

Abstract
The goal of the present investigation was to determine the persistence of striatal dopaminergic dysfunction after a mild chemically-induced hypoxic event in Fisher 344 rats. To this end, we gave a single injection of the mitochondrial complex II inhibitor 3-nitropropionic acid (3-NP; 16.5 mg/kg, i.p.) to 2 month old male F344 rats and measured various indices of striatal dopaminergic functioning and lipid peroxidation over a 3 month span. Separate groups of rats were used to measure rod walking, evoked dopamine (DA) release, DA content, MDA accumulation, DA receptor binding, and tyrosine hydroxylase activity. The results showed that 3-NP exposure reduced most measures of DA functioning including motoric ability, DA release, and D2 receptor densities for 1 to 3 months post drug administration. Interestingly, DA content was reduced 1 week after 3-NP exposure, but rose to 147% of control values 1 month after 3-NP treatment. MDA accumulation, a measure of lipid peroxidation activity, was increased 24 hr and 1 month after 3-NP treatment. 3-NP did not affect tyrosine hydroxylase activity, suggesting that alterations in DA functioning were not the result of nigrostriatal terminal loss. These data demonstrate that a brief mild hypoxic episode caused by 3-NP exposure has long-term detrimental effects on the functioning of the nigrostriatal DA system.
Keywords: Striatum, 3-NP, dopamine release, D1 and D2 receptors, fast scan voltammetry, HPLC
Introduction
Neuronal dopamine (DA) systems are extremely sensitive to hypoxia caused by a reduction in oxygen delivery or exposure to mitochondrial toxins (Büyükuysal and Mete, 1999; Orset et al., 2006; Toner and Stamford, 1999). When oxygen or ATP is limited, DA neurons release DA into the extracellular space by reversing the DA transporter (DAT) and through vesicular exocytosis (Büyükuysal and Mete, 1999; Fernagut et al., 2002a). This increase in synaptic DA levels, and the subsequent oxidation of DA, leads to the formation of hydroxyl radicals, increased lipid peroxidation, and decreased Na+-K+-ATPase activity (Pereyra-Muñoz et al., 2006; Ste-Marie et al., 2000). As a result of the oxidative stress produced by DA metabolism, dopaminergic neurons especially those in the substantia nigra degenerate and die (Beal, 1995; Fernagut et al., 2002b; Jenner and Olanow, 1998).
As a primary target of DA afferents, the striatum is vulnerable to the consequences of hypoxia-induced DA release and associated DA oxidation (Beal et al., 1993a, b; Fancellu et al., 2003; Fernagut et al., 2002b; Gu et al., 2004; Hashimoto et al., 1994; Kim and Chan, 2002; Klivenyi et al., 2000; Lancelot et al., 1995; Pang et al., 2002; Reynolds et al., 1998; Toner and Stamford, 1999). For example, oxygen or ATP deprivation increases the formation of reactive oxygen species and lipid peroxidation in the striatum, thus inducing apoptotic cell death (Beal 1994, 1995; Gu et al., 2004; Hashimoto et al., 1994; Lancelot et al., 1995; Reynolds et al., 1998). Indeed, the striatal pathology resulting from stroke or exposure to mitochondrial toxins can be mimicked by direct injection of DA into the striatum (Hastings et al., 1996), while prior elimination or reduction of striatal DA reduces the risk for subsequent stroke or toxin-induced striatal lesions (Hashimoto et al., 1994; Ren et al., 1997; Reynolds et al., 1998).
Importantly, while the aforementioned studies assessed the role of DA release after extreme hypoxic events, little is known about the effects of mild or brief hypoxic events on striatal dopaminergic activity. Interestingly, in one of the few studies investigating this issue it was found that a very low dose of the mitochondrial complex II inhibitor 3-nitropropionic acid (3-NP), administered via drinking water, increased striatal DA content (Johnson et al., 2000). Moreover, we previously demonstrated that a single systemic injection of 3-NP increased corticostriatal excitation for 24 hr, apparently via a dopamine depletion-related mechanism (Akopian et al., 2008). Thus, it appears that 3-NP exposure causes short-term changes in striatal functioning. In the present study, we examined the persistence of striatal alterations after a single mild hypoxic event. Specifically, we measured striatal DA release, DA content, DA receptor densities and affinities, tyrosine hydroxylase activity, and rod-walking (a DA-dependent behavior) for 3 months after a single injection of 3-NP (16.5 mg/kg). In addition, we assessed malondialdehyde (MDA) accumulation (a measure of lipid peroxidation) at several time points.
Materials and Methods
Animals and drug treatment
Subjects were 336 young adult male F344 rats (Charles Rivers) that were between 180-230 g at time of injection. Rats were ordered at 2 months and allowed to acclimate to the vivarium for 1 week. Rats were then given a single injection of 3-NP (16.5 mg/kg. i.p.) or saline in the home cage. 3-NP was obtained from Sigma-Aldrich (St. Louis, MO) and was dissolved in saline and neutralized with NaOH. Subjects were treated according to the “Guide for the Care and Use of Mammals in Neuroscience and Behavioral Research” (National Research Council, 2003) under a research protocol approved by the Institutional Animal Care and Use Committee of CSUSB or USC.
Rod walking behavioral analysis
Rod walking was assessed using a 60 cm long wooden dowel, 5 cm in diameter that was grooved and suspended between two platforms 60 cm above a padded surface. Rats were placed in the center of the rod facing one platform and latencies to reach the escape platform were measured. Each rat was tested 3 times, with a maximum limit of 120 s. A rod walking score was calculated for each rat using the following scale: 0-fall, 1-clasp, 2-all paws on top, 3-takes steps, 4-reachs the platform (Friedmann and Gerhardt, 1992). The rat was then given a score that reflected the highest score of the three trials.
Two rod walking experiments were conducted. In the first experiment, baseline levels of rod walking were determined before all rats were injected with 3-NP. Separate groups of rats were then tested for rod walking performance 24 hr, 48 hr, or 1 week post-injection. In the second experiment, rats were injected with 3-NP or saline and tested 2 weeks, 1 month, or 3 months after injection. In both experiments, rats were killed immediately after behavioral testing and brains were used for fast scan cyclic voltammetry.
Brain slice preparation
Rats were anesthetized with halothane and decapitated immediately after behavioral testing (i.e., 24 hr, 48 hr, 1 week, 2 weeks, 1 month, or 3 months after 3-NP treatment). Brains were removed and placed in cooled (1-4° C), modified-oxygenated artificial cerebrospinal fluid (aCSF). In the modified aCSF, some of the sodium was replaced with sucrose to reduce tissue excitability during brain slice cutting (sucrose 124 mM, NaCl 62 mM). This solution maintained the osmotic balance found in normal aCSF. Normal aCSF contained (concentrations in mM) NaCl 124, MgSO4 1.3, KCl 3.0, NaH2PO4 1.25, NaHCO3 26, CaCl2 2.4, glucose 10.0, equilibrated with a 95% O2 - 5% CO2 mixture to obtain a pH value of 7.3 - 7.4.
Hemi-coronal striatal slices were cut at a thickness of 400 μm using a Vibratome1000 (Vibratome Co., St. Louis, MO). The slices were immediately placed in an oxygenated aCSF solution and were slowly brought to room temperature (23° C). Single slices were transferred to a recording chamber (Haas ramp style gas interface chamber), and bathed continuously with the oxygenated aCSF solution maintained at a temperature of 32° C.
