

Abstract
Here, we describe a simple system in which human keratinocytes can be redirected to an alternative differentiation pathway. We transiently transfected freshly isolated human skin keratinocytes with the single transcription factor OCT4. Within two days these cells displayed expression of endogenous embryonic genes and showed reduced genomic methylation. More importantly, these cells could be specifically converted into neuronal and contractile mesenchymal cell types. Redirected differentiation was confirmed by expression of neuronal and mesenchymal cell mRNA and protein, and via a functional assay in which the newly differentiated mesenchymal cells contracted collagen gels as efficiently as authentic myofibroblasts. Thus, to generate patient-specific cells for therapeutic purposes, it may not be necessary to completely reprogram somatic cells into induced pluripotent stem (iPS) cells before altering their differentiation and grafting them into new tissues.
Keywords: keratinocyte, tissue repair, epidermal cells, stem cells, reprogramming
INTRODUCTION
In June, 2006, Yamanaka reported that mouse embryonic fibroblasts could be reprogrammed into embryonic stem-like cells by retroviral-mediated transduction of four transcription factors, Oct4, Sox2, c-Myc, and Klf4.1 These induced pluripotent stem (iPS) cells proved that differentiated, perhaps nondividing cells, could be reprogrammed into highly proliferative embryonic cells. Since then, a multitude of reports have surfaced showing that iPS cells can be produced from a variety of mouse and human cells (for review see.2,3 In a proof of principle experiment, mouse iPS cells were used to a sickle cell gene defect in mice,4 thereby demonstrating their great therapeutic potential. They have also been used to make patient specific cell lines for human studies.5 However, the iPS method, which completely reprograms the cells, cannot be used to produce cells for human therapy. Integrating retroviruses or lentiviruses were used to delivery the reprogramming factors. These viruses may cause several problems, including cancer if the integration sites affect oncogenes or tumor suppressor genes. The number of integration sites in the transduced fibroblasts was as high as twenty per clone, resulting in tumor formation in iPS cell-derived mice.3,6 Furthermore, this complete reprogramming approach requires the delivery of multiple genes, enhancing the possibility of deleterious effects. Although several groups since 2007 have now shown that iPS cells can be generated without the use of viral vectors and with fewer factors (for review see7,8), this technology still exhibits inefficient induction and a long production time, both major limitations if iPS cells are to be used for human therapy.
Skin keratinocytes offer advantages over other cell types for reprogramming. The skin is the largest organ in the body, and the epidermis provides the greatest number of easily obtainable proliferative cells. Moreover, keratinocytes are one hundred fold more efficient than fibroblasts when used in the various iPS cell protocols.9 In addition, our laboratory has shown that mouse epidermal stem cells, a subset of keratinocytes, can populate tissues derived from all three germ layers when injected into the developing blastocyst.10 Thus, keratinocytes offer high reprogramming and therapeutic potential. Another advantage of keratinocytes is that they are readily obtained from human skin. A recent review emphasized that signaling in the reprogrammed cells varies greatly amongst species.11 Thus, it is critical to test reprogramming in human cells if human tissues are to be replaced. Additionally, if patient-specific cells are needed, then rapid cell production may also be critical.
Keeping these two points in mind, we explored the possibility that it might not be necessary to completely reprogram human cells to iPS cells in order to use them to replace damaged human tissues. In our present study, we tested the potential of using freshly isolated human skin keratinocytes for rapid alternative tissue replacement. We transiently transfected keratinocytes with the human OCT4 transcript, exposed these cells to alternative differentiating conditions, and tested the functionality of these new cell types. The results presented here demonstrate that a transient exposure to OCT4 is sufficient to effect changes, that allow directed differentiation of the human keratinocytes into functioning mesodermally-derived cell types. This method could be used to provide customized patient-specific cells for therapeutic purposes.
RESULTS
OCT4 protein is temporally expressed in human keratinocytes
In order to demonstrate that a transient exposure to OCT4 is sufficient to allow a change in differentiation of the human keratinocytes, we needed to show that the OCT4 transcription factor was expressed for a short amount of time, and when expressed, it moved to the nucleus where it could transactivate its target genes. We transiently introduced a plasmid carrying the full-length human OCT4 transcript into human keratinocytes and determined where and for how long the OCT4 protein was expressed. Using immunocytochemical analysis, we determined that the OCT4 protein localized to the nuclei of the transfected keratinocytes 48 hours after transfection (Fig. 1B). Untransfected control keratinocytes showed no expression of OCT4 (Fig. 1A). To investigate the temporal expression pattern, we counted the numbers of OCT4-expressing keratinocytes over six days (Fig. 1C). Twenty-four hours after transfection, ~24% of cells expressed OCT4. This percentage peaked at 48 hours with ~32% of cells expressing OCT4. With each day thereafter, the percentage of keratinocytes expressing OCT4 decreased with most lost by 6 days post-transfection. Thus, pcDNA3-OCT4 transiently produced the OCT4 protein that localized to the nuclei of the keratinocytes.
FIGURE 1.
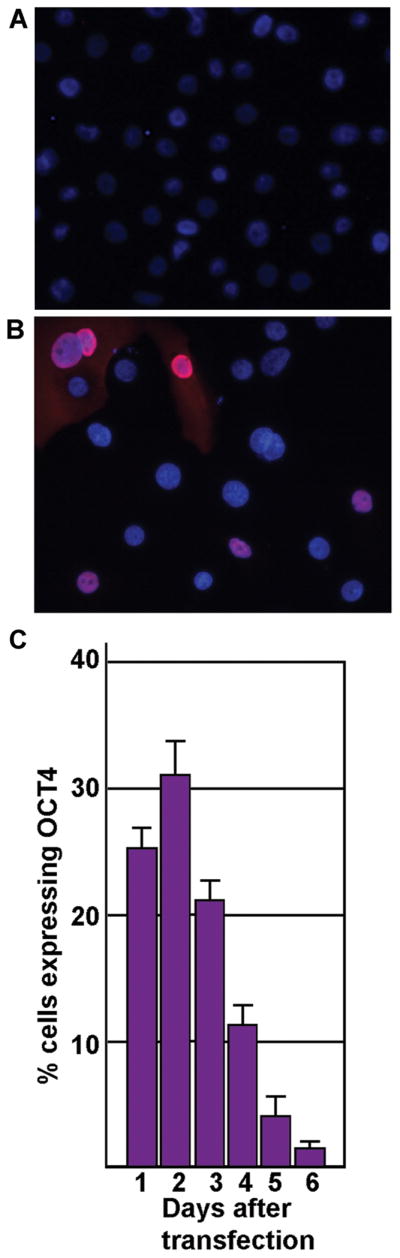
Transient expression of OCT4 in human keratinocytes. A) Immunofluorescent image showing that untransfected human skin keratinocytes do not express OCT4 (20x). Cells were stained with both DAPI (blue) to identify all nuclei and with antibody to OCT4 (red). B) Immunofluorescent image of keratinocytes 48 hours after transient transfection with pcDNA-OCT4 (40x). Nuclei are identified by DAPI (blue). Transfected cells are identified by antibody to OCT4 (red/pink). C) Graph depicting temporal expression of OCT4 in human keratinocytes. Shown is the percentage of cells expressing OCT4 protein, from 1 through 6 days after transient transfection with pcDNA3-OCT4.
