryo-EM data [5] (orange, triangles down) and one obtained by X-ray crystallography [20] (red, circles). Distances and structure for the MDFF fit into a simulated cryo-EM map generated from the tRNA from the crystal structure [20] are shown in blue (triangles up).
Complex formation between the ribosome and the protein-conducting channel
The ribosome is responsible for all protein synthesis in the cell, the nascent polypeptide often exiting into and folding directly in the cytoplasm. However, in many cases, such as for membrane proteins or those secreted to a different cellular compartment or outside the cell, the ribosome-nascent-chain (RNC) complex is directed to a membrane-bound channel shortly after synthesis begins. This protein-conducting channel (PCC), also referred to as SecYEG (bacterial) or Sec61αβγ (eukaryotic), serves to aid the passage of the nascent chain across a membrane or into it, the direction depending on the polypeptide [21]. To carry out this task, in which translocation of the nascent chain into the channel occurs concomitantly with translation, the ribosome must dock to the PCC, forming a complex. Visualization of this ribosome-channel complex has been limited to cryo-EM maps, most of them at a resolution too low to definitively determine the orientation or even the number of channels present. Recently, however, cryo-EM maps resolving a monomer of SecY/Sec61 bound to the ribosome have been obtained [22, 23, 7]. Two of these maps in particular [22, 7] served as the basis for MDFF and subsequent modeling studies of ribosome-channel complexes (Fig. 3A) [6, 7].
Fig. 3.
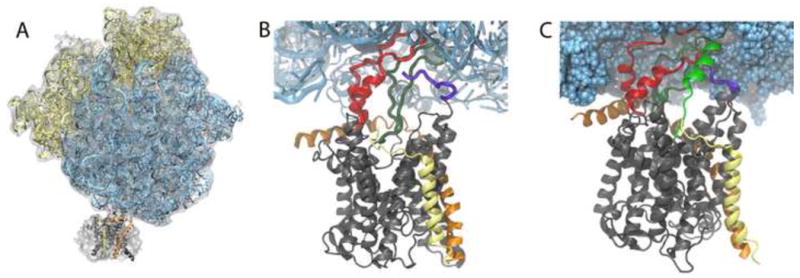
Ribosome-SecY complex formation. (A) Cryo-EM map from [22] of an inactive bacterial ribosome-SecY-monomer complex with the MDFF-fitted atomic model. (B) Close-up of the interactions between bacterial ribosome and channel. The four primary connections involve SecY's cytoplasmic loops (red and dark green), SecY's C-terminus (purple) and SecE (orange). (C) Simulated translocation of a polypeptide. The polypeptide (green) is seen exiting the ribosomal tunnel and entering SecY.
Atomic-scale structures of ribosome-channel complexes resulting from MDFF have revealed much about how these complexes are formed and maintained. In the case of a non-translating, bacterial ribosome-SecY-monomer complex [22], two cytoplasmic loops of SecY are seen to insert into the ribosome's polypeptide exit tunnel, shown in Fig. 3B [22, 6]. Two additional connections were also observed, between the C-terminus of SecY and ribosomal protein L24 and between the accessory protein SecE and L23 and L29 in the ribosome [6]. All four connections involve conserved residues in the channel and, furthermore, three of them share analogues in the crystal structure of SecYEG with SecA, a post-translational translocase [24]. Further support for this binding mode came in the form of another MDFF-derived structure, this one of an actively translating, mammalian ribosome-Sec61-monomer complex [7]. The ribosome-channel connections observed in the mammalian structure are quite similar to those found in the bacterial one, further supporting the universality of ribosome-PCC complex formation [7].
