

Abstract
1,2,3,4-Diepoxybutane (DEB) is a carcinogenic metabolite of 1,3-butadiene (BD), an important industrial and environmental chemical present in urban air and in cigarette smoke. DEB is considered the ultimate carcinogenic species of BD due to its potent genotoxicity and mutagenicity attributed to its ability to form DNA-DNA cross-links and exocyclic nucleoside adducts. Mutagenesis studies suggest that DEB adducts formed at adenine bases may be critically important, as it induces large numbers of A → T transversions. We have recently identified three types of exocyclic DEB-dA lesions: N6,N6-(2,3-dihydroxybutan-1,4-diyl)-2′-deoxyadenosine (N6,N6-DHB-dA), 1,N6-(2-hydroxy-3-hydroxymethylpropan-1,3-diyl)-2′-deoxyadenosine (1,N6-γ-HMHP-dA), and 1,N6-(1-hydroxymethyl-2-hydroxypropan-1,3-diyl)-2′-deoxyadenosine (1,N6-α-HMHP-dA) (Seneviratne et al Chem. Res. Toxicol. 2010, 23, 118-133). In the present work, a post-synthetic methodology for preparing DNA oligomers containing stereo- and site-specific N6,N6-DHB-dA and 1,N6-γ-HMHP-dA adducts was developed. DNA oligomers containing site specific 6-chloropurine were coupled with optically pure 1-amino-2-hydroxy-3,4-epoxybutanes to generate oligomers containing N6-(2-hydroxy-3,4-epoxybut-1-yl)adenine adducts, followed by their spontaneous cyclization to 1,N6-γ-HMHP-dA lesions. N6,N6-DHB-dA containing strands were prepared analogously by coupling 6-chloropurine containing DNA with 3S,4S or 3R,4R pyrrolidine-3,4-diols. Oligodeoxynucleotide structures were confirmed by ESI- MS, exonuclease ladder sequencing, and HPLC-MS/MS of enzymatic digests. UV melting and CD spectroscopy studies of DNA duplexes containing N6,N6-DHB-dA and 1,N6-γ-HMHP-dA revealed that both lesions lower the thermodynamic stability of DNA. Interestingly, structurally modified DNA duplexes were more thermodynamically stable when adenine residue was placed opposite 1,N6-γ-HMHP-dA instead of thymine, suggesting that these adducts may preferentially pair with dA.
Introduction
1,2,3,4-Diepoxybutane (DEB) is a genotoxic intermediate produced upon the metabolic activation of 1,3-butadiene (BD), a common industrial and environmental chemical classified as a human carcinogen (1,2). Available experimental evidence suggests that DEB may be responsible for many of the adverse biological effects of BD. DEB is 50-100 fold more genotoxic and mutagenic than structurally analogous monoepoxide metabolites of BD, e.g. 3,4-epoxy-1-butene (EB) and 3,4-epoxy-1,2-butanediol (EBD), probably due to its ability to modify two nucleophilic sites within cellular biomolecules (3,4). Efficient metabolic conversion of BD to DEB in tissues of laboratory mice has been proposed to cause the increased susceptibility of this species to tumor induction by BD (5,6).
Initial DNA alkylation by DEB produces 2-hydroxy-3,4-epoxybut-1-yl (HEB) lesions which contain an inherently reactive oxirane group. The latter can hydrolyze to the corresponding 2,3,4-trihydroxybutyl monoadducts, alkylate neighboring nucleobases within DNA duplex to form DNA-DNA lesions, or interact with nucleophilic side chains within DNA binding proteins to form DNA-protein cross-links (7-15). Alternatively, DEB can form potentially promutagenic exocyclic lesions by alkylating two sites of the same DNA base, e.g. 7,8-dihydroxy-3-(2-deoxy-β-D-erythro-pentofuranosyl)-3,5,6,7,8,9-hexahydro-1,3-diazepino[1,2-a]purin-11(11H)one, N6,N6-(2,3-dihydroxybutan-1,4-diyl)-2′-deoxyadenosine, 1,N6-(1-hydroxymethyl-2-hydroxypropan-1,3-diyl)-2′-deoxyadenosine, and 1,N6-(2-hydroxy-3-hydroxymethylpropan-1,3-diyl)-2′-deoxyadenosine (16-19).