Transfected cells show transient expression of endogenous OCT4 target genes
Since the OCT4 protein translocated to the nucleus, we hypothesized that the keratinocytes would show transient reactivation of some of the endogenous OCT4 targets. Using RT-PCR, we investigated expression of SOX2, NANOG, and REX-1 in the transiently transfected keratinocytes (Fig. 2). The OCT4 primers detected the OCT4 transcript from either the plasmid or the endogenous gene for five days after transfection. This correlated with the protein expression pattern described above (Fig. 1C). We found a temporal expression pattern of endogenous OCT4 target genes. SOX2 expression was clearly evident on days one and two after OCT4 transfection, with less expression on the following days. NANOG was expressed for as long as OCT4 was expressed. REX-1, on the other hand, was not detected until day three after transfection, which is not so surprising as it is normally activated only in the presence of both OCT4 and SOX2. Keratinocytes transfected with the control pcDNA3-GFP plasmid or untransfected keratinocytes showed no expression of these genes (Fig. 2 bottom gel). Moreover, once the cells transiently transfected with OCT4 were induced to differentiate (see experiments described below), neither OCT4 or its target genes were detectable.
FIGURE 2.

Transient expression of OCT4 induces endogenous expression of OCT4 target genes in human skin keratinocytes. Shown in top four gels are RT-PCR gels for expression of OCT4 and the endogenous OCT4 target genes, SOX2, NANOG, and REX-1, from 1 through 5 days after transient transfection with pcDNA3-OCT4. Bottom gel demonstrates that untransfected HSKs do not express OCT4 RNA. 100bp = 100 base pair ladder.
Transient expression of OCT4 alters methylation states of OCT4 and NANOG promoters
Although demethylation of the promoter region of the OCT4 gene has been shown in iPS cells (for review see12), the methylation state of the endogenous OCT4 promoter after transient transfection has not been reported. Thus, we sequenced bisulfite-treated genomic DNA from the human keratinocytes that had been transiently transfected with pcDNA3-OCT4. We ensured that only OCT4-transfected cells were examined by using a fusion product of OCT4 and GFP which we inserted into the pcDNA3 vector. This allowed us to use FACS to sort the transfected from the untransfected cells by sorting for GFP positive cells. We analyzed human keratinocytes transfected for 48 hours with pcDNA3-OCT-GFP or with the control plasmid pcDNA3-GFP. As a hypomethylated control, we used the NTERA-2 cell line. The OCT4 promoter region of the NTERA-2 cells was mostly demethylated (Fig. 3). This was expected since NTERA-2 cells are embryonic in character and the OCT4 gene is continually expressed. Both the human keratinocytes transfected with GFP alone or with OCT4-GFP showed greater methylation as compared to the NTERA-2 cells. However, the OCT4-transfected keratinocytes showed a consistent demethylation of two cytosines (positions 74 and 80). We confirmed that the OCT4-GFP was effecting endogenous changes by examining the methylation state of NANOG, which is an embryonic pluripotent gene and importantly, a gene that was not exogenously introduced into the human keratinocytes. Forty-eight hours after transient transfection with OCT4-GFP, the NANOG gene also showed partial and consistent demethylation at positions −649, −611, −565, −516, and −378 upstream of the translational start site (Fig. 3). This is consistent with our finding for the endogenous OCT4 promoter and corresponds to published demethylated areas in fully converted iPS cells. 13 Thus, transient expression of OCT4 results in partial demethylation of the endogenous OCT4 and NANOG promoters.
FIGURE 3.

Transient expression of OCT4 produces a change in methylation state of the endogenous OCT4 and NANOG promoters in the transfected human skin keratinocytes (HSK-OCT4-GFP). Shown are methylation states of CpG islands −929 to +421 around the transcription start site of the endogenous OCT4 promoter and upstream areas −1091 to −299 of the NANOG promoter for the NTERA-2 cell line (positive control), human skin keratinocytes (HSK-GFP) 48 hours after transient transfection with pcDNA3-GFP (negative control), and human skin keratinocytes (HSK-OCT4-GFP) 48 hours after transient transfection with pcDNA3-OCT4-GFP. Transfected keratinocytes were sorted from untransfected keratinocytes based on GFP; thus the analysis was done only on the transfected cells. Note, demethylation of OCT4 was consistent at +74 and +80, and demethylation of NANOG was consistent at −649, −611, −565, −516, and −378. Numbers relative to translational start site.
Transient expression of OCT4 effects a decrease in global genomic methylation
Global changes in DNA methylation patterns throughout the genome occur when a cell’s differentiation or developmental state is changed. However, it was not known whether transient expression of OCT4 could alter the overall level of DNA methylation. Using a modified ELISA assay, we investigated the global methylation level of human keratinocytes transiently transfected with OCT4. Controls were GFP-transfected keratinocytes, untransfected keratinocytes, and the undifferentiated NTERA-2 cells. Examining the control cells, we found that global methylation levels between untransfected keratinocytes and GFP-transfected keratinocytes were not significantly different, whereas methylation states between GFP-transfected keratinocytes and NTERA-2 cells were significantly different (Fig. 4). Keratinocytes transiently expressing the OCT4-GFP fusion protein showed a significant decrease in global methylation as compared to untransfected and GFP-transfected keratinocytes. The OCT4-transfected cells exhibited a methylation level as low as that found in the NTERA-2 cells (Fig. 4). Thus, transient transfection of OCT4 causes a generalized demethylation of the DNA in keratinocytes, suggesting an overall reprogramming in the keratinocytes that might make them susceptible to a directed change in their differentiation pathway.
FIGURE 4.

Transient expression of OCT4 produces an overall decrease in global methylation in the transfected human skin keratinocytes (HSK). Shown is a graph depicting percent global methylation in four cellular groups: Untransfected HSKs (HSK); HSKs transiently transfected with pcDNA3-GFP (HSK-GFP) for 48 hours; HSKs transiently transfected with pcDNA3-OCT4-GFP fusion (HSK-OCT4) for 48 hours; and the NTERA-2 positive control cell line that continually expresses OCT4. Note, transfection of OCT4 significantly decreased the global methylation state of HSK (p=0.002) as compared to transfection with GFP alone.