MD simulations of the MDFF-derived structure of the bacterial ribosome-SecY complex in its native water-lipid environment permitted a functional analysis of the complex [6]. While SecY is known to be water-tight in its closed state [25, 26, 27], it does become permeable to ions and small molecules upon RNC binding and nascent chain removal [28, 29, 30]. This suggests that ribosome binding is already sufficient to open the channel, at least partially. In agreement with this suggestion, simulation of the ribosome-SecY complex also indicated a destabilizing effect of ribosome binding on the PCC, particularly the channel-blocking plug [6]. MD simulations also proved that despite the partial occlusion of the exit tunnel by SecY's loops, the complex is still competent for translocation. By pulling a nascent polypeptide through the exit tunnel and into SecY, it was shown that the tunnel could accommodate an α helix without breaking interactions between SecY and the ribosome (see Fig. 3C) [6].
TnaC-mediated translational stalling
There exist several known nascent peptide chains that can regulate ribosome function while they are still being synthesized. A classical example is TnaC, a leader peptide of the tna operon in E. coli responsible for tryptophan (Trp) degradation [31]. The presence of a nascent TnaC peptide inside the ribosomal exit tunnel creates a Trp binding site in the ribosome [32]. High concentrations of free Trp then lead to inhibition of TnaC translation termination, which eventually leads to expression of the tna operon through an intricate gene regulatory mechanism [31]. A 5.8-Å cryo-EM reconstruction of the 70S ribosome stalled by TnaC was recently obtained, showing that this nascent chain has a distinct conformation and engages in several interactions with the exit tunnel [8]. MDFF was employed to obtain an atomic model of the 70S:TnaC complex and to determine the mechanism of TnaC-mediated translational stalling (Fig. 4A–D).
Fig. 4.
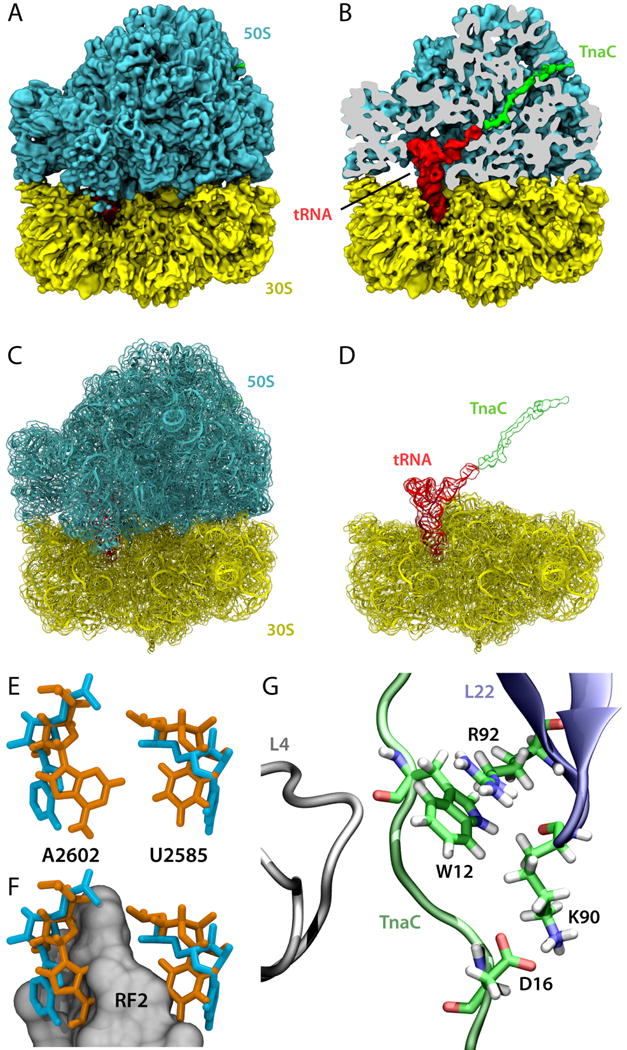
TnaC-mediated translational stalling. (A) 5.8-Å cryo-EM reconstruction of a 70S:TnaC complex. (B) Same as in (A), but with the 50S subunit sliced to show the TnaC-tRNA molecule inside the ribosome. (C) MDFF-derived 70S:TnaC atomic model with the cryo-EM map shown in transparent surface. (D) Same as in (C), but with the 50S subunit removed for clarity. (E) Conformations of the 23S rRNA residues A2602 and U2585 in the 70S:TnaC model (orange) and a 70S:RF2 crystal structure (blue; PDB 2WH1/2WH2 [47]). (F) Same as (E), but with a region of RF2 represented as a gray surface, showing that the conformations of A2602 and U2585 seen in the cryo-EM map are incompatible with proper positioning of RF2. (G) Recognition of critical TnaC residues by the ribosome as discovered by MD simulations.