Fast scan cyclic voltammetry (FSCV) quantification of striatal DA release
Disc carbon fiber electrodes (CFE) were made from 7 mm unsized carbon fibers (Goodfellow Corporation, Devon, PA) by electrophoretic anodic deposition of paint (ALA Scientific Instruments, Inc., Westbury, NY) (Schulte and Chow, 1996). Extracellular DA was monitored at the carbon fiber microelectrode every 100 msec by applying a triangular waveform (-0.4 to +1.0 Volt vs. Ag/AgCl, 300 Volt/second). Currents were recorded with a modified VA-10× Voltammetric and Amperometric Amplifier (NPI Electronic, Tamm, Germany). Data acquisitions were controlled by Clampex 7.0 software (Axon Instruments, Foster City, CA). Electrical stimulation of the brain slice surface across a twisted, bipolar, nichrome electrode was used to evoke DA release. Single constant current pulses of 250 μA and 0.1 msec duration were obtained by using an A360R Constant Current Stimulus Isolator (WPI, Sarasota, FL) and a Master-8 pulse generator (A.M.P.I., Jerusalem, Israel). Stimulus intervals between pulses were not less than 5 min. Each site was stimulated three times and the maximum DA release from the three samples was used. The CFE's were inserted 75 to 100 μm into brain slices at a position 100 to 200 μm from the stimulating electrode pair (Miles et al., 2002). Each slice was sampled for DA at 5 sites, which represented medial to lateral and dorsal to ventral dimensions (Akopian et al., 2008; Petzinger et al., 2007). A single DA release value was then calculated as the average from these 5 sites in each brain slice. Changes in extracellular DA were determined by monitoring the current over a 200 mV window at the peak oxidation potential for DA (for review, see Patel and Rice, 2006). Subtracting the current obtained before stimulation from the current obtained in the presence of DA created background-subtracted cyclic voltammograms. Electrodes were calibrated with 5 μM DA solutions in aCSF following each experiment to convert the oxidation current to DA concentration. Previous work has demonstrated that this method causes action potential mediated DA release that is TTX sensitive (Patel et al., 2003; Patel and Rice, 2006; Petzinger et al., 2007).
High performance liquid chromatography (HPLC) analysis of striatal DA and DOPAC
Rats were decapitated, and their striata removed 2 hr, 4 hr, 8 hr, 12 hr, 24 hr, 48 hr, 1 week, 1 month, or 3 months after 3-NP or saline administration. Following the procedure of Crawford et al. (2000), frozen striatal sections were sonicated in 10 volumes of 0.1 N HClO4 and then centrifuged at 20,000g for 20 min at 4°C. The supernatant was then filtered through a 0.22 mm centrifugation unit for 5 min at 2,000g at 4°C. Twenty microliters of the resulting extracts were assayed for DA content using HPLC (ESA, Chelmsford, MA; 582 pump with a MD-150 column) with electrochemical detection (ESA, Coulochem II EC detector). The mobile phase consisted of 75 mM NaH2PO4, 1.4 mM 1-octane sulfonic acid (OSA), 10 mM EDTA, and 10% acetonitrile at a pH of 3.1 (MD-TM Mobile Phase, ESA) and was pumped at a rate of 0.5 ml/min.
DA D1 and D2 homogenate receptor binding assay
Rats were decapitated, and their striata removed 24 hr, 1 week, 1 month, or 3 months after 3-NP or saline administration. On the day of assay, striatal tissue was thawed on ice and crude membrane homogenates were made using the following protocol. Tissue from each rat was homogenized in 100 volumes of 50 mM Tris-HCl buffer (pH 7.4) for approximately 20 s using a Brinkmann Polytron. Homogenates were then centrifuged at 20,000g for 30 min. The pellet was resuspended in 100 volumes of the same buffer and centrifuged again at 20,000g for 30 min. The final pellet was suspended in approximately 30 volumes of buffer (pH 7.4). Protein concentrations for the final pellet were determined using the Bio-Rad Protein Assay with BSA as the standard.
Tissue suspensions (50–100 μg/protein) were added to duplicate tubes containing 50 mM Tris, 2 mM NaCl2, 5 mM KCl, 1 mM MgSO4, and 2 mM CaCl2 (pH 7.4) at a final volume of 1 ml. For the D1 assay, tubes included [3H]-SCH 23390 in concentrations ranging from 0.1 to 5 nM. Non-specific binding was determined in the presence of 10 μM (+)-butaclamol. To prevent binding of [3H]-SCH 23390 to serotonin receptors 100 nM mianserin was added to all tubes. For the D2 assay, tubes included [3H]-spiperone in concentrations ranging from 0.05 to 0.8 nM. Non-specific binding was determined in the presence of 10 μM (−)-sulpiride. Because of the specificity of sulpiride, a serotonin antagonist was not used in the D2 assay. Incubation time for both assays was 30 min at 37°C. Incubation was terminated by vacuum filtration over glass fiber filters (Whatman GF/B, pretreated with 0.1% polyethylenimine). Filters were washed twice with ice-cold Tris-HCl buffer and radioactivity was measured by liquid scintillation spectrometry. DA D1 and D2 binding sites (Bmax) and affinity (Kd) were determined using nonlinear regression using Prism (GraphPad Software).
D1 receptor binding autoradiography
Rats were decapitated 1 week or 1 month after 3-NP or saline treatment and their brains were removed and frozen in liquid isopentane at -30°C. Eighteen μm sections were cut, thaw mounted on electrostatically coated slides (Superfrost Plus, Fisher Scientific), air-dried under vacuum, and stored at -80°C until assay. On the day of assay, slide-mounted brain sections were thawed for 5 min and pre-incubated in 50 mM Tris HCl (pH 7.4) for 30 min at room temperature. Slides were incubated in 50 mM Tris buffer containing 2 nM [3H]-SCH 23390 for 30 min at room temperature. Non-specific binding for all assays was determined in the presence of 10 μM (+)-butacamol. After labeling, sections were washed in ice-cold Tris buffer (3 washes for 20 s); sections were then dried under a stream of cold air. Brain sections were apposed to [3H]-sensitive film along with calibrated standards ([3H] microscales, Amersham) for 4 weeks at -20°C. The relative density of receptors in fmol/mg tissue was quantified using a computer-assisted image analyzer (MCID, InterFocus Imaging Ltd, Cambridge, England). Specific binding for D1 receptors was determined by subtracting total binding from non-specific binding.
Tyrosine hydroxylase protein content assays
For the immunohistochemical assay, rats were anesthetized (pentobarbital, 30 mg/kg) and perfused transcardially with 100 ml of ice-cold 0.9% saline followed by 250 ml of 4% paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.2) (called PFA/PBS) 24 hr, 48 hr, 1 week, or 1 month after 3-NP treatment. Brains were removed, post-fixed in PFA/PBS at 4°C for 24 hr, immersed in 20% sucrose/PBS, frozen in isopentane cooled on dry ice, and stored at -80°C until sectioning. Each brain was sectioned using a sliding microtome cryostat (Leica CM-1900) at 25 μm thickness encompassing the entire striatum. Sections were collected free-floating in PBS containing thimerosal and stored at 4°C until used for Nissl or immunohistochemical staining. Tissue sections at the level of the mid-striatum (+0.60 relative to Bregma) were selected for immunohistochemical staining for tyrosine hydroxylase (TH) (polyclonal antibody made in rabbit, Chemicon, Temecula, CA). Coordinate is from Paxinos and Watson (1998). Briefly, sections were rinsed in TBS (50 mM Tris pH 7.2 with 0.9% NaCl), quenched in 10% methanol/3 % H2O2/50 mM Tris pH 7.2, blocked in 4% normal serum, exposed to antibody (concentration of 1:1000) for 48 hr at 4°C, rinsed in TBS, and then exposed to secondary antibody using the ABC Elite Kit (Vectastain, Burlingame, CA). Antibody staining was visualized by development in 0.1% diaminobenzoic acid/3%H2O2. The relative optical density (OD) due to immunostaining within the striatum was determined by densitometry following subtraction of background determined from staining within the adjacent corpus callosum.