Transient expression of OCT4 allows differentiation of human keratinocytes into human neuronal-like cells
Encouraged by the changes in global methylation of the transiently transfected human keratinocytes, we assessed the susceptibility of these cells to be redirected into a neuronal differentiation pathway. We transfected human skin keratinocytes with pcDNA3-OCT4, and at 48 hours switched their media to one of two types of neuronal differentiation media. Medium #1 was specifically modified for human cells from a mouse protocol.14 Medium #2 contained Noggin and SB-431542, inhibitors of the type I TGFβ receptor, shown to block phosphorylation and nuclear translocation of SMADs and to promote differentiation of embryonic stem cells and iPS cells into dopaminergic neurons.15 Controls for these experiments were untransfected human keratinocytes grown in the keratinocyte medium (KSFM), untransfected keratinocytes grown in one of the neuronal media, and a neuroepithelioma cell line (SK-N-MC). At the time of switching the media, all cells exhibited the typical cobblestone shape found in proliferating keratinocyte cultures. Twelve days later, the keratinocytes transiently transfected with OCT4 and the untransfected keratinocytes kept in KSFM still resembled proliferative keratinocytes (Fig. 5A). The untransfected keratinocytes switched to either neuronal media #1 or #2 became large, flat cells that lifted off the dish (data not shown). This was likely due to the neuronal media containing high calcium levels, which is known to halt proliferation and induce terminal differentiation of skin keratinocytes.16 In contrast, keratinocytes that had been transfected with pcDNA3-OCT4 48 hours before switching to the neuronal media exhibited neuronal phenotypic changes. These cells no longer showed the typical keratinocyte cobblestone appearance, but instead formed tight clusters of elongated cells, with triangular cell bodies and lengthy projections (for an example, see Fig. 5B).
FIGURE 5.

Human skin keratinocytes (HSKs) transiently transfected with pcDNA3-OCT4 can be differentiation into neuronal-like cell types. A) Phase image of HSKs transfected with pcDNA3-OCT4 for 48 hrs, then grown for 12 days in KSFM. B) Phase image of HSKs transfected with pc-DNA3-OCT4 for 48 hrs, then grown for 12 days in neuronal medium from protocol #1. C) RT-PCR showing expression of neuronal nuclear Fox-3 splicing protein (NeuN), neuron-specific class III β Tubulin (Tuj1), tyrosine hydroxylase (TH), keratin 14 (K14), and GAPDH. Lanes 1 = Untransfected HSKs grown for 22 days in KSFM; 2 = Untransfected HSKs grown for 22 days in neuronal medium from protocol #1; 3 = Neuroepithelioma SK-N-MC cell line; 4 = HSKs transfected with OCT4 for 48 hrs, then grown for 22 days in neuronal medium #1; 5 = HSKs transfected with OCT4 for 48 hrs, then grown for 12 days in neuronal medium #2. D) Western blots showing expression of NeuN protein. Lanes 1 = HSKs transfected with OCT4 for 48 hrs, then grown for 22 days in neuronal medium #1; 2 = HSKs transfected with OCT4 for 48 hrs and grown in KSFM for 22 days; 3 = untransfected HSKs grown in neuronal medium #1 for 22 days; 4 = untransfected HSKs grown in KSFM for 22 days. Note, only cells in lane 1 show expression of NeuN protein. KSFM = keratinocyte serum free medium.
To confirm that a change from keratinocyte to neuronal cell type had occurred, we examined the cultures for loss of keratin 14 (K14), an intermediate filament protein found in proliferating keratinocytes. We also probed for a concomitant expression of the following neuronal markers: Tuj1, the neural progenitor marker βIII tubulin; NeuN, the neuronal nuclear Fox-3 splicing protein found in cultured neurons; and tyrosine hydroxylase (TH), the rate limiting enzyme in dopaminergic neurons. RNA and protein were collected from cultures twelve and twenty-two days after switching media. RT-PCR revealed that the transiently transfected cells that were differentiated in medium #1 expressed NeuN, Tuj1 and tyrosine hydroxylase, while the same cells grown in medium #2 expressed Tuj1 and tyrosine hydroxylase, but no NeuN (Fig. 5C), suggesting that two different types of neuronal cells were produced. These cultures also showed very light expression of K14, indicating that some keratinocytes were present. This was not totally unexpected as we had not achieved 100% transfection efficiency (see Fig. 1). Untransfected control keratinocytes expressed K14, but did not express any of neuronal markers, whether grown in KSFM or either of the neuronal media (Fig. 5C). The neuroepithelioma cell line expressed Tuj1 and no K14. Western blot analysis for NeuN supported the RT-PCR results (Fig. 5D). The phenotypic changes, as well as the changes in gene and protein expression, were consistent and repeatable, suggesting that a 48-hour transient expression of OCT4 is sufficient to render human keratinocytes susceptible to changing their differentiation pathway into cells derived from the neuroectoderm.
Human keratinocytes can be redirected into human mesenchymal cells
To determine that transient transfection of OCT4 is sufficient to allow redirection of keratinocytes into mesodermally-derived cell types, we modified a published protocol used to differentiate multipotent progenitor cells into smooth muscle cells.17 Forty-eight hours after transfection with pcDNA3-OCT4, human keratinocytes were transferred into the mesenchymal differentiation medium containing TGFβ-1, PDGF-BB, and 100 μM ascorbic acid. After four days in this medium, the transfected keratinocytes began change their phenotype. By twelve days, none of the cells exhibited the typical cobblestone shape of keratinocytes. Instead they were all elongated rod-shaped cells, with parallel orientation, and typical characteristics of cultured smooth muscle or myofibroblast cells from a mesenchymal cell lineage18 (Fig. 6B). The untransfected keratinocytes grown in KSFM for twelve days retained their keratinocyte morphology (Fig. 6A). However, if grown in the mesenchymal medium, these cells terminally differentiated due to the high level of calcium in the medium.16 In general, both the RT-PCR (Lane 1 in Fig. 6D) and the Western blot analysis (Lane 1 in Fig. 6E) confirmed that the cells exhibited a mesenchymal phenotypic change. No K14 was found in the keratinocytes transfected with OCT4 and grown in the mesenchymal medium for 12 days, suggesting that no keratinocytes remained in these cultures (lane 1 Fig. 6D and E). Both the untransfected keratinocytes and the keratinocytes transfected with OCT4 and grown in KSFM expressed K14, but did not express any of the mesenchymal markers (Lanes 2 and 3 in Fig. 6D and E). Taken together, the changes seen in phenotype, gene expression and protein expression, suggest that transient transfection with OCT4 is sufficient to allow keratinocytes to be differentiated into mesenchymal cells.
FIGURE 6.

Human skin keratinocytes (HSKs) transiently transfected with pcDNA3-OCT4 can be differentiated into myofibroblasts or smooth muscle-type cells. A) Phase image of HSKs grown in KSFM for 12 days. B) Phase image of OCT4-transfected HSKs grown in smooth muscle differentiation medium for 11 days. C) RT-PCR showing expression of calponin, SM22α, and keratin 14 (K14). Lanes 1 = HSKs transfected with OCT4 for 48 hrs, then grown in mesenchymal differentiation medium for 12 days; 2 = HSKs transfected with OCT4 for 48 hrs and grown in KSFM for 12 days; 3 = untransfected HSKs grown in KSFM for 12 days; 4 = human aorta cDNA positive control. E) Western blot analysis of vimentin, SM22α, myocardin, and actin proteins. Lanes 1 = HSKs transfected with OCT4 for 48 hrs, then grown in mesenchymal differentiation medium for 12 days; 2 = HSKs transfected with OCT4 for 48 hrs and grown in KSFM for 12 days; 3 = untransfected HSKs grown in KSFM for 12 days; 4 = WPMY-1 myofibroblast cell line. Note, myocardin has three isoforms, 73 kD, 95.7 kD, and 101 kD 34. KSFM = keratinocyte serum free medium.