Under normal conditions, when the ribosome reaches the TnaC stop codon, release factor 2 (RF2) catalyzes hydrolysis of the ester bond between TnaC and the P-site tRNA at the peptidyl transferase center (PTC) terminating translation. At high concentrations of Trp, however, the reaction catalyzed by RF2 is inhibited [31]. Analysis of the 70S:TnaC atomic model shows that two 23S rRNA conserved residues within the PTC, namely A2602 and U2585, adopt distinct conformations incompatible with binding of RF2, providing a structural basis for understanding TnaC-mediated PTC silencing [8]. Figures 4E and 4F present a superposition of the 70S:TnaC atomic model and a 70S:RF2 crystal structure, showing that the position of the two aforementioned PTC residues would clash with RF2 [8]. The free Trp that binds somewhere at the PTC in the 70S:TnaC complex may provide further steric hindrance to proper RF2 positioning within the ribosome, but the exact location of the Trp binding site is still unknown.
Even though the register of TnaC in the ribosomal exit tunnel could be satisfactorily obtained from the cryo-EM data, the precise positioning of side chains and their interactions with the exit tunnel could not be directly derived from EM data at the present resolution. To further investigate the recognition of critical TnaC residues by the ribosome, extensive MD simulations of TnaC inside the exit tunnel were performed using MDFF-derived models as starting conditions [9]. The simulations, supported by quantum chemistry calculations, indicate that the critical TnaC residue Trp12 is recognized by the ribosome via a cation-π interaction with Arg92 of ribosomal protein L22 (Fig. 4G). Furthermore, the critical TnaC residue Aspl6 is seen to form salt bridges with ribosomal proteins, in particular with Lys90 of L22, also an essential residue for TnaC stalling (Fig. 4G). This work illustrates that not only can MDFF-derived models help interpret cryo-EM data, but they can also readily enable further investigations by means of MD simulations.
Membrane curvature induced by the bacterial photosynthetic core complex dimer
The presented applications of MDFF focused hitherto on ribosome-related investigations. In this section, a different MDFF application is showcased, namely, the morphogenesis of cellular membranes. Interest in how cellular membranes obtain high-curvature forms has emerged in recent years [33, 34, 35], as membrane curvature is essential for vital processes like cell division, fusion, and compartmentalization. One suggested membrane-bending mechanism involves integral membrane proteins [33, 34, 35]. For determination of the precise molecular mechanisms that drive the dynamic process of membrane bending, all-atom MD simulation has proven to be a practical technique [36, 37, 11, 38, 39, 40, 12].
One of the few integral membrane proteins implicated in membrane bending is the bacterial photosynthetic core complex dimer, which is a molecular assembly containing three types of proteins (Fig. 5A). The light-harvesting complex 1 (LH1), which absorbs sunlight, is arranged into an S-shaped array surrounding two reaction centers (RC) that perform the initial processing of the light energy. Besides LH1 and RC, the bacterial photosynthetic core complex dimer (from the species Rhodobacter sphaeroides) includes also an additional transmembrane polypeptide, termed PufX, which is responsible for the dimerization of the core complex [41] and potentially plays a role in the overall core complex geometry as well [42]. The full core complex dimer is about 20 nm by 10 nm in the membrane plane.
Fig. 5.