For the western blotting assay, rats were killed 1 day, 2 days, 1 week, or 1 month after 3-NP treatment and tissue from the mid-striatum (+1.6 to -0.26 relative to Bregma) was dissected and homogenized in buffer (25 mM Tris pH 7.4, 1 mM EDTA, 100 uM PMSF). Coordinates are from Paxinos and Watson (1998). Debris was removed by brief centrifugation. Protein concentration was determined by the BCA method (Pierce Inc., Rockford, IL) in microtiter dishes and read on a Biotek plate reader. Equal amounts of protein (20 ug) were loaded per well on 10 ×10 cm Tris-glycine minigels and proteins were separated by the method of Laemmli (Laemmli, 1970). Proteins were transferred to nitrocellulose filters by electroblotting in Towbin buffer (50 mA for 12 hr). Filters were then blocked in Licor Blocking Buffer, exposed to primary antibody against TH (AB152, made in rabbit, Millipore, Inc., 1:2500) and alpha-tubulin (05-829, made in mouse, Millipore, Inc., 1:5000) washed, exposed to secondary antibody conjugated to either Alexafluor 680 (anti-mouse) or Alexafluor 800 (anti-rabbit) and scanned on a Licor Odyssey fluorescent imaging workstation. The relative intensity of bands was determined using computer assisted image analysis (Bioquant Inc., Nashville, TN).
Lipid peroxidation assay of brain slices
Lipid peroxidation in cortex and striatum was evaluated by measuring thiobarbituric acid reactive substances (TBARS) according to the thiobarbituric acid (TBA) test described by Garcia et al. (2005) with modifications. Cortex and striatum were separated from corticostriatal slices and homogenized in 500 μl ice-cold deionized water containing 5 mM BHT. From this homogenate, 20 μl was taken to determine the amount of proteins using Lowry's assay (Lowry et al., 1951) and 350 μl were precipitated with 250 μl of 40% TCA and centrifuged at 3,000g for 15 min. Five hundred microliters of TBA reagent (0.67% TBA and 0.05N NaOH) was added to 500 μl of the resulting supernatant. This solution was placed under a stream of nitrogen at 4°C for 1 min and was afterwards heated at 100°C for 15 min. Clear solutions were obtained for direct spectrophotometric measurement at 532 nm. All procedures were performed on ice at 4°C and all solutions were prepared on deionized water previously gassed with 100% N2 to remove oxygen. For quantitation, calibration curves were constructed using 1,1,3,3-tetramethoxypropane (TMP, malondialdehyde-bis-(dimethylacetal)) as a standard, ranging from 0.05 to 1 nmol for absorption measurements. The formation of TBARS was expressed as malondialdehyde equivalents per mg protein.
Statistics
Data from the rod walking experiment were analyzed with separate paired t-tests at the 24 hr and 48 hr time points. Data from the later time points (1 week, 1 month, and 3 months) were analyzed using a 2 × 3 (drug treatment × time point) ANOVA. Fast scan cyclic voltammetry data from the 24 hr, 48 hr, and 1 week time points were analyzed using a one-way ANOVA with a single saline control. Data from the remaining two time points (2 week, 1 month and 3 months) were analyzed using a 2 × 3 (drug × time point) ANOVA. Lipid peroxidation data from the 2 hr, 4 hr, 8 hr, 12 hr, 24 hr, and 48 hr time points were analyzed with a one-way ANOVA using a single saline control. Data from the remaining two time points (2 week, 1 month and 3 months) were analyzed using a 2 × 3 (drug × time point) ANOVA. HPLC data from the 2 hr, 4 hr, 8 hr, 12 hr, 24 hr, and 48 hr time points were analyzed with a one-way ANOVA using a single saline control. Data from the remaining three time points (1 week, 1 month, and 3 months) were analyzed by a 2 × 3 (drug × time point) ANOVA. Separate saline controls were used at each age to control for age-dependent changes in DA content. Bmax and Kd values from the homogenate binding assay were analyzed using separate 2 × 4 (drug treatment × time point) ANOVAs. Specific binding in the autoradiagraphy experiment was analyzed by a 2 × 2 (drug treatment × time point) ANOVA. Data from the tyrosine hydroxylase study were analyzed using one-way ANOVAs. Planned comparisons were made using independent t-tests and post hoc analyses were done using Dunnett or Tukey tests, P<0.05.
Results
Rod-walking performance
When compared to pre-injection performance, rod walking was impaired 24 hr (n = 20) and 48 hr (n = 7) after 3-NP administration [t (19) = 7.26, P<0.001; t (6) = 3.58, P<0.01, respectively] (see Fig. 1). Post-injection rod walking performance was not different from pre-injection performance at the 1 week time point. Statistical analysis of data from the later time points (2 weeks, 1 month, and 3 months) showed that 3-NP treatment impaired the motoric ability of rats (n = 4-8) when compared to saline-treated controls [drug treatment main effect, F (1, 53) = 13.13, P< 0.001] (see Fig. 1).
Fig 1.

Mean (± SEM) behavioral score of male F344 rats on a rod walking test. Rats in the 24 Hr, 48 Hr and 1 week group were tested on the rod walking test before (control) and after being injected with 3-NP (16.5 mg/kg, i.p). The remaining groups were treated with 3-NP (16.5 mg/kg, i.p.) or saline and tested 2 weeks, 1 month, or 3 months post injection. *Significantly different from control or saline-treated rats.
Fast scan cyclic voltammetry
Evoked DA release (n = 8-15) was reduced both 24 hr and 1 week after 3-NP administration [group effect, F (3, 42) = 11.53, P<0.001, Dunnett tests, P<0.05] (see Fig. 2). At 48 hr, the 3-NP group showed a nonsignificant elevation in evoked DA release when compared to a saline-treated control group (P=0.056). When assessed at the later three time points (2 weeks, 1 month, and 3 months) DA release varied according to drug treatment and time point [drug treatment × time point interaction, F (1, 69) = 7.64, P <0.005]. Specifically, DA release was significantly reduced 3 months after 3-NP treatment (to 62% of age-matched controls) [Tukey tests, P<0.05], but the saline- and 3-NP-treated rats did not differ significantly at the 2 week and 1 month time point. When the saline control groups were compared (i.e., 0, 1, and 2 weeks, 1 and 3 months post 3-NP), age-dependent differences in DA release were apparent (See Fig. 2). Specifically, rats tested at 1 month and 3 months had greater evoked DA release than rats tested immediately or 2 weeks after saline injection [time main effect, F (3, 48) = 21.66, P<0.0001, Tukey tests, P<0.05].
Fig 2.

Evoked DA release in male F344 rats. DA release was averaged over all 5 striatal regions and was induced by a single intrastriatal stimulus (200 A, 0.1 msec). Rats were treated with 3-NP (16.5 mg/kg, i.p.) or saline and tested 24 hr, 48 hr, 1 week, 2 weeks, 1 month, or 3 months post injection. The same saline-treated control group (2 month old) was used for rats tested at 24 hr, 48 hr, and 1 week. Rats at the later time points were compared to age-matched saline controls. Rats are the same as those presented in Fig. 1. *Significantly different from saline controls. **Significantly different from rats tested 24 hr or 1 week after saline injection. Insert shows example voltammogram (left) and time course of evoked DA signal (right).
Striatal DA and DOPAC content
3-NP treatment significantly altered striatal DA content in rats (n = 6-8) tested at early and late time points (see Fig. 3). DA levels did not vary at the 2 hr time point; however, DA levels were reduced to about 87% of control values when measured 4 hr after 3-NP administration and remained depressed through the 24 hr time point [group effect, F (6, 45) = 11.22, P<0.001, Dunnett tests, P<0.05]. By 48 hr, DA levels of the 3-NP- and saline-treated controls did not differ. At 1 week, DA levels of 3-NP-treated rats were significantly reduced (66%) relative to control values, but at the 1 month time point DA levels were significantly higher (147%) than saline-treated controls [drug × time interaction, F (2, 34) = 4.06, P<0.05, Tukey tests, P<0.05]. At 3 months, DA levels of 3-NP-treated rats did not differ from saline controls.
Fig 3.