Mesenchymal cells differentiated from keratinocytes transiently transfected with OCT4 function as contractile cells
To further asses the mesenchymal cells, we used a classic contraction assay to determine if they could function as contractile cells. Keratinocytes were transfected with pcDNA3-OCT4 for 48 hours, then grown in the mesenchymal medium for ten days. After this, the cells were removed from the culture dish and embedded in collagen type I gels. Control cells were: untransfected keratinocytes grown in mesenchymal medium or KSFM, primary human fibroblasts, and the WPMY-1 myofibroblast cell line. Function was assessed by determining how well the cells contracted the gel. The amount of contraction was determined by measuring the diameters of the collagen discs over time. The OCT4-transfected keratinocytes grown in the mesenchymal medium contracted the collagen type I gels at a rate similar to the freshly isolated dermal fibroblasts (2.3 mm/day vs. 2.2 mm/day) and slightly more rapidly than the WPMY-1 myofibroblast cell line (1.7 mm/day) (see 1, 4, and 5 in Fig. 7A). There was no significant difference in the total amount of contraction (~50%) among these three cell types (Fig. 7B). The untransfected keratinocytes whether grown in KSFM or in the mesenchymal medium did not contract the collagen gels at all (see 2 and 3 in Fig. 7A). Thus, transient transfection of OCT4 is sufficient and necessary to allow human skin keratinocytes to be differentiated into functioning alternative cell types.
FIGURE 7.
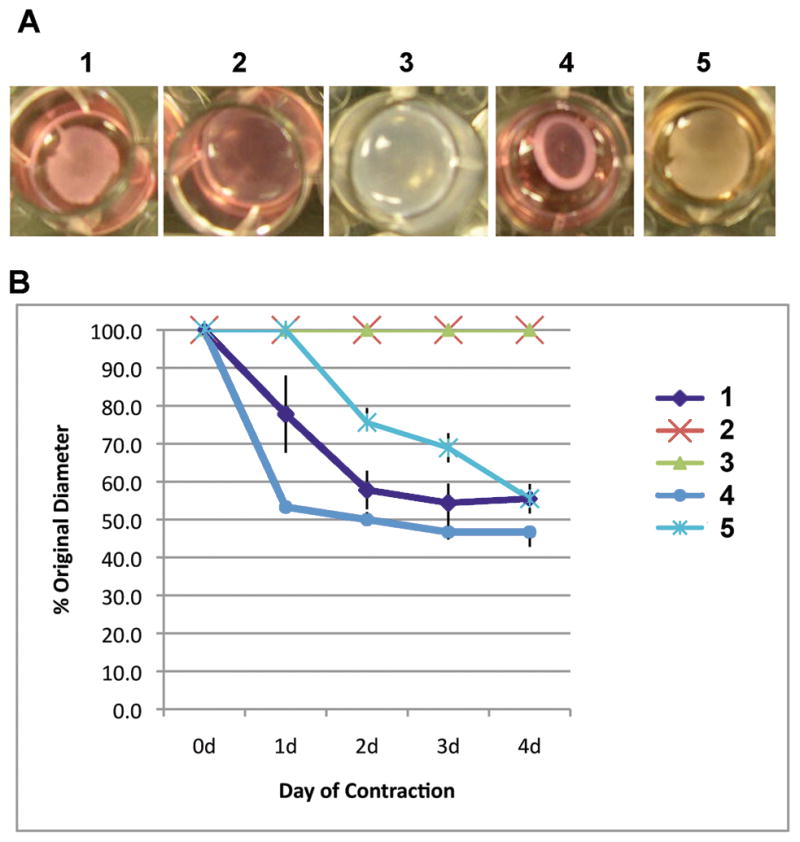
Transient expression of OCT4 allows human keratinocytes (HSKs) to be directed to differentiate into contractile cells. Cells were embedded in collagen type I in 12-well culture plates and allowed to contract the collagen gels for four days. A) Photographs of collagen gels at 3 days after cells were embedded into the gels. B) Graph showing amount of contraction by the various cells over 4 days. Note, HSKs transfected with OCT4 for 48 hrs, then grown in mesenchymal differentiation medium for 12 days contracted the collagen gels as well as the fibroblasts and the WPMY-1 myofibroblast cell line contracted the gels, whereas untransfected HSKs showed no contractility whether grown in KSFM or in mesenchymal differentiation medium. 1 = HSKs transfected with OCT4 for 48 hrs, grown in mesenchymal differentiation medium for 12 days, then embedded in the collagen type I gel; 2 = untransfected HSKs grown in mesenchymal differentiation medium for 12 days, then embedded in the collagen gel; 3 = untransfected HSKs grown in KSFM for 12 days, then embedded in the collagen gel; 4 = primary human fibroblasts grown in DMEM then embedded in the collagen gel; 5 = WPMY-1 myofibroblast cell line grown in DMEM then embedded in the collagen gel. KSFM = keratinocyte serum free medium.
DISCUSSION
In 1990, Oct4 was discovered to be a transcription factor that was essential for mammalian early embryonic development.19,20 Since then, Oct4 has been proved necessary to maintain pluripotency of embryonic stem cells in that it activates other pluripotency genes and represses differentiation genes.21 Thus, Oct4 is considered to be the master regulator of pluripotency. Two genes that Oct4 activates are Sox2 and Nanog. Along with Oct4, these genes are required to maintain cells in the pluripotent state.22,23 However, in order for the pluripotent cells to differentiate, all three of these genes must be turned off.24,25 Thus, we reasoned that to increase the potential of somatic cells, Oct4 would first need to be introduced into the cells, maintained just long enough to reactivate the endogenous Sox2 and Nanog genes, then all three genes would have to be turned off in the cells in order to redirect the cell’s differentiation into an alternative cell type. Very recently, it was shown that each species’ cells react differently to reprogramming and differentiation signals, such that it is imperative that the correct species cells be used to obtain the correct subsequent differentiation signals.11 Since we hoped to develop a technology that could be translated to human therapy, we transiently expressed the human OCT4 cDNA in freshly isolated human skin keratinocytes. This resulted in reactivation of the endogenous SOX2 and NANOG genes in the cells, but only for four to five days. We discovered that If these cells were exposed to new growth factors and cytokines during this four day period, they could be stimulated to change their differentiation pathway. Furthermore, we could define the type of new cell by exposing the transiently transfected keratinocytes to specific differentiation factors designed for specific cell types. This strategy allowed us to direct the keratinocytes to differentiate into dopaminergic neurons, which expressed tyrosine hydroxylase, and into mesenchymal cells, which showed contractile function. Thus, this simple technology could allow rapid development of replacement cells for human therapy.