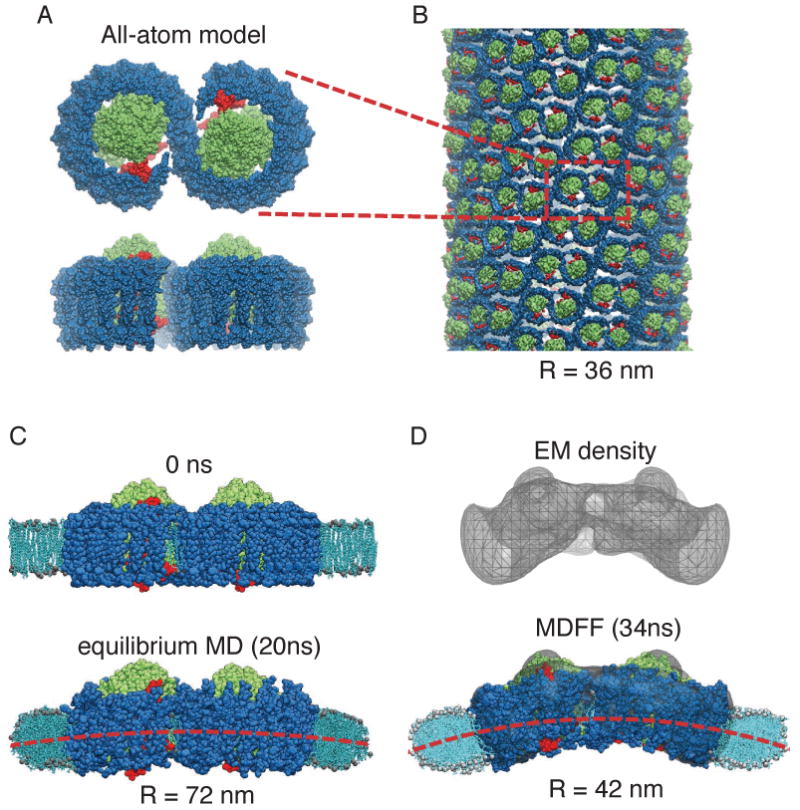
Membrane-bending properties of the bacterial photosynthetic core complex. (A) Top (perpendicular to the membrane plane) and side (along the membrane plane) views of an all-atom model of the core complex dimer [36] (RC: green; LH1: blue; PufX: red). (B) Example of a tubular photosynthetic membrane observed in mutant Rba. sphaeroides [44]. The core complex dimers can be seen to arrange helically in the tubular vesicle, and one complex is outlined in red for identification. This tubular membrane has a radius of 36 nm. (C) Setup of the membrane-protein system for equilibrium MD of the all-atom model. The membrane is shown in light blue; the water box included in the simulation is not shown for clarity. During an equilibrium MD simulation, a small bending was seen in the core complex dimer and a shallow curvature in the membrane emerged [36]. (D) 25Å-resolution EM reconstruction obtained through single-particle analysis [44]. The all-atom model was fitted into the EM map employing MDFF; during the MDFF simulation the surrounding membrane developed a more prominent curvature [11] that remained stable in an equilibrium simulation.
The core complex dimer has been hypothesized to induce membrane curvature since it resides in a highly curved membrane region, and aggregation of the dimers can form tubular vesicles of radius 30-60 nm in certain mutants of Rba. sphaeroides [43, 44] (Fig. 5B). However, lack of a crystal structure for the full complex hinders an understanding of how the core complex shapes the membrane. An all-atom model was therefore constructed for the core complex by assembling partial structural data and homology modeling [36]. Subsequent equilibrium MD simulation of the modeled core complex structure embedded in a membrane patch and a solvent environment showed that the core complex dimer exhibits an intrinsic tendency to bend about its dimerization junction (Fig. 5C). Bending of the core complex induces the surrounding membrane to curve along with it, but the resulting curvature is too shallow to explain the observed size of tubular vesicles.