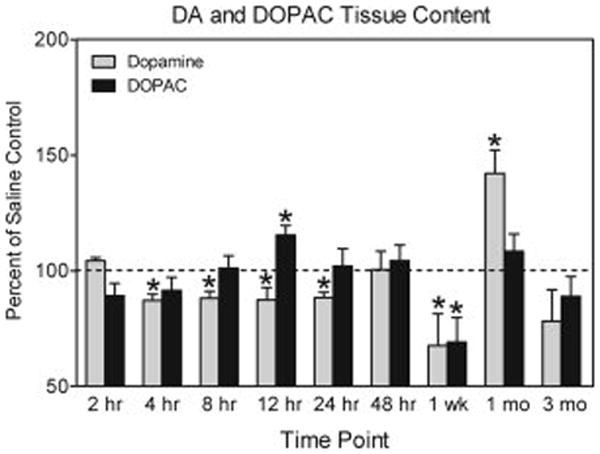
DA and DOPAC content in the striatum of male F344 rats presented as percent of control (± SEM). Rats were treated with 3-NP (16.5 mg/kg, i.p.) or saline and tested 2 hr, 4 hr, 8 hr, 12 hr, 24 hr, 48 hr, 1 week, 1 month, or 3 months post injection. Rats tested at 2 hr, 4 hr, 8 hr, 12 hr, 24 hr, and 48 hr were compared to a single saline-treated control group (DA: x̄ = 7.614 ng/mg ± 0.543; DOPAC: x̄ = 1.067 ng/mg ± 0.187). Rats at the later time points were compared to age-matched saline controls (1 week, DA: x̄ = 7.385 ng/mg ± 0.322; DOPA: x̄ = 1.005 ng/mg ± 0.201; 1 month, DA: x̄ = 8.56 ng/mg ± 1.27; DOPAC: x̄ = 1.107 ng/mg ± 0.244; 3 month, DA: x̄ = 9.13 ng/mg ± 1.184; DOPAC: x̄ = 1.325 ng/mg ± 0.216). Raw data are expressed as ng/mg wet weight tissue. *Significantly different from saline controls (represented by the dashed line).
DOPAC levels were altered by 3-NP in a time dependent manner. When assessed 2-8 hr post-injection, DOPAC levels did not vary according to group. At the 12 hr time point, DOPAC levels of 3-NP-treated rats were significantly elevated (115%) when compared to the saline-treated group [group effect, F (6, 45) = 3.91, P<0.01, Dunnett tests, P<0.05], but DOPAC levels did not differ at the 24 hr time point. DOPAC levels of the 3-NP group were significantly reduced (69%) relative to the control group at the 1 week time point [drug × time interaction, F (2, 34) = 6.76, P<0.001, Dunnett tests, P<0.05], but there were no differences between groups at the 1 and 3 month time points.
Lipid peroxidation assay
No significant changes in MDA levels (n = 4-7) were detected in striatal samples until 24 hr after 3-NP administration, when lipid peroxidation was 777.9% of control values [group effect, F (5, 31) = 6.04 P<0.001, Dunnett tests, P <0.05] (see Fig. 4). No group differences in lipid peroxidation were detected at the 48 hr time point. 3-NP treated rats were still at control levels at the 2 week time point but by 1 month a significant elevation in MDA levels was noted in the 3-NP treated rats (476% of control levels) [time × drug interaction, F (1, 33) = 37.78, P<0.00, Tukey tests, P <0.05]. This increase in lipid peroxidation however disappeared by the 3 month time point [time × drug interaction, Tukey test, P <0.05]. Comparisons of the four saline control groups (i.e., at 0, 2 weeks, 1 month, and 3 months) revealed no age-dependent differences in lipid peroxidation.
Fig 4.

MDA accumulation in male F344 rats presented as percent of control (± SEM). Rats were treated with 3-NP (16.5 mg/kg, i.p.) or saline and tested 2 hr, 4 hr, 24 hr, 48 hr, 2, week, 1 month, or 3 months post injection. The same saline-treated control group (2 month old) was used for rats tested at 2 hr, 4 hr, 24 hr, and 48 hr (x̄ =0.0867 nm/mg protein ± 0.033). Rats at the later time points were compared to age-matched saline controls (2.5 month : x̄ = 0.116 nm/mg protein ± 0.011; 3 month: x̄ = 0.162 nm/mg protein ± 0.047; 5 month; x̄ = 0.116 nm/mg protein ± 0.016). *Significantly different from saline controls represented by dashed line.
D1 and D2 receptor binding
As assessed by homogenate binding assays (n = 6), D1 receptor densities were not altered by 3-NP treatment at 24 hr, 1 week, 2 week, 1 month, and 3 months (see Fig. 6). 3-NP treatment did affect D1 receptors, however, because the affinity of D1 receptors was decreased, as evidenced by a large increase in Kd values at the 1 week and 2 week time points [drug treatment × time point interaction, F (4, 50) = 5.48, P<0.01; Dunnett tests, P<0.05] (see Fig. 5). Because a change in affinity could mask changes in receptor densities, we also measured D1 receptor binding using autoradiography at the 1 week and 1 month time points. Similar to our homogenate binding data, D1 receptor densities were not altered by 3-NP at either time point (data not shown).
Fig 6.

Mean (± SEM) striatal Bmax values from D1 and D2 homogenate binding assays. Rats were treated with 3-NP (16.5 mg/kg, i.p.) or saline and assayed 24 hr, 1 week, 2 weeks, 1 month, or 3 months after injection. *Significantly different from saline controls.
Fig 5.

Mean (± SEM) striatal Kd values from D1 and D2 homogenate binding assays. Rats were treated with 3-NP (16.5 mg/kg, IP) or saline and assayed 24 hr, 1 week, 2 weeks, 1 month, or 3 months after injection. *Significantly different from saline controls.
In contrast to D1 receptors, 3-NP decreased the density of D2 binding sites [drug treatment main effect, F (1, 50) = 4.99, P<0.01] (see Fig. 6 and Fig. 7). This 3-NP-induced reduction in D2 binding sites was significant at only the 1 and 3 month time points [drug treatment × time point interaction, F (4, 50) = 4.59, P<0.01; Dunnett tests, P<0.05]. 3-NP exposure did not affect the affinity of D2 receptors sites.
Fig 7.

Representative saturation isotherms for striatal DA D2 receptors in saline- and 3-NP-injected rats at 1 month post injection.
Analysis of striatal tyrosine hydroxylase protein
Both immunohistochemical staining and western immunoblotting were used to assess changes in the pattern of expression of tyrosine hydroxylase protein in the striatum of rats exposed to 3-NP (Fig. 8). Immunohistochemical staining with an antibody against TH protein and analysis of TH immunoreactive fiber density using computer-assisted image analysis was carried out on coronal sections from the mid-striatum. This analysis showed no significant depletion of TH immunoreactivity in 3-NP-exposed rats at 24 hr, 48 hr, 1 day, 1 week, or 1 month after toxin exposure compared to saline-injected control rats (n=3, each post 3-NP time point and age-matched control). The optical density for TH immunostaining of control rats (27.0 ± 2.1, mean relative optical density units ± SEM) did not differ from 3-NP-treated rats at the 24 hr (27.9 ± 2.1), 48 hr (33.0 ± 5.9), 1 week (30.5 ± 2.2), or 1 month (26.8 ± 2.0) time points. To verify immunohistochemical staining results, western immunoblot analysis was carried out on tissue homogenates from the mid-striatum of saline and 3-NP-treated rats one month after exposure. Similar to findings with immunohistochemical staining there was no significant difference in TH protein expression between saline and 3-NP treated groups (saline 1.00 ± 0.26 compared to 3-NP 1.12 ± 0.16 arbitrary relative OD units, n=5 each group).
Fig 8.