Recently, much emphasis has been on making induced pluripotent stem (iPS) cells by fully dedifferentiating somatic cells. In 2006, Takahashi and Yamanaka demonstrated that mouse fibroblasts could be reprogrammed into fully functional pluripotent stem cells that resembled embryonic stem cells.1 To do this, they genomically incorporated four factors (Oct4, Sox2, Klf4, and c-myc) into the mouse fibroblasts using retroviral vectors. Although exciting, two limitations preclude this technology from being easily translated to humans: genomic retroviral integration and the very low induction efficiency to form the functional iPS cells.5 Even keratinocytes which were reported to be 100-fold more receptive than fibroblasts, formed iPS cells at a rate of only 0.7%.9 Varas, et al.26 examined retroviral insertion sites in clones of iPS cells derived from mouse fibroblasts and determined that although there were several insertion sites in numerous genes, none of them were specifically common among the iPS clones. They concluded that the low induction efficiency could not be explained by insertional mutagenesis of the retroviruses. Thus, it is still unclear why this technology is so inefficient in inducing iPS cells.
More recently, several reports described alternative ways to make iPS cells. Most were designed to result in transgene-free iPS cells that could be used for future therapeutic applications. Sommer, et al.27 described a lentiviral expression vector that carried Oct4, Sox2, Klf4, and c-myc in a single vector with 2A peptide and internal ribosome entry site technology. This system has the advantage over the initial retroviral system by allowing expression of all four factors with a single viral insertion, thereby decreasing the potential of insertional mutagenesis. Yusa et al.28 used the piggyBac transposon, to integrate the four factors into mouse embryonic fibroblasts. This transposon is a mobile element from insects, which integrates into mammalian genomes at TTAA sites. The advantage over viral integration is that the transposon can be removed from the host with little to no change in nucleotide sequences because the integration sites are repaired upon excision.28 Stadtfeld et al.29 generated iPS cells using a non-integrating adenoviral vector. In this system the iPS cells showed the expected characteristic embryonic changes, i.e. expression of endogenous pluripotency genes, formation of teratomas, and demethylation of DNA. Another system used continual transfection of plasmid vectors carrying the four genes into mouse embryonic fibroblasts (MEFs).8 This produced virus-free and functional iPS cells, which was an improvement over the viral vector systems; however, it still took several months to make the iPS cells. Although both the adenoviral transduction and repeated plasmid transfection successfully generated integration-free iPS cells, the efficiency of reprogramming using these methods was 100–1000-fold lower than the retroviral method, thereby reducing induction efficiency to 0.001–0.0001%. Since genetic alterations could occur during the reprogramming process, the retroviral and lentiviral systems are problematic for human use. Of the insertional technologies, the piggyBac system offers the best alternative as it allows correction of most of the insertion sites. One concern of this system was shown in mouse embryonic stem cells, where approximately 5% of the excision sites showed microdeletions, which might be mutagenic.28 It will be critical to maintain genomic stability if we wish to use the cells in humans. The success of the adenoviral system and the multi-plasmid transfection confirms that insertional mutagenesis is not a requirement for cellular reprogramming. Our system goes one step further. Instead of reprogramming the somatic cells completely to embryonic stem cells, we show that a transient expression of OCT4 is enough to allow the keratinocytes to change their differentiation pathway.
It is clear that manipulating genetic expression of embryonic pluripotency genes is critical to reprogramming iPS cells. However, it may not be necessary to introduce four exogenous genes into the cells. For example, CD45+ cord blood cells were successfully differentiated into several other cell types by exposing the cells to culture medium containing several growth factors. Such cells showed transient expression of Oct4 and Nanog with concomitant changes in H3 and H4 histone acetylation.30 This has some limitations as not everyone will have access to cord blood cells. Using skin keratinocytes is a relevant alternative. It may be that keratinocytes are unique among somatic cell types. Since keratinocytes are responsive to the external environment and are continually replacing themselves,31,32 it may be that they are more readily responsive to partial reprogramming than are fibroblasts. Furthermore, our previously published data demonstrate that mouse epidermal stem cells can be reprogrammed by transfer into the developing blastocyst, which suggests that keratinocyte stem cells might be amenable to reprogramming without external transfection.10 It would advantageous if we could directly translate the mouse findings to human keratinocyte stem cells. However, we have been unsuccessful in using the mouse sorting technology that we developed to isolate human keratinocyte stem cells, which suggests that human skin and mouse skin keratinocyte stem cells differ at some as yet unknown fundamental property.
The data presented here suggest that a brief introduction of the single factor OCT4 into human skin keratinocytes is sufficient to alter the global methylation of the cells. Furthermore, if these cells are then exposed to differentiating factors, they respond by not only changing their phenotype and gene and protein expression profiles, but also their function. Thus, our findings suggest that it is not necessary to fully dedifferentiate human keratinocytes into iPS or embryonic stem cells in order to use them in translational therapies.
MATERIALS AND METHODS
Cell isolation, culture, and transfection
Primary adult human skin keratinocytes (HSKs) and fibroblasts (HFbs) were isolated from normal skin, obtained from the Surgical Pathology Department at The University of Iowa Hospitals and Clinics with full approval of The University of Iowa’s IRB. The skin specimens were not diseased or pathological; they were from amputations after accidents, mammary reductions, or from surgical reductions for obesity. The ages of the donors were noted for each experiment. To ensure sterility, strips of skin were soaked for 1 hour in medium containing 10% Antibiotic-antimycotic (Invitrogen, Carlsbad, CA). Epithelial sheets were mechanically separated from the connective tissue after overnight incubation of skin in Dispase II (24.0 U/ml, Roche, Indianapolis, IN) at 4°C. HSKs were isolated from the epithelial sheets by treatment in 0.25% trypsin (Invitrogen) for 30 minutes at 37°C. Cells were plated at a concentration of 8×104 cells/ml in keratinocyte serum free medium (KSFM, Invitrogen) + 1.5% Antibiotic-antimycotic on culture dishes coated with 0.25μg/cm2 collagen type IV (Collaborative Biomedical). Fibroblasts were obtained by treating the connective tissue in 0.4% collagenase for 1 hour at 37°C. HFbs were plated at a concentration of 8×104 cells/ml in DMEM + 10% FBS + 1.5% Antibiotic-antimycotic. For transfection, cells were grown as a monolayer culture at 37°C in a 5% CO2 humidified incubator until 50–60% confluent, then transiently transfected with pcDNA3-OCT4, pcDNA3-OCT4-GFP, or pcDNA3-GFP using Turbofection (OriGene, Rockville, MD) at a concentration of 1 μg of plasmid to 2μl of Turbofection per cm2. The transfection solution was left on the cells for 6–24 hours, then replaced with complete medium
Control cell lines, obtained from the ATCC and cultured according to ATCC instructions, were the following: NTERA-2, a human teratocarcinoma cell line with characteristics and gene expression profiles similar to cultured human embryonic cells; WPMY-1, a human myofibroblast cell line with characteristics similar to smooth muscle cells; and SK-N-MC, a neuroepithelioma cell line with characteristics similar to cultured proliferative neuronal cells.