The first three-dimensional structural data for the core complex dimer became available through a low-resolution (25 Å) cryo-EM reconstruction obtained from single-particle analysis. The density map shows a prominent bend in the overall geometry of the core complex [44] (Fig. 5D, top). Employing MDFF, this new density map was integrated with the previous all-atom model [11, 10]. During the MDFF simulation, the originally only slightly bent model became more strongly bent as defined by the EM map, the surrounding membrane adapting again to the shape of the complex, forming a highly curved patch (Fig. 5D, bottom) [11]. The radius of curvature of the resulting membrane patch, that remained stable when the influence of the density map was removed from the simulation, matches well the size of the observed tubular vesicles. In addition, the local curvature properties of the core complex seen in the simulation offer a microscopic rationalization for a helical arrangement of the complexes in the tubular vesicles as seen in EM images [11, 44, 12] (Fig. 5B). Thus, even at a limited resolution, the incorporation of EM data resulted in an improved atomic model, demonstrating the wide applicability of MDFF.
Conclusions and Outlook
The resolution of atomic-level structures of biomolecules, e.g., proteins and RNA, has permitted great insight into their function and profoundly transformed the life sciences. Similarly, lower-resolution density maps of large biomolecular complexes have provided a glimpse of how cells organize themselves into an assortment of large structures and organelles. However, both approaches have limits to their applicability: atomic-resolution methods are limited in the size of the complexes they can be applied to, while cryo-EM typically cannot reach atomic resolution. While both techniques are continuously challenging these limits, e.g., atomic-level ribosome structures have become routine [45] and cryo-EM recently broke the atomic-resolution barrier [46], hybrid methods suited to merge the detail of atomic-scale structures with the overall architecture of complexes captured in density maps will be crucial for imaging cellular components at the atomic scale. We have developed MDFF [1, 4], a hybrid method that employs MD simulations to combine structural data from X-ray crystallography and cryo-EM. MDFF has been successfully applied to several research problems, namely control of GTP hydrolysis by elongation factor Tu upon ribosome binding [5], structural and regulatory aspects of ribosome-translocon complexes [6, 7], recognition of the regulatory nascent chain TnaC by the ribosome [9] and TnaC-mediated translational stalling [8], analysis of intermediate states relevant to tRNA:mRNA translocation through the ribosome [13], protein-induced membrane curvature in photosynthetic chromatophores [10, 11], characterization of the actin-myosin interface [14], and recovery of atomic details from coarse-grained structural models [15].
Since MDFF is an MD-based method, any atomic system that can be simulated with standard MD force fields can also be studied with MDFF. In particular, systems containing proteins, nucleic acids, water, ions, and lipids are supported. MDFF simulations are performed with NAMD [2], a highly scalable parallel MD simulation package, which means that the structure of large, i.e., megadalton or multi-million-atom, assemblies can be modeled with MDFF. Of special interest is that MDFF-derived structures can readily permit further investigations by means of MD simulations and related techniques. In fact, nearly all applications of MDFF thus far benefited from MD simulations initiated from MDFF-derived atomic models. Ongoing developments of MDFF include optimization of parameters using a large test set of atomic structures in different conformations, use of implicit solvent models, combination with enhanced sampling techniques, implementation of symmetry restraints, correlation-based MDFF, and interactive MDFF.
As any other hybrid modeling method, MDFF has certain drawbacks. Since it is based on MD simulations, all but the simplest applications require relatively sophisticated modeling knowledge, which represents a challenge to making the method easily applicable by experimental structural biologists. Another disadvantage is that MDFF has one of the largest computational costs compared to competing methods, especially when explicit solvent is employed. An intrinsic limitation of the method is the difficulty in describing rotations of structural elements. Take, for instance, a protein helix placed into its density but requiring a 180-degree rotation around its axis; the MDFF potential is unable to induce the required rotation. Moreover, the conservative use of secondary structure restraints to avoid overfitting prevents conformational changes involving folding/refolding of secondary structure elements to be modeled. Even in the absence of such restraints, the time scale probed by MDFF simulations is presently limited due to lack of computational power and is, thus, likely insufficient to capture such conformational changes.