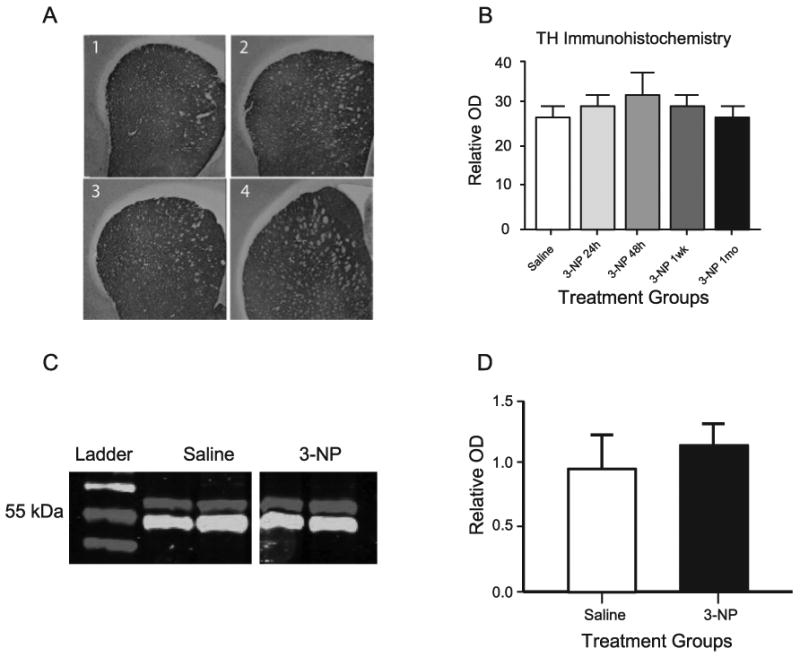
Analysis of striatal TH protein expression. (A) Representative coronal sections at mid-striatum of immunohistochemical staining for TH protein, Panel 1, saline injected; Panel 2, 24 Hr post-3-NP injection; Panel 3, 1 week post-3-NP injection; Panel 4, 1 month post-3-NP injection. (B) Tissue sections were analyzed for relative OD in the mid-striatum as an indicator of TH immunoreactive fiber density in coronal sections from rats injected with saline or 3-NP at 24 hours, 48 hours, 1 week, or 1 month. (C) Representative data from western immunoblotting showing bands for TH (upper band at 62 kDa) and α-tubulin (lower band at 50 kDa) for proteins extracted from age-matched saline treated rats (left 2 lanes) and 1 month post 3-NP treatment (right 2 lanes). (D) Quantification of relative TH expression determined by western immunoblot data derived from 5 rats from each group showing no difference between saline and 3-NP (1 month) groups.
Discussion
The plant mycotoxin, 3-NP, produces dystonia and putaminal necrosis in humans though inhibition of mitochondrial succinate dehydrogenase (for review, see Ludolph et al., 1991). Rodents and primates given multiple high doses of 3-NP experience large lesions in the striatum and a pattern of cell loss mimicking that seen in Huntington's disease and hypoxia (Beal, 1994; Beal et al., 1993b; Borlongan et al., 1995; Hamilton and Gould, 1987; Nishino et al., 2000). Moreover, the cell loss induced by 3-NP is dependent on DA release (Beal et al., 1993b, 1994; Fernagut et al., 2002a; Reynolds et al., 1998). Recently, we demonstrated that a low dose of 3-NP was capable of altering striatal functioning for at least 48 hr in the absence of any cell loss (Akopian et al., 2008). In the current investigation, we examined the long-lasting effects on striatal dopaminergic functioning using the same low dose of 3-NP. In general, we found that a single injection of 3-NP (16.5 mg/kg) led to striatal dysregulation that was apparent 1 to 3 months post drug administration. Specifically, 3-NP reduced performance on the rod walking task and decreased evoked DA release and the density of D2 receptors for 3 months. Interestingly, DA content and lipid peroxidation were both increased 1 month after 3-NP administration.
Motor impairment is a characteristic symptom of striatal cell loss and is found in neurodegenerative disorders, such as Huntington's disease, and after hypoxic/ischemic events (Bhidayasiri and Truong, 2004; Cowper-Smith et al., 2008; Gil and Rego, 2008; Meade et al., 2000). This striatal cell loss and accompanying motor impairment can also be produced through repeated administration of 3-NP (Brouillet et al., 2005; Kumar and Kumar, 2009). Importantly, in the present study the mild hypoxic event induced by a single 3-NP injection produced motoric impairment that persisted for 3 months without the development of striatal lesions. This finding suggests that impairments of striatal-mediated motor behavior are not necessarily the result of cell loss, but may be mediated by disordered striatal functioning. Indeed, the decline in rod walking performance, which occurred 1 month after 3-NP treatment, was temporally related to signs of altered striatal functioning (i.e, elevated DA tissue content and lipid peroxidation). A similar hypothesis (i.e., that cell loss is not necessary for motoric impairment) has been proposed for many of the symptoms of Hungtington's disease, in which extreme neuronal dysfunction precedes the loss of striatal neurons (Levine et al., 2004).
As mentioned above, persistent changes in striatal DA tissue content and DA release were detected in the present investigation; however, these alterations were not stable over the 3 month assessment period. During the initial 24 hr period DA content and release were reduced. Acute 3-NP in vivo, much like in vivo ischemia, triggers a large release of DA and thus the decrease in DA release and DA content we found at 24 hours likely reflects a temporary depletion of DA in nigrostriatal terminals (Kraft et al, 2009; Nishino et al, 1997; Yang et al, 2002) DA content and release recovered to approximate the levels seen in saline injected controls by 48 hours after 3-NP. This finding is in agreement with previous studies showing that DA content recovers within 48 hr of 3-NP administration (Marti et al., 2003; Reynolds et al.,1998). A similar time frame of recovery is observed following stroke-related depletion of striatal DA (Decker et al., 2005). Although the mechanism responsible for this 3-NP-induced reduction in DA release is unclear, one possibility is that 3-NP indirectly activates ATP-sensitive potassium channels by increasing H2O2 production (Bao et al., 2005). Alternatively, 3-NP may reduce the packaging of catecholamines into synaptic vesicles through ATP depletion in a manner silimar to the R6/2 transgenic mouse model for Huntington's disease (Johnson et al., 2007). Recovery to baseline values was not permanent, because both DA content and release were reduced 1 week after 3-NP treatment. After 1 month, the striatal DA content of 3-NP-treated rats was significantly greater than saline controls, while baseline values were reached at 3 months. DA release showed a different pattern of effects, because the striatal DA release of 3-NP treated rats did not differ from saline controls at 1 month, but showed a significant decline at 3 months. Although 3-NP is known to cause a loss of nigrostriatal terminals, even at doses that do not cause cell loss (Blum et al., 2004), it is unlikely that our present results reflect terminal loss because we found no changes in striatal TH immunoreactivity or protein content.
Systemic 3-NP administration increases DA release in the striatum for up to 6 hr (Nishino et al., 1997). Because enhanced extracellular levels of DA are believed to play a pathological role in lipid and protein oxidation (Hastings et al., 1996), it was not surprising that elevated lipid peroxidation activity was detected in the current study. MDA accumulation was significantly increased at the 24 hr time point (when both DA release and DA content were decreased), thus indicating a lack of correspondence between elevated synaptic DA and lipid peroxidation. Interestingly, a similar temporal disparity has been reported between methamphetamine-induced striatal DA depletion and elevated lipid peroxidation (Gluck et al., 2001). While the reason for the temporal disparity in our study is unclear, it is possible that the increase in MDA accumulation was not directly related to DA oxidation, but was the result of toxicity induced by the DA metabolite, DOPAC, which was elevated 12 hr after the 3-NP administration. It is also possible that other 3-NP-induced effects, such as mitochondrial dysfunction, excitotoxicity, or ATP depletion, could be responsible (Alexi et al., 1998; Beal, 1994; Brouillet et al., 2005). The increased MDA accumulation that occurred 1 month after 3-NP administration coincided with an increase in DA content and may be the direct result of DA oxidation. This possibility is supported by studies showing that DA toxicity is augmented in neurons that have been previously weakened by prior lipid peroxidation (Fancellu et al., 2003; Jakel and Maragos, 2000; Rees et al., 2007). Thus, while early MDA accumulation may not be a direct result of DA release, the initial increase in lipid peroxidation at 24 hr may have increased the toxicity of DA oxidation which, in turn, resulted in lipid peroxidation at 1 month.