RNA extraction and RT-PCR
Total RNA was extracted from cultured cells using Trizol or the RNAqueous-4PCR kit (Invitrogen) according to manufacturer’s instructions. cDNA was generated through a reverse transcription reaction consisting of the following: 1.5 μg of RNA; 50 μM random primers; 5x FS RT Buffer; 10 mM dNTP mix; 0.1M DTT; 40 IU RNasin; 200 IU RTase superscript III; and nuclease-free DEPC water. RNA, random primers, dNTPs, and water were combined and heated at 65°C for 5 min., then chilled on ice. Following addition of the other components, the RT reaction was incubated at 25°C for 5 min., 55°C for 1 hour, then 70°C for 15 min., using a GeneMate Genius (ISC Bioexpress, Kaysville, UT) or a MultiGene (LabNet, Edison, NJ) thermocycler, and then chilled on ice. For PCR, 1 μl of cDNA template was added to 24 μl of the following reaction mixture: 10x PCR Buffer; 25 mM MgCl2, nuclease-free DEPC water; 10 mM dNTPs (Invitrogen); 5 IU Taq polymerase (Qiagen, Valencia, CA); and 10 μM of each primer. PCR was performed for 35 cycles using the following conditions: denaturation at 94°C for 0.5–2 min., annealing at the specified Tm (see below) for 1 min., elongation at 72°C for 1 min/kb. Positive controls were RNA isolated from neuroepithelioma SK-N-MC cells or cDNA from aorta (purchased from ScienCell, Carlsbad, CA).
| Forward primer | Reverse primer | Tm | bp | |
| hOCT4 | CAGTGCCCGAAACCCACAC | GGAGACCCAGCAGCCTCAAA | 59 | 161 |
| hSOX2 | CAGGAGTTGTCAAGGCAGAGAAGA | GCCGCCGCCGATGATTGTTATTAT | 60 | 320 |
| hNANOG | TTTGGAAGCTGCTGGGGAAG | GATGGGAGGAGGGGAGAGGA | 59 | 194 |
| hREX-1 | CAGATCCTAAACAGCTCGCAGAAT | GCGTACGCAAATTAAAGTCCAGA | 56 | 310 |
| hNeuN | AGCGACAGTTACGGCAGAGT | ACTTGGACTTGGTTGGATGC | 57 | 235 |
| TuJ1 | GGCCAAGTTCTGGGAAGTCA | CGAGTCGCCCACGTAGTTG | 57 | 235 |
| TH | TTGCTGAGATCGCCTTCCAGTACA | AGTACAGCGTGGACAGCTTCTCAA | 60 | 495 |
| Calponin | AGGCTCCGTGAAGAAGATCA | CCACGTTCACCTTGTTTCCT | 55 | 214 |
| SM22α | CGCGAAGTGCAGTCCAAAATCG | GGGCTGGTTCTTCTTCAATGGGG | 59 | 906 |
| K14 | TTCTGAACGAGATGCGTGAC | GCAGCTCAATCTCCAGGTTC | 59 | 188 |
| GAPDH | GAAGGTGAAGGTCGGAGTC | GAAGATGGTGATGGGATTTC | 55 | 226 |
Protein isolation and Western blot analysis
Protein was extracted from experimental and control cells by scraping in 0.5 ml RIPA buffer (Thremo Fisher Scientific (Pierce), Rockford, IL) per 100 mm culture dish. After 30 min. on ice, the slurry was spun at 13,000 rpm, protease inhibitor added (Roche, Indianapolis, IN), and the supernatant collected. Protein concentration was determined using the Bradford Assay (BioRad, Hercules, CA). All proteins were detected by Western blot with commercially available antibodies. Proteins (20 μg protein per lane) were resolved using 12% polyacrilamide gels (BioRad), then transferred to pure nitrocellulose membrane (0.045 μM, Bio-Rad) using the semidry transfer Trans Blot-SD system (BioRad). Membranes were washed in TBS/0.1% Tween 20, blocked for 2 hours in 5% low fat milk in TBS/0.1% Tween, and incubated with primary antibodies over night at 4°C on a shaker, then rinsed and incubated with IR (infra red) conjugated secondary antibodies (IRDye™800 anti-mouse IgG (1:2500), Rockland Inc, Philadelphia, PA, USA). Protein immunoblots were scanned using the Odyssey, LI-COR-infrared imager. Protein extracted from control cell lines (WPMY-1, SK-N-MC) were treated similar to the experimental cells. Primary antibodies were: mouse anti-NeuN (1:500, Millipore, Billerica, MA), mouse anti-SM22α (1:1000, Abcam, Cambridge, MA), mouse anti-Myocardin (1:1000, Santa Cruz, Santa Cruz, CA), mouse anti-Vimentin (1:1000, BD Biosciences, San Jose, CA), and mouse anti-Actin (1:1000, Sigma-Aldrich, St. Louis, MO).
FACS
Cells were transiently transfected with pcDNA3-OCT4-GFP or pcDNA3-GFP, harvested at 48 hours using 0.5% trypsin/EDTA, pelleted by centrifugation, and resuspended at 2×106 cells/ml in PBS. Propidium iodide was added to a final concentration of 0.1 μg/ml and the cells filtered through a 70 μm cell strainer prior to sorting. Cells were live sorted using a FacsDiva in the University of Iowa’s Flow Cytometry Core Facility. Transiently transfected live cells were selected by negative staining of propidium iodide and positive staining for GFP. For experiments measuring DNA methylation, cells were lysed and genomic DNA harvested following the spin-column protocol (DNeasy Blood and Tissue kit, Qiagen).
Global DNA methylation
Global DNA methylation was measured with a modified ELISA assay, using the Methylamp Global DNA Methylation kit (Epigentek, Brooklyn, NY) according to the manufacturer’s protocol. Replicate samples of genomic DNA were bound in 96 well strip-wells. Samples were blocked, then labeled using an anti-methyl-cytosine primary antibody and an enzyme conjugated secondary antibody. Following addition of the substrate solution for 2–5 minutes, the reaction was halted and absorbance at 450 nm measured. To quantitate DNA methylation, samples were background subtracted and normalized to 100% methylated DNA control samples.