Over the last years, several different flexible fitting methods have been proposed (for a recent account, see [4]). However, a systematic comparison between the different methods is still lacking. It will be valuable to evaluate the performance of the available methods on test sets under comparable conditions. Such a comparison will help users choose the most appropriate hybrid method for the problem at hand, and will also drive further method development. During the next few years, we envision that lessons learned from one method will be adapted to improve other methods. In this context, the VMD-NAMD platform, due to its wide availability and use, will provide a suitable framework to build a comprehensive hybrid modeling toolkit. Furthermore, analogous to the well-established set of methods currently availabl to interpret X-ray crystallography, multi-method protocols can be designed, taking advantage of the specific strengths of different methodologies, improving the overall quality of atomic models obtained from cryo-EM data.
Acknowledgments
The authors thank Joachim Frank, Roland Beckmann, Chris Akey, and Neil Hunter for fruitful collaborations. This work was supported by the National Institutes of Health (P41-RR005969) and the National Science Foundation (PHY0822613). E.S. is supported by a fellowship from the Alexander von Humboldt Foundation. E.V. is supported by a Marie Curie International Incoming Fellowship within the 7th European Community Framework Programme.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comp Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Humphrey W, Dalke A, Schulten K. VMD – Visual Molecular Dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 4.Trabuco LG, Villa E, Schreiner E, Harrison CB, Schulten K. Molecular Dynamics Flexible Fitting: A practical guide to combine cryo-electron microscopy and X-ray crystallography. Methods. 2009;49:174–180. doi: 10.1016/j.ymeth.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Villa E, Sengupta J, Trabuco LG, LeBarron J, Baxter WT, Shaikh TR, Grassucci RA, Nissen P, Ehrenberg M, Schulten K, Frank J. Ribosome-induced changes in elongation factor Tu conformation control GTP hydrolysis. Proc Natl Acad Sci USA. 2009;106:1063–1068. doi: 10.1073/pnas.0811370106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gumbart J, Trabuco LG, Schreiner E, Villa E, Schulten K. Regulation of the protein-conducting channel by a bound ribosome. Structure. 2009;17:1453–1464. doi: 10.1016/j.str.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Becker T, Bhushan S, Jarasch A, Armache JP, Funes S, Jossinet F, Gumbart J, Mielke T, Berninghausen O, Schulten K, Westhof E, Gilmore R, Mandon EC, Beckmann R. Structure of monomeric yeast and mammalian Sec61 complexes interacting with the translating ribosome. Science. 2009;326:1369–1373. doi: 10.1126/science.1178535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seidelt B, Innis CA, Wilson DN, Gartmann M, Armache JP, Villa E, Trabuco LG, Becker T, Mielke T, Schulten K, Steitz TA, Beckmann R. Structural insight into nascent polypeptide chain-mediated translational stalling. Science. 2009;326:1412–1415. doi: 10.1126/science.1177662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trabuco LG, Harrison CB, Schreiner E, Schulten K. Recognition of the regulatory nascent chain TnaC by the ribosome. Structure. 2010;18:627–637. doi: 10.1016/j.str.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sener MK, Hsin J, Trabuco LG, Villa E, Qian P, Hunter CN, Schulten K. Structural model and excitonic properties of the dimeric RC-LH1-PufX complex from Rhodobacter sphaeroides. Chem Phys. 2009;357:188–197. doi: 10.1016/j.chemphys.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsin J, Gumbart J, Trabuco LG, Villa E, Qian P, Hunter CN, Schulten K. Protein-induced membrane curvature investigated through molecular dynamics flexible fitting. Biophys J. 2009;97:321–329. doi: 10.1016/j.bpj.