Striatal DA receptors displayed dynamic alterations over the 3-month recovery period. D2 receptors showed a small, insignificant reduction in Bmax values 2 weeks after the 3-NP injection. At 1 month post 3-NP administration, however, D2 receptor densities were significantly reduced and they remained reduced 3 months after 3-NP exposure. These reductions in D2 receptors are similar to those observed after stroke, because D2 receptor densities are decreased for 1-2 months after the ischemic event (Filloux et al., 1996; Kostic et al., 1991; Przedborski et al., 1991; Zouakia et al., 1997). Exposure to multiple injections of amphetamine also causes an enduring reduction in D2 receptor Bmax values (Chen et al.,2008; Ginovart et al., 2004; Seemen et al., 2002; Sun et al., 2003). In this case, receptor internalization appears to be responsible for the amphetamine-induced decline in D2 receptors (Seemen et al., 2002), and it is possible that the same process may be initiated by 3-NP and other hypoxic/ischemic events. While repeated amphetamine exposure reduces the total number of D2 receptors, there is nearly a 4-fold increase in the number of D2 receptors existing in the high affinity state (Giordano et al., 2006; Seemen et al., 2007). At this point it is unknown whether 3-NP causes a similar alteration in affinity state, but such a change would impact various D2 receptor-mediated effects like long-term synaptic depression at corticostriatal synapses (Centonze et al., 2004).
D1 receptors were also affected by 3-NP treatment, but the response was different than for D2 receptors. Exposure to 3-NP decreased the affinity of D1 receptors (i.e., Kd values increased) from 1 to 2 weeks post injection, which is suggestive of a reduced binding capacity. D1 receptor Bmax values did not increase at any post 3-NP time point. In agreement with our findings, Kostic et al. (1991) reported a decline in striatal D2 receptor Bmax values 1 month after perinatal hypoxia without an accompanying change in D1 receptors.
In summary, the current study indicates that DA nigrostriatal synapses are particularly sensitive to metabolic challenges and that even a brief episode of chemical hypoxia produces significant short- and long-term changes in the functioning of nigrostriatal DA synapses. The short-term synaptic changes, taking place during the first 24 hr after 3-NP treatment, may contribute to the generation of striatal lesions that occur after more intense episodes of hypoxia (Hashimoto et al., 1994; Ren et al., 1997; Reynolds et al., 1998). The long-term alterations in DA functioning may represent latent damage as well as adaptive steps to restore normal function during a prolonged period of recuperation. In any event, these data indicate that a low dose of 3-NP, insufficient to produce cell loss, causes alterations in DA functioning that are capable of inducing motoric dysfunction.
Acknowledgments
This research was supported by a grant from the NIH (AG 21937) and a gift from Ms. Anita Wilson.
Footnotes
No conflict of interest exists for this study.
References
- Akopian G, Crawford C, Beal MF, Cappelletti M, Jakowec MW, Petzinger G, Gheorghe SL, Chow R, Walsh JP. Decreased striatal dopamine release underlies increased expression of long-term synaptic potentiation at corticostriatal synapses 24 hours after 3-nitropropionic acid induced chemical hypoxia. J Neurosci. 2008;28:9585–9597. doi: 10.1523/JNEUROSCI.5698-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexi T, Hughes PE, Faull RL, Williams CE. 3-Nitropropionic acid's lethal triplet: cooperative pathways of neurodegeneration. Neuroreport. 1998;9:R57–R64. doi: 10.1097/00001756-199808030-00001. [DOI] [PubMed] [Google Scholar]
- Bao L, Avshalumov MV, Rice ME. Partial mitochondrial inhibition causes striatal DA release suppression and medium spiny neuron depolarization via H2O2 elevation, not ATP depletion. J Neurosci. 2005;25:10029–10040. doi: 10.1523/JNEUROSCI.2652-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF. Neurochemistry and toxin models in Huntington's disease. Curr Opin Neurol. 1994;7:542–547. doi: 10.1097/00019052-199412000-00012. [DOI] [PubMed] [Google Scholar]
- Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993a;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins B, Henshaw R, Rosen B, Hyman BT. Age-dependent striatal excitotoxic lesions produced by the endogenous mitochondrial inhibitor malonate. J Neurochem. 1993b;61:1147–1150. doi: 10.1111/j.1471-4159.1993.tb03633.x. [DOI] [PubMed] [Google Scholar]
- Bhidayasiri R, Truong DD. Chorea and related disorders. Postgrad Med J. 2004;80:527–534. doi: 10.1136/pgmj.2004.019356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum D, Galas MC, Cuvelier L, Schiffmann SN. Chronic intoxication with 3-nitropropionic acid in rats induces the loss of striatal DA terminals without affecting nigral cell viability. Neurosci Lett. 2004;354:234–238. doi: 10.1016/j.neulet.2003.10.034. [DOI] [PubMed] [Google Scholar]
- Borlongan CV, Koutouzis TK, Randall TS, Freeman TB, Cahill DW, Sanberg PR. Systemic 3-nitropropionic acid: behavioral deficits and striatal damage in adult rats. Brain Res Bull. 1995;36:549–556. doi: 10.1016/0361-9230(94)00242-s. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Jacqard C, Bizat N, Blum D. 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington's Disease. J Neurochem. 2005;95:1521–1540. doi: 10.1111/j.1471-4159.2005.03515.x. [DOI] [PubMed] [Google Scholar]
- Büyükuysal RL, Mete B. Anoxia-induced dopamine release from rat striatal slices: involvement of reverse transport mechanism. J Neurochem. 1999;72:1507–1515. doi: 10.1046/j.1471-4159.1999.721507.x. [DOI] [PubMed] [Google Scholar]
- Centonze D, Usiello A, Costa C, Picconi B, Erbs E, Bernardi G, Borrelli E, Calabresi P. Chronic haloperidol promotes corticostriatal long-term potentiation by targeting dopamine D2L receptors. J Neurosci. 2004;24:8214–8222. doi: 10.1523/JNEUROSCI.1274-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JHJ, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarina L, Davies SW, Penney JB, Bates GP, Young AB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. Proc Nat'l Acad Sci USA. 1998;95:6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Ding Y, Cagniard B, Van Laar AD, Mortimer A, Chi W, Hastings TG, Kang UJ, Zhuang X. Unregulated cytosolic DA causes neurodegeneration associated with oxidative stress in mice. J Neurosci. 2008;28:425–433. doi: 10.1523/JNEUROSCI.3602-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowper-Smith CD, Anger GJ, Magal E, Norman MH, Robertson GS. Delayed administration of a potent cyclin dependent kinase and glycogen synthase kinase 3 beta inhibitor produces long-term neuroprotection in a hypoxia-ischemia model of brain injury. Neuroscience. 2008;155:864–875. doi: 10.1016/j.neuroscience.2008.05.051. [DOI] [PubMed] [Google Scholar]
- Crawford CA, Zavala AR, Karper PE, McDougall SA. Long-term effects of postnatal amphetamine treatment on striatal protein kinase A activity, dopamine D1-like and D2-like binding sites, and dopamine content. Neurotox Teratol. 2000;22:799–804. doi: 10.1016/s0892-0362(00)00109-4. [DOI] [PubMed] [Google Scholar]