Site-specific DNA methylation determination
Bisulfite sequencing was used to measure the methylation of specific sites surrounding the promoter region of OCT4. Nonmethylated cytosines were converted to uracils by a two-step chemical/bisulfite reaction using (EZDNA methylation kit, Zymo Research, Orange, CA). The region surrounding the OCT4 start site of the converted DNA was PCR amplified using three sets of primers covering the range −910 to 535 (numbers relative to translational start site). For NANOG, region −1091 to −299 was PCR amplified. Primers for OCT4 and NANOG were the following sets:
| Forward primer | Reverse primer | |
| OCT4-1 | GGATTTGGTTAAGTTTTTAAGGTTTTT | CCAAAACAACTAACCCTACCTACTC |
| OCT4-2 | TATTTAGGAGGTTGGAGTAGAAGGAT | AAACCTTAAAAACTTAACCAAATCC |
| OCT4-3 | TGGGTAGATGGTGTTAGGTATTTAG | ACCCAAACTAATCTTAAATTCCTATCC |
| NANOG | AGAGTTAGAGGGAAAAAGTTAGAAGT | AACCCACCCTTATAAATTCTCAATTA |
Cloning of PCR products was done using TOPO TA Cloning kit (Invitrogen). PCR products were legated into pCR2.1-TOPO, and used to transform DH5α competent cells following the manufacturer’s protocol. Transformed cells were spread on selective agar plates containing 50 μg/ml ampicillin, spread with 40 μl of 40 mg/ml X-gal and incubated at 37°C overnight. White colonies were picked, grown at 37°C overnight in LB medium containing 50 μg/ml ampicillin. Plasmid DNA from each sample was harvested and purified using the PureLink Quick Plasmid Miniprep Kit (Invitrogen) following the manufacturers instructions. Plasmid preparations were tested for the presence of the inserted PCR product by restriction enzyme analysis using EcoR1. All positive preparations were sequenced at the University of Iowa DNA Facility using the M13 reverse primer in pCR2.1-TOPO.
Neuronal cell differentiation
HSKs were plated and transiently transfected with pcDNA3-OCT4 as described above. Forty-eight hours later, the cells were exposed to neuronal medium using one of two protocols. Conditions for Protocol #1 (for general neural differentiation) were modified for human cells from the procedure of Grinnell, et al.,14 which was based upon the procedure from Jiang, et. al.33 The base medium consisted of 60 ml DMEM-low glucose (Invitrogen), 40 ml MCDB-201 (Sigma), 1 ml Insulin-Transferrin-Selenium (Sigma), 1 ml linoleic, Acid-Albumin (Sigma), 3 mg Ascorbic Acid 2-phosphate (Sigma), 1 ml 100 μM dexamethasone (Sigma), 1 ml Antibiotic-antimycotic (Invitrogen). This combination was stirred, filter sterilized, and then frozen into aliquots at 20°C. The base medium was supplemented with 100 ng/ml basic fibroblast growth factor (b-FGF, Sigma), 100 ng/ml Sonic Hedgehog (SHH, Sigma), 10 ng/ml fibroblast growth factor-8 (FGF-8, Cell Sciences, Canton, MA), and 10 ng/ml brain-derived neurotrophic factor (BDNF, Cell Sciences). Cells were then cultured for 22 days, with the medium renewed every 2nd day. Cells were photographed every 2 days. On day 22, RNA and protein were extracted for RT-PCR and Western blot, respectively.
Conditions for Protocol #2 (for dopaminergic neuron differentiation) were modified from Chambers, et al.15 Neuronal differentiation of transfected HSKs was initiated by adding KSR medium containing 10 μM SB431542 (Tocris, Ellisville, MO), an inhibitor of SMAD phosphorylation, and 500 ng/ml Noggin (R&D systems, Minneapolis, MN). The combination of these two molecules inhibited SMAD signaling and promoted neuronal differentiation.15 The KSR medium consisted of 820 ml of Knockout DMEM (Invitrogen), 15% KSR serum (Invitrogen), 1% L-glutamine 200 mM (Invitrogen), 1% antibiotic-antimycotic (Invitrogen) 1% MEM NEAA (Invitrogen), and 1 ml of 2-mercaptoethanol (Sigma). On day 3 of differentiation, the KSR was replaced with fresh KSR containing SB431542 and Noggin. On day 5, KSR was replaced with a 3:1 mixture of KSR/N2 (Invitrogen) media containing SB431542 10 μM, Noggin 500 ng/ml, and 200 ng/ml SHH (Sigma). On day 7, the medium was replaced with KSR/N2 media 1:1 containing SB431542, Noggin, and SHH. On day 9, the medium was replaced with KSR/N2 media 1:3 containing SB431542, Noggin, SHH, and 100 ng/ml FGF-8 (Cell Sciences), 20 ng/ml BDNF (Cell Sciences), and 0.2 mM ascorbic acid (Sigma). On Day 11, the medium was replaced with N2 media containing SB431542, Noggin, SHH, FGF-8, BDNF, and AA. On day 12, addition of Noggin and SB431542 was ceased. Thereafter, the medium was replaced every two days with N2 media containing 20 ng/ml BDNF, 10–20 ng/ml glial cell line-derived neurotrophic factor (GDNF), and 0.2 mM AA, 0.5–1.0 mM dibutyryl camp, 1 ng/ml transforming growth factor type β3 (TGF-β3, R&D Systems). Cells were photographed every 2 days. RNA and protein were extracted for RT-PCR and Western blot, respectively, on day 12.
Contractile mesenchymal cell differentiation
HSKs were plated and transfected with pcDNA3-OCT4 as described above. At 48h after transfection, medium was replaced with smooth muscle medium containing: DMEM with 10% FBS (Thermo Scientific HyClone, Logan, UT), 2.5 ng/ml TGF-β1 (R&D Systems), 5 ng/ml PDGF-BB (R&D Systems), and 100 μM ascorbic acid (Sigma). The medium and protocol was modified from Ross et al.17 Cells were photographed and medium was changed every two days for 12 days. On day 12, RNA and protein were extracted for RT-PCR and Western blot, respectively.
Contraction of collagen type I gels
To test function, cells were added to collagen gels and rates of contraction determined. Gels were made as follows: DMEM (high glucose, Invitrogen) +10% FBS with 7×104 cells was added to rat tail collagen type I (BD Biosciences) to a concentration of 1.25 mg/ml. The pH was raised to neutral with 0.1 N NaOH. Cells and collagen were allowed to gel at room temperature for 15 min. in 12-well culture plates, then 1 ml of DMEM + 10 % FBS + fresh vitamin C (50 μg/ml) was added. A sterile pipette tip was passed around the solidified collagen in the well to detach the collagen disk from the culture dish and allow it to float unimpeded, and the plates were placed in a 37°C 5% CO2 incubator. Medium was replaced each day. Experimental and control cells were plated in triplicate and the diameter of each collagen disk was photographed and measured every day for 4 days. Mean diameters were calculated and standard deviations determined. Results were graphed.