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsin J, Chandler DE, Gumbart J, Harrison CB, Sener M, Strumpfer J, Schulten K. Self-assembly of photosynthetic membranes. ChemPhysChem. 2010;11:1154–1159. doi: 10.1002/cphc.200900911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trabuco LG, Schreiner E, Eargle J, Cornish P, Ha T, Luthey-Schulten Z, Schulten K. The role of L1 stalk-tRNA interaction in the ribosome elongation cycle. J Mol Biol. doi: 10.1016/j.jmb.2010.07.056. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lorenz M, Holmes KC. The actin-myosin interface. Proc Natl Acad Sci USA. 2010;107:12529–12534. doi: 10.1073/pnas.1003604107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim H, Hsin J, Liu Y, Selvin PR, Schulten K. Formation of salt bridges mediates internal dimerization of myosin VI medial tail domain. Structure. doi: 10.1016/j.str.2010.09.011. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fasano O, Vendittis ED, Parmeggiani A. Hydrolysis of GTP by elongation factor Tu can be induced by monovalent cations in the absence of other effectors. J Biol Chem. 1982;257(6):3145–3150. [PubMed] [Google Scholar]
- 17.Kjeldgaard M, Nissen P, Thirup S, Nyborg J. The crystal structure of elongation factor EF-Tu from Thermus aquaticus in the GTP conformation. Structure. 1993;1:35–50. doi: 10.1016/0969-2126(93)90007-4. [DOI] [PubMed] [Google Scholar]
- 18.Berchtold H, Reshetnikova L, Reiser CO, Schirmer NK, Sprinzl M, Hilgenfeld R. Crystal structure of active elongation factor Tu reveals major domain rearrangements. Nature. 1993;365:126–132. doi: 10.1038/365126a0. [DOI] [PubMed] [Google Scholar]
- 19.Daviter T, Wieden HJ, Rodnina MV. Essential role of histidine 84 in elongation factor Tu for the chemical step of GTP hydrolysis on the ribosome. J Mol Biol. 2003;332:689–699. doi: 10.1016/s0022-2836(03)00947-1. [DOI] [PubMed] [Google Scholar]
- 20.Schmeing TM, Voorhees RM, Kelley AC, Gao YG, Murphy FV, Weir JR, Ramakrishnan V. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science. 2009;326:688–694. doi: 10.1126/science.1179700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rapoport TA. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature. 2007;450:663–669. doi: 10.1038/nature06384. [DOI] [PubMed] [Google Scholar]
- 22.Ménétret JF, Schaletzky J, Clemons WM, Jr, Osborne AR, Skånland SS, Denison C, Gygi SP, Kirkpatrick DS, Park E, Ludtke SJ, Rapoport TA, Akey CW. Ribosome binding of a single copy of the SecY complex: implications for protein translocation. Mol Cell. 2007;28:1083–1092. doi: 10.1016/j.molcel.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 23.Ménétret JF, Hegde RS, Agular M, Gygi SP, Park E, Rapoport TA, Akey CW. Single copies of Sec61 and TRAP associate with a nontranslating mammalian ribosome. Structure. 2008;16:1126–1137. doi: 10.1016/j.str.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zimmer J, Nam Y, Rapoport TA. Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature. 2008;455:936–943. doi: 10.1038/nature07335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gumbart J, Schulten K. Molecular dynamics studies of the archaeal translocon. Biophys J. 2006;90:2356–2367. doi: 10.1529/biophysj.105.075291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saparov SM, Erlandson K, Cannon K, Schaletzky J, Schulman S, Rapoport TA, Pohl P. Determining the conductance of the SecY protein translocation channel for small molecules. Mol Cell. 2007;26:501–509. doi: 10.1016/j.molcel.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 27.Gumbart J, Schulten K. The roles of pore ring and plug in the SecY protein-conducting channel. J Gen Physiol. 2008;132:709–719. doi: 10.1085/jgp.200810062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simon S, Blobel G. A protein-conducting channel in the endoplasmic-reticulum. Cell. 1991;65:371–380. doi: 10.1016/0092-8674(91)90455-8. [DOI] [PubMed] [Google Scholar]
- 29.Heritage D, Wonderlin WF. Translocon pores in the endoplasmic reticulum are permeable to a neutral, polar molecule. J Biol Chem. 2001;276:22655–22662. doi: 10.1074/jbc.M102409200. [DOI] [PubMed] [Google Scholar]