- Decker MJ, Jones KA, Solomon IG, Keating GL, Rye DB. Reduced extracellular DA and increased responsiveness to novelty: neurochemical and behavioral sequelae of intermittent hypoxia. Sleep. 2005;28:169–176. doi: 10.1093/sleep/28.2.169. [DOI] [PubMed] [Google Scholar]
- Fancellu R, Armentero MT, Nappi G, Blandini F. Neuroprotective effects mediated by DA receptor agonists against malonate-induced lesion in the rat striatum. Neurol Sci. 2003;24:180–181. doi: 10.1007/s10072-003-0119-x. [DOI] [PubMed] [Google Scholar]
- Fernagut PO, Diguet E, Jaber M, Bioulac B, Tison F. DA transporter knock-out mice are hypersensitive to 3-nitropropionic acid-induced striatal damage. Eur J Neurosci. 2002a;15:2053–2056. doi: 10.1046/j.1460-9568.2002.02047.x. [DOI] [PubMed] [Google Scholar]
- Fernagut PO, Diguet E, Stefanova N, Biran M, Wenning GK, Canioni P, Bioulac B, Tison F. Subacute systemic 3-nitropropionic acid intoxication induces a distinct motor disorder in adult C57Bl/6 mice: behavioural and histopathological characterization. Neuroscience. 2002b;114:1005–1017. doi: 10.1016/s0306-4522(02)00205-1. [DOI] [PubMed] [Google Scholar]
- Filloux FM, Adair J, Narang N. The temporal evolution of striatal DA receptor binding and mRNA expression following hypoxia-ischemia in the neonatal rat. Dev Brain Res. 1996;94:81–91. doi: 10.1016/0165-3806(96)00053-3. [DOI] [PubMed] [Google Scholar]
- Friedmann MN, Gerhardt GA. Regional effects of aging on DA function in the Fisher-344 rat. Neurobiol Aging. 1992;13:325–332. doi: 10.1016/0197-4580(92)90046-z. [DOI] [PubMed] [Google Scholar]
- Garcia O, Massieu L. Glutamate uptake inhibitor L-trans-pyrrolidine 2,4-dicarboxylate becomes neurotoxic in the presence of subthreshold concentrations of mitochondrial toxin 3-nitropropionate: involvement of mitochondrial reducing activity and ATP production. J Neurosci Res. 2003;749:956–966. doi: 10.1002/jnr.10825. [DOI] [PubMed] [Google Scholar]
- Garcia YJ, Rodrıguez-Malaver AJ, Penaloza N. Lipid peroxidation measurement by thiobarbituric acid assay in rat cerebellar slices. J Neurosci Meth. 2005;144:127–135. doi: 10.1016/j.jneumeth.2004.10.018. [DOI] [PubMed] [Google Scholar]
- Gil JM, Rego AC. Mechanisms of neurodegeneration in Huntington's disease. Eur J Neurosci. 2008;27:2803–2820. doi: 10.1111/j.1460-9568.2008.06310.x. [DOI] [PubMed] [Google Scholar]
- Ginovart N, Wilson AA, Houle S, Kapur S. Amphetamine pretreatment induces a change in both D2-receptor density and apparent affinity: a [11C]raclopride positron emission tomography study in cats. Biol Psychiatry. 2004;55:1188–1194. doi: 10.1016/j.biopsych.2004.02.019. [DOI] [PubMed] [Google Scholar]
- Giordano TP, 3rd, Satpute SS, Striessnig J, Kosofsky BE, Rajadhyaksha AM. Up-regulation of DA D2L mRNA levels in the ventral tegmental area and dorsal striatum of amphetamine-sensitized C57BL/6 mice: role of Cav1.3 L-type Ca2+ channels. J Neurochem. 2006;99:1197–1206. doi: 10.1111/j.1471-4159.2006.04186.x. [DOI] [PubMed] [Google Scholar]
- Gluck MR, Moy LY, Jayatilleke E, Hogan KA, Manzino L, Sonsalla PK. Parallel increases in lipid and protein oxidative markers in several mouse brain regions after methamphetamine treatment. J Neurochem. 2001;79:152–160. doi: 10.1046/j.1471-4159.2001.00549.x. [DOI] [PubMed] [Google Scholar]
- Gu W, Zhao H, Yenari MA, Sapolsky RM, Steinberg GK. Catalase over-expression protects striatal neurons from transient focal cerebral ischemia. Neuroreport. 2004;15:413–416. doi: 10.1097/00001756-200403010-00006. [DOI] [PubMed] [Google Scholar]
- Hamilton BF, Gould DH. Nature and distribution of brain lesions in rats intoxicated with 3-nitropropionic acid: a type of hypoxic (energy deficient) brain damage. Acta Neuropathol. 1987;72:286–297. doi: 10.1007/BF00691103. [DOI] [PubMed] [Google Scholar]
- Hashimoto N, Matsumoto T, Mabe H, Hashitani T, Nishino H. DA has inhibitory and accelerating effects on ischemia-induced neuronal cell damage in the rat striatum. Brain Res Bull. 1994;33:281–288. doi: 10.1016/0361-9230(94)90195-3. [DOI] [PubMed] [Google Scholar]
- Hastings TG, Lewis DA, Zigmond MJ. Role of oxidation in the neurotoxic effects of intrastriatal DA injections. Proc Natl Acad Sci USA. 1996;93:1956–1961. doi: 10.1073/pnas.93.5.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakel RJ, Maragos WF. Neuronal cell death in Huntington's disease: a potential role for dopamine. Trends Neurosci. 2000;23:239–245. doi: 10.1016/s0166-2236(00)01568-x. [DOI] [PubMed] [Google Scholar]
- Jenner P, Olanow CW. Understanding cell death in Parkinson's disease. Ann Neurol. 1998;44:S72–S84. doi: 10.1002/ana.410440712. [DOI] [PubMed] [Google Scholar]
- Johnson MA, Villanueva M, Haynes CL, Speipel AT, Buhler LA, Wightman RM. Catecholamine exocytosis is diminished in R6/2 Huntington's disease model mice. J Neurochem. 2007;103:2102–2110. doi: 10.1111/j.1471-4159.2007.04908.x. [DOI] [PubMed] [Google Scholar]
- Johnson JR, Robinson BL, Ali SF, Binienda Z. DA toxicity following long term exposure to low doses of 3-nitropropionic acid (3-NPA) in rats. Toxicol Lett. 2000;116:113–118. doi: 10.1016/s0378-4274(00)00214-9. [DOI] [PubMed] [Google Scholar]
- Kim GW, Chan PH. Involvement of superoxide in excitotoxicity and DNA fragmentation in striatal vulnerability in mice after treatment with the mitochondrial toxin, 3-nitropropionic acid. J Cereb Blood Flow Metab. 2002;22:798–809. doi: 10.1097/00004647-200207000-00005. [DOI] [PubMed] [Google Scholar]
- Klivenyi P, Andreassen OA, Ferrante RJ, Dedeoglu A, Mueller G, Lancelot E, Bogdanov M, Andersen JK, Jiang D, Beal MF. Mice deficient in cellular glutathione peroxidase show increased vulnerability to malonate, 3-nitropropionic acid, and 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. J Neurosci. 2000;20:1–7. doi: 10.1523/JNEUROSCI.20-01-00001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic VS, Przedborski S, Jackson-Lewis V, Cadet JL, Burke RE. Effect of unilateral perinatal hypoxic-ischemic brain injury on striatal DA uptake sites and D1 and D2 receptors in adult rats. Neurosci Lett. 1991;129:197–200. doi: 10.1016/0304-3940(91)90460-b. [DOI] [PubMed] [Google Scholar]
- Koutouzis TK, Borlongan CV, Scorcia T, Creese I, Cahill DW, Freeman TB, Sanberg PR. Systemic 3-nitropropionic acid: long-term effects on locomotor behavior. Brain Res. 1994;646:242–246. doi: 10.1016/0006-8993(94)90085-x. [DOI] [PubMed] [Google Scholar]
- Kraft JC, Osterhaus GL, Ortiz AN, Garris PA, Johnson MA. In vivo dopamine release and uptake impairments in rats treated with 3-nitropropionic acid. Neurosci. 2009;161:940–949. doi: 10.1016/j.neuroscience.2009.03.083. [DOI] [PubMed] [Google Scholar]
- Kumar P, Kumar A. Possible role of sertraline against 3-nitropropionic acid induced behavioral, oxidative stress and mitochondrial dysfunctions in rat brain. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:100–108. doi: 10.1016/j.pnpbp.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Lancelot E, Callebert J, Revaud ML, Boulu RG, Plotkine M. Detection of hydroxyl radicals in rat striatum during transient focal cerebral ischemia: possible implication in tissue damage. Neurosci Lett. 1995;197:85–88. doi: 10.1016/0304-3940(95)11901-8. [DOI] [PubMed] [Google Scholar]