In our previous studies, DNA strands containing site specific N6-(2-hydroxy-3,4-epoxybut-1-yl)-2′-deoxyadenosine adducts (N6-HEB-dA, 1 in Chart 1) were prepared (20). We found that under physiological conditions, N6-HEB-dA lesions spontaneously converted to exocyclic DEB-dA adducts 1,N6-(2-hydroxy-3-hydroxymethylpropan-1,3-diyl)-2′-deoxyadenosine (1,N6-γ-HMHP-dA, lesion 3), and 1,N6-(1-hydroxymethyl-2-hydroxypropan-1,3-diyl)-2′-deoxyadenosine (1,N6-α-HMHP-dA, lesion 4) (Chart 1). Another exocyclic adduct, N6,N6-(2,3-dihydroxybutan-1,4-diyl)-2′-deoxyadenosine (N6,N6-DHB-dA, lesion 2), was produced under anhydrous conditions in the presence of organic base (19). Adducts 3 and 4 interconvert under physiological conditions by a reversible reaction favoring 4 as a more thermodynamically stable lesion (Scheme 1) (19). Compounds 3 and 4 are expected to be strongly mispairing by analogy with structurally analogous etheno and propano adducts induced by vinyl chloride and α,β-unsaturated aldehydes (21-23). The availability of site specifically modified DNA oligomers containing structurally defined DEB derived lesions is critical for experiments examining the effects of these adducts on DNA structure and polymerase fidelity. It was therefore the goal of the present work to prepare DNA oligomers containing stereo- and site-specific exocyclic DEB-dA adducts 2 and 3 and to examine their effects on DNA structure and stability.
Chart 1.

Scheme 1.
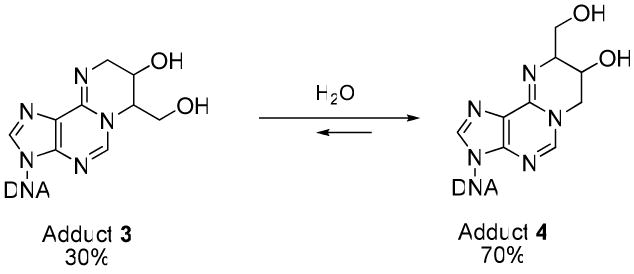
Interconversion of adducts 3 and 4 under physiological conditions.
Materials and Methods
5′-O-(4,4′-Dimethoxytrityl)-3′-O-(2-cyanoethyl)-N,N-diisopropylphosphoramidite of 6-chloropurine-2′-deoxyriboside was purchased from ChemGenes Corporation (Wilmington, MA). Micro Bio-Spin 6 chromatograpy columns were obtained from Bio-Rad Laboratories (Hercules, CA). Water was purified with a Milli-Q Ultrapure water filtration system from Millipore (Bedford, MA). Coaster Spin X centrifuge tube filters (0.45 μm pore cellulose acetate membrane) were from Corning Life Sciences. p-iPr-PAC-dG-CE, PAC-dA-CE, Ac-dC-CE, dT phosphoramidites, iPr-PAC-dG-CPG ABI, Ac-dC-CPG ABI columns, and the THF/Py/Pac2O capping mix A were purchased from Glen research (Sterling, VA). Bovine spleen exonuclease (PDE I) and snake venom exonuclease (PDE II) were acquired from Worthington Biochemical corporation (Lakewood, NJ). Illustra NAP-5 and NAP-25 columns were from GE Healthcare Biosciences (Pittsburgh, PA). SPE Carbograph cartridges were obtained from Grace Davison Discovery Science (Deerfield, IL). V-vials were purchased from Wheaton (Millville, NJ). Dowex® 50WX8-200 ion exchange resin was purchased from Sigma-Aldrich Chemical Co. (Milwaukee, WI) and activated by an overnight incubation with 1M ammonium acetate.
Stereoisomers of N-Fmoc-1-amino-2-hydroxy-3,4-epoxybutane, R,R and R,S-N6-(2-hydroxy-3,4-epoxybut-1-yl)-2′-deoxyadenosine (R,R and R,S-N6-HEB-dA, compound 1a and 1b), R,R and S,S-N6,N6-(2,3-dihydroxybutan-1,4-diyl)-2′-deoxyadenosine (R,R and S,S-N6,N6-DHB-dA, compound 2a and 2b), R,S and R,R-1,N6-(2-hydroxy-3-hydroxymethylpropan-1,3-diyl)-2′-deoxyadenosine, (R,S and R,R-1,N6-γ-HMHP-dA, compound 3a and 3b), and S,R and R,R-1,N6-(1-hydroxymethyl-2-hydroxypropan-1,3-diyl)-2′-deoxyadenosine (S,R and R,R-1,N6-α-HMHP-dA, compounds 4a and 4b) were synthesized in our laboratory as described previously (19). All other chemicals and enzymes were purchased from Sigma-Aldrich Chemical Co. (Milwaukee, WI).
Instrumentation
UV melting temperatures were determined using a Varian Cary 100 Bio UV-Vis spectrophotometer (Varian Inc., Palo Alto, CA). CD data were obtained with a JACSO J-815 CD spectropolarimeter (Easton, MD). MALDI-TOF experiments were performed with a Bruker Reflex III MALDI-TOF instrument (Bruker Daltonics).
HPLC-UV Analyses
HPLC separations were carried out with an Agilent Technologies model 1100 HPLC system equipped with a photodiode array UV detector (Wilmington, DE). Unless specified otherwise, UV absorbance was monitored at 260 nm and at 25 °C. Columns and solvent elution systems were as follows:
System 1
A 250 mm × 4.6 mm Jupiter Proteo 90 Å C12 column (Phenomenex, Torrance, CA) was eluted at a flow rate of 1.0 mL/min with a linear gradient of acetonitrile (B) in 100 mM TEAA, pH 7.0 (A). Solvent composition was changed from 3% to 11% B in 10 minutes, then further to 11.7% B in 21 minutes, and finally to 50% B in 2 minutes.