- 30.Wonderlin WF. Constitutive translation-independent opening of the protein-conducting channel in the endoplasmic reticulum. Pflug Arch Eur J Physiol. 2009;457:917–930. doi: 10.1007/s00424-008-0545-y. [DOI] [PubMed] [Google Scholar]
- 31.Yanofsky C. RNA-based regulation of genes of tryptophan synthesis and degradation in bacteria. RNA. 2007;13:1141–1154. doi: 10.1261/rna.620507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gong F, Yanofsky C. Instruction of translating ribosome by nascent peptide. Science. 2002;297:1864–1867. doi: 10.1126/science.1073997. [DOI] [PubMed] [Google Scholar]
- 33.McMahon HT, Gallop JL. Membrane curvature and mechanisms of dynamic cell membrane remodeling. Nature. 2005;438:590–596. doi: 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]
- 34.Zimmerberg J, Kozlov MM. How proteins produce cellular membrane curvature. Nat Rev Mol Cell Biol. 2006;7:9–19. doi: 10.1038/nrm1784. [DOI] [PubMed] [Google Scholar]
- 35.Kozlov MM. Biophysics: Joint effort bends membrane. Nature. 2010;463:439–440. doi: 10.1038/463439a. [DOI] [PubMed] [Google Scholar]
- 36.Chandler D, Hsin J, Harrison CB, Gumbart J, Schulten K. Intrinsic curvature properties of photosynthetic proteins in chromatophores. Biophys J. 2008;95:2822–2836. doi: 10.1529/biophysj.108.132852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chandler DE, Gumbart J, Stack JD, Chipot C, Schulten K. Membrane curvature induced by aggregates of LH2s and monomeric LH1s. Biophys J. 2009;97:2978–2984. doi: 10.1016/j.bpj.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arkhipov A, Yin Y, Schulten K. Four-scale description of membrane sculpting by BAR domains. Biophys J. 2008;95:2806–2821. doi: 10.1529/biophysj.108.132563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin Y, Arkhipov A, Schulten K. Simulations of membrane tubulation by lattices of amphiphysin N-BAR domains. Structure. 2009;17:882–892. doi: 10.1016/j.str.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arkhipov A, Yin Y, Schulten K. Membrane-bending mechanism of amphiphysin N-BAR domains. Biophys J. 2009;97:2727–2735. doi: 10.1016/j.bpj.2009.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frese R, Olsen J, Branvall R, Westerhuis W, Hunter C, van Grondelle R. The long-range supraorganization of the bacterial photosynthetic unit: A key role for PufX. Proc Natl Acad Sci USA. 2000;97:5197–5202. doi: 10.1073/pnas.090083797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hsin J, Chipot C, Schulten K. A glycophorin A-like framework for the dimerization of photosynthetic core complexes. J Am Chem Soc. 2009;131:17096–17098. doi: 10.1021/ja905903n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siebert CA, Qian P, Fotiadis D, Engel A, Hunter CN, Bullough PA. Molecular architecture of photosynthetic membranes in Rhodobacter sphaeroides: the role of PufX. EMBO J. 2004;23:690–700. doi: 10.1038/sj.emboj.7600092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qian P, Bullough PA, Hunter CN. Three-dimensional reconstruction of a membrane-bending complex: The RC-LH1-PufX core dimer of Rhodobacter sphaeroides. J Biol Chem. 2008;283:14002–14011. doi: 10.1074/jbc.M800625200. [DOI] [PubMed] [Google Scholar]
- 45.Schmeing TM, Ramakrishnan V. What recent ribosome structures have revealed about the mechanism of translation. Nature. 2009;461:1234–1242. doi: 10.1038/nature08403. [DOI] [PubMed] [Google Scholar]
- 46.Zhang X, Jin L, Fang Q, Hui WH, Zhou ZH. 3.3 Å cryo-EM structure of a nonenveloped virus reveals a priming mechanism for cell entry. Cell. 2010;141:472–482. doi: 10.1016/j.cell.2010.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weixlbaumer A, Jin H, Neubauer C, Voorhees RM, Petry S, Kelley AC, Ramakrishnan V. Insights into translational termination from the structure of RF2 bound to the ribosome. Science. 2008;322:953–956. doi: 10.1126/science.1164840. [DOI] [PMC free article] [PubMed] [Google Scholar]