Acknowledgments
We thank the members of the Bickenbach lab for helpful discussion, Matthew Fitzgerald and Dr. Frederick Domann for assistance with the bisulfite technique, and members of the UI Flow Cytometry Core. This research was funded in part by grants from the National Institutes of Health to JRB (R21AR053936 and R01AR053619) and by a grant from the Maryland Stem Cell Research Fund to RLE and JRB (MSCRFII-0178-01).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Zhao R, Daley GQ. From fibroblasts to iPS cells: induced pluripotency by defined factors. J Cell Biochem. 2008;105:949–955. doi: 10.1002/jcb.21871. [DOI] [PubMed] [Google Scholar]
- 3.Yamanaka S. Induction of pluripotent stem cells from mouse fibroblasts by four transcription factors. Cell Prolif. 2008;41(Suppl 1):51–56. doi: 10.1111/j.1365-2184.2008.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanna J, Wernig M, Markoulaki S, Sun C-W, Meissner A, Cassady JP, et al. Treatment of Sickle Cell Anemia Mouse Model with iPS Cells Generated from Autologous Skin. Science (Science Express on 6 December 2007) 2007;318:1920–1923. doi: 10.1126/science.1152092. [DOI] [PubMed] [Google Scholar]
- 5.Park I-H, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. EPub 10 January 2008. [DOI] [PubMed] [Google Scholar]
- 6.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 7.Amabile G, Meissner A. Induced pluripotent stem cells: current progress and potential for regenerative medicine. Trends Mol Med. 2009;15:59–68. doi: 10.1016/j.molmed.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 8.Okita K, Hong H, Takahashi K, Yamanaka S. Generation of mouse-induced pluripotent stem cells with plasmid vectors. Nat Protoc. 2010;5:418–428. doi: 10.1038/nprot.2009.231. [DOI] [PubMed] [Google Scholar]
- 9.Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotech. 2008;26:1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- 10.Liang L, Bickenbach JR. Somatic epidermal stem cells can produce multiple cell lineages during development. Stem Cells. 2002;20:21–31. doi: 10.1634/stemcells.20-1-21. [DOI] [PubMed] [Google Scholar]
- 11.Schnerch A, Cerdan C, Bhatia M. Distinguishing between mouse and human pluripotent stem cell regulation: the best laid plans of mice and men. Stem Cells. 2010;28:419–430. doi: 10.1002/stem.298. [DOI] [PubMed] [Google Scholar]
- 12.Deng J, Shoemaker R, Xie B, Gore A, LeProust EM, Antosiewicz-Bourget J, et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol. 2009;27:353–360. doi: 10.1038/nbt.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell (EPub Nov 30, 2007) 2007;131:834–835. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 14.Grinnell KL, Yang B, Eckert RL, Bickenbach JR. De-differentiation of mouse interfollicular keratinocytes by the embryonic transcription factor Oct-4. J Invest Dermatol. 2007;127:372–380. doi: 10.1038/sj.jid.5700531. [DOI] [PubMed] [Google Scholar]
- 15.Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hennings H, Michael D, Cheng C, Steinert P, Holbrook K, Yuspa SH. Calcium regulation of growth and differentiation in mouse epidermal cells in culture. Cell. 1980;19:245–254. doi: 10.1016/0092-8674(80)90406-7. [DOI] [PubMed] [Google Scholar]
- 17.Ross JJ, Hong Z, Willenbring B, Zeng L, Isenberg B, Lee EH, et al. Cytokine-induced differentiation of multipotent adult progenitor cells into functional smooth muscle cells. J Clin Invest. 2006;116:3139–3149. doi: 10.1172/JCI28184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lau HK. Regulation of proteolytic enzymes and inhibitors in two smooth muscle cell phenotypes. Cardiovasc Res. 1999;43:1049–1059. doi: 10.1016/s0008-6363(99)00136-4. [DOI] [PubMed] [Google Scholar]
- 19.Scholer H, Ruppert S, Suzuki N, Chowdhury K, Gruss P. New type of POU domain in germ line-specific protein Oct-4. Nature. 1990;344:435–439. doi: 10.1038/344435a0. [DOI] [PubMed] [Google Scholar]
- 20.Scholer HR, Dressler GR, Balling R, Rohdewohld H, Gruss P. Oct-4: a germline-specific transcription factor mapping to the mouse t-complex. EMBO J. 1990;9:2185–2195. doi: 10.1002/j.1460-2075.1990.tb07388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ovitt C, Scholer H. The molecular biology of Oct-4 in the early mouse embryo. Mol Hum Reprod. 1998;4:1021–1031. doi: 10.1093/molehr/4.11.1021. [DOI] [PubMed] [Google Scholar]
- 22.Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 2003;17:126–140. doi: 10.1101/gad.224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, et al. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003;13:631–642. doi: 10.1016/s0092-8674(03)00393-3. [DOI] [PubMed] [Google Scholar]
- 24.Kuroda T, Tada M, Kubota H, Kimura H, Hatano SY, Suemori H, et al. Octamer and Sox elements are required for transcriptional cis regulation of Nanog gene expression. Mol Cell Biol. 2005;25:2475–2485. doi: 10.1128/MCB.25.6.2475-2485.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodda D, Chew J, Lim L, Loh Y, Wang B, Ng H, et al. Transcriptional regulation of nanog by OCT4 and SOX2. J Biol Chem. 2005;280:24731–24737. doi: 10.1074/jbc.M502573200. [DOI] [PubMed] [Google Scholar]
- 26.Varas F, Stadtfeld M, de Andres-Aguayo L, Maherali N, di Tullio A, Pantano L, et al. Fibroblast-derived induced pluripotent stem cells show no common retroviral vector insertions. Stem Cells. 2009;27:300–306. doi: 10.1634/stemcells.2008-0696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sommer CA, Stadtfeld M, Murphy GJ, Hochedlinger K, Kotton DN, Mostoslavsky G. Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem Cells. 2009;27:543–549. doi: 10.1634/stemcells.2008-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yusa K, Rad R, Takeda J, Bradley A. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nat Methods. 2009;6:363–369. doi: 10.1038/nmeth.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322:945–949. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong CJ, Casper RF, Rogers IM. Epigenetic changes to human umbilical cord blood cells cultured with three proteins indicate partial reprogramming to a pluripotent state. Exp Cell Res. 2010 doi: 10.1016/j.yexcr.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 31.Halprin KM. Epidermal “turnover time”--a re-examination. Br J Dermatol. 1972;86:14–19. doi: 10.1111/j.1365-2133.1972.tb01886.x. [DOI] [PubMed] [Google Scholar]
- 32.Webb A, Kaur P. Epidermal stem cells. Front Biosci. 2006;11:1031–1041. doi: 10.2741/1861. [DOI] [PubMed] [Google Scholar]
- 33.Jiang Y, Henderson D, Blackstad M, Chen A, Miller R, Verfaillie C. Neuroectodermal differentiation from mouse multipotent adult progenitor cells. Proc Natl Acad Sci USA. 2003;100:11854–11860. doi: 10.1073/pnas.1834196100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Torrado M, Lopez E, Centeno A, Medrano C, Castro-Beiras A, Mikhailov AT. Myocardin mRNA is augmented in the failing myocardium: expression profiling in the porcine model and human dilated cardiomyopathy. J Mol Med. 2003;81:566–577. doi: 10.1007/s00109-003-0470-7. [DOI] [PubMed] [Google Scholar]