- Levine MS, Cepeda C, Hickey MA, Fleming SM, Chesselet MF. Genetic mouse models of Huntington's and Parkinson's diseases: illuminating but imperfect. Trends Neurosci. 2004;27:691–697. doi: 10.1016/j.tins.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosenbrough NG, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;93:265–275. [PubMed] [Google Scholar]
- Ludolph AC, He F, Spencer PS, Hammerstad J, Sabri M. 3-Nitropropionic acid-exogenous animal neurotoxin and possible human striatal toxin. Can J Neurolog Sci. 1991;18:492–498. doi: 10.1017/s0317167100032212. [DOI] [PubMed] [Google Scholar]
- Marti M, Mela F, Ulazzi L, Hanau S, Stocchi S, Paganini F, Beani L, Bianchi C, Morari M. Differential responsiveness of rat striatal nerve endings to the mitochondrial toxin 3-nitropropionic acid: implications for Huntington's disease. Eur J Neurosci. 2003;18:759–767. doi: 10.1046/j.1460-9568.2003.02806.x. [DOI] [PubMed] [Google Scholar]
- Meade CA, Figueredo-Cardenas G, Fusco F, Nowak TS, Jr, Pulsinelli WA, Reiner A. Transient global ischemia in rats yields striatal projection neurons and interneuron loss resembling that in Huntington's Disease. Exp Neurol. 2000;166:307–323. doi: 10.1006/exnr.2000.7530. [DOI] [PubMed] [Google Scholar]
- Miles PR, Mundorf ML, Wightman RM. Release and uptake of catecholamines in the bed nucleus of the stria terminalis measured in the mouse brain slice. Synapse. 2002;44:188–197. doi: 10.1002/syn.10069. [DOI] [PubMed] [Google Scholar]
- National Research Council. Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research. Washington, DC: National Academy Press; 2003. [PubMed] [Google Scholar]
- Nishino H, Hida H, Kumazaki M, Shimano Y, Nakajima K, Shimizu H, Ooiwa T, Baba H. The striatum is the most vulnerable region in the brain to mitochondrial energy compromise: a hypothesis to explain its specific vulnerability. J Neurotrauma. 2000;17:251–260. doi: 10.1089/neu.2000.17.251. [DOI] [PubMed] [Google Scholar]
- Nishino H, Kumazaki M, Fukuda A, Fujimoto I, Shimano Y, Hida H, Sakurai T, Deshpande SB, Shimizu H, Morikawa S, Inubushi T. Acute 3-nitropropionic acid intoxication induces striatal astrocytic cell death and dysfunction of the blood-barrier: involvement of dopamine toxicity. Neurosci Res. 1997;27:343–355. doi: 10.1016/s0168-0102(97)01170-x. [DOI] [PubMed] [Google Scholar]
- Orset C, Parrot S, Sauvinet V, Cottet-Emard JM, Pequignot JM, Denoroy L. NMDA receptors inhibit the mild hypoxia-induced dopamine efflux in the rat striatum. Synapse. 2006;59:458–461. doi: 10.1002/syn.20260. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Ling GY, Gajendiran M, Xu ZC. Asymmetrical changes of excitatory synaptic transmission in DA-denervated striatum after transient forebrain ischemia. Neuroscience. 2002;114:317–326. doi: 10.1016/s0306-4522(02)00309-3. [DOI] [PubMed] [Google Scholar]
- Patel J, Mooslehner KA, Chan PM, Emson PC, Stamford JA. Presynaptic control of striatal DA neurotransmission in adult vesicular monoamine transporter 2 (VMAT2) mutant mice. J Neurochem. 2003;85:898–910. doi: 10.1046/j.1471-4159.2003.01732.x. [DOI] [PubMed] [Google Scholar]
- Patel J, Rice ME. Monitoring DA release in brain slices. In: Grimes CA, Dickey EC, Pishko MV, editors. Encyclopedia of Sensors. Stevenson Ranch, CA: American Scientific Publishers; 2006. pp. 313–334. [Google Scholar]
- Paxinos G, Watson C. The rat brain in sterotaxic coordinates. San Diego, CA: Academic Press; 1998. [Google Scholar]
- Pereyra-Muñoz N, Rugerio-Vargas C, Angoa-Pérez M, Borgonio-Pérez G, Rivas-Arancibia S. Oxidative damage in substantia nigra and striatum of rats chronically exposed to ozone. J Chem Neuroanat. 2006;31:114–123. doi: 10.1016/j.jchemneu.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Petzinger GM, Walsh JP, Akopian G, Hogg E, Abernathy A, Arevalo P, Turnquist P, Fisher BE, Togasaki D, Jakowec ME. Effects of treadmill exercise on dopaminergic transmission in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-(MPTP)-lesioned mouse model of basal ganglia injury. J Neurosci. 2007;27:5291–5300. doi: 10.1523/JNEUROSCI.1069-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przedborski S, Kostic V, Jackson-Lewis V, Cadet JL, Burke RE. Effect of unilateral perinatal hypoxic-ischemic brain injury in the rat on DA D1 and D2 receptors and uptake sites: a quantitative autoradiographic study. J Neurochem. 1991;57:1951–1961. doi: 10.1111/j.1471-4159.1991.tb06409.x. [DOI] [PubMed] [Google Scholar]
- Ren Y, Li X, Xu ZC. Asymmetrical protection of neostriatal neurons against transient forebrain ischemia by unilateral DA depletion. Exp Neurol. 1997;146:250–257. doi: 10.1006/exnr.1997.6525. [DOI] [PubMed] [Google Scholar]
- Rees JN, Florang VR, Anderson DG, Doorn JA. Lipid peroxidation products inhibit DA catabolism yielding aberrant levels of a reactive intermediate. Chem Res Toxicol. 2007;20:1536–1542. doi: 10.1021/tx700248y. [DOI] [PubMed] [Google Scholar]
- Reynolds DS, Carter RJ, Morton AJ. DA modulates the susceptibility of striatal neurons to 3-nitropropionic acid in the rat model of Huntington's disease. J Neurosci. 1998;18:10116–10127. doi: 10.1523/JNEUROSCI.18-23-10116.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte A, Chow RH. A simple method for insulating carbon-fiber microelectrodes using anodic electrophoretic deposition of paint. Anal Chem. 1996;68:3054–3058. doi: 10.1021/ac960210n. [DOI] [PubMed] [Google Scholar]
- Seeman P, McCormick PN, Kapur S. Increased DA D2high receptors in amphetamine-sensitized rats, measured by the agonist [3H](+)PHNO. Synapse. 2007;61:263–267. doi: 10.1002/syn.20367. [DOI] [PubMed] [Google Scholar]
- Seeman P, Tallerico T, Ko F, Tenn C, Kapur S. Amphetamine-sensitized animals show a marked increase in DA D2 high receptors occupied by endogenous DA, even in the absence of acute challenges. Synapse. 2002;46:235–239. doi: 10.1002/syn.10139. [DOI] [PubMed] [Google Scholar]
- Ste-Marie L, Vachon P, Vachon L, Bémeur C, Guertin MC, Montgomery J. Hydroxyl radical production in the cortex and striatum in a rat model of focal cerebral ischemia. Can J Neurol Sci. 2000;27:152–159. [PubMed] [Google Scholar]
- Sun W, Ginovart N, Ko F, Seeman P, Kapur S. In vivo evidence for DA-mediated internalization of D2-receptors after amphetamine: differential findings with [3H]raclopride versus [3H]spiperone. Mol Pharmacol. 2003;63:456–462. doi: 10.1124/mol.63.2.456. [DOI] [PubMed] [Google Scholar]
- Toner CC, Stamford JA. Effects of metabolic alterations on dopamine release in an in vitro model of neostriatal ischaemia. Brain Res Bull. 1999;48:395–399. doi: 10.1016/s0361-9230(99)00016-7. [DOI] [PubMed] [Google Scholar]
- Yang ZJ, Camporesi C, Yang X, Wang J, Bosco G, Lok J, Reza G, Schelper RL, Camporesi EM. Hyperbaric oxygen mitigates focal cerebral injury and reduces striatal dopamine release in a rat model of transient middle cerebral artery occlusion. Eur J Appl Physiol. 2002;87:101–107. doi: 10.1007/s00421-002-0601-9. [DOI] [PubMed] [Google Scholar]
- Zouakia A, Guilloteau D, Zimmer L, Besnard JC, Chalon S. Evolution of DA receptors in the rat after neonatal hypoxia-ischemia: autoradiographic studies. Life Sci. 1997;60:151–162. doi: 10.1016/s0024-3205(96)00605-4. [DOI] [PubMed] [Google Scholar]