System 2
A 250 mm × 4.6 mm Jupiter Proteo 90 Å C12 column (Phenomenex, Torrance, CA) was eluted at a flow rate of 1.0 mL/min with a linear gradient of acetonitrile (B) in 100 mM TEAA, pH 7.0 (A). Solvent composition was changed from 3% to 9% B in 10 minutes, then further to 10.3% B in 21 minutes, and finally to 70% B in 2 minutes.
System 3
A 250 mm × 10.0 mm Jupiter Proteo 90 Å C12 column (Phenomenex, Torrance, CA) was eluted at a flow rate of 2.5 mL/min with a linear gradient of acetonitrile (B) in 100 mM TEAA, pH 7.0 (A). Solvent composition was changed from 3% to 9% B in 10 min, then further to 10.3% B in 21 min, and finally to 70% B in 2 min.
System 4
A 250 mm × 10.0 mm Jupiter Proteo 90 Å C12 column (Phenomenex, Torrance, CA) was eluted at a flow rate of 2.5 mL/min with a linear gradient of acetonitrile (B) in 100 mM TEAA, pH 7.0 (A). Solvent composition was changed from 3% to 9% B in 10 min, then further to 11.7% B in 15 min, 12.6 % B in 7 min, 13.0 % B in 1 min, and finally to 60% B in 3 min.
System 5
A 150 mm × 2.1 mm Zorbax Oligo column, 5 μm particle size (Agilent Technologies, Inc.; Wilmington, DE) was eluted at a flow rate of 0.3 mL/min with a linear gradient of 20 mM NaH2PO4 buffer, pH 7.0 containing 20% of acetonitrile (A) and 20 mM NaH2PO4 buffer, pH 7.0, containing 1M sodium chloride and 20% of acetonitrile (B). Solvent composition was changed from 5 to 20% B in 30 min and to 60% B in 28 min.
System 6
A 250 mm × 4.6 mm Supelcosil LC-C18-DB column was eluted at a flow rate of 1.0 mL/min with a linear gradient of acetonitrile (B) in water (A). Solvent composition was kept at 1% B for 8 minutes and then changed linearly from 1% B to 60% B over 2 min and kept at 60% B for 8 min.
Oligodeoxynucleotide Synthesis
Synthetic DNA oligonucleotides containing site-specific 6-chloropurine residue (Oligomers 5, 12, and 17 in Table 1) were synthesized using an ABI Applied Biosystems model 394 DNA synthesizer. Oligonucleotide synthesis was conducted on a 1 μmol scale using PAC protected dG and dA phosphoramidites (ABI) and THF/Py/Pac2O (Glen Research) as the capping mix A. 6-Chloropurine containing oligomers were cleaved from the solid support using 0.1 M NaOH at room temperature (72 h in the dark), followed by desalting with NAP-25 size exclusion cartridges. Synthetic oligomers were purified using HPLC systems 1 and 4 described above and desalted by passing through NAP-25 cartridges and HPLC system 6. Desalted DNA was stored at − 70 °C prior to the coupling reactions to generate adducts 2 and 3.
Table 1.
Mass spectrometric characterization of synthetic oligodeoxynucleotides prepared in this study.
| Oligonucleotide | Molecular weight | ||
|---|---|---|---|
| Calculated | Observed | ||
| 5 | 5′-CGG ACX AGA AG-3′ X = 6-Cl-Purine | 3418.2 | 3418.4 |
| 6 | 5′-CGG ACX AGA AG-3′ X = 1a | 3485.2 | 3484.8 |
| 7 | 5′-CGG ACX AGA AG-3′ X = 1b | 3485.2 | 3484.9 |
| 8 | 5′-CGG ACX AGA AG-3′ X = 2a | 3485.2 | 3485.0 |
| 9 | 5′-CGG ACX AGA AG-3′ X = 2b | 3485.2 | 3484.8 |
| 10 | 5′-CGG ACX AGA AG-3′ X = 3a | 3485.2 | 3485.1 |
| 11 | 5′-CGG ACX AGA AG-3′ X = 3b | 3485.2 | 3485.0 |
| 12 | 5′-GAA GAC CTX GGC GTC C-3′ X = 6-Cl-Purine | 4910.7 | 4910.3 |
| 13 | 5′-GAA GAC CTX GGC GTC C-3′ X = 1a | 4977.3 | 4977.4 |
| 14 | 5′-GAA GAC CTX GGC GTC C-3′ X = 2a | 4977.3 | 4976.6 |
| 15 | 5′-GAA GAC CTX GGC GTC C-3′ X = 2b | 4977.3 | 4976.7 |
| 16 | 5′-GAA GAC CTX GGC GTC C-3′ X = 3a | 4977.3 | 4977.5 |
| 17 | 5′-TCA TXG AAT CCT TCC CCC-3′ X = 6-Cl-Purine | 5373.4 | 5374.2 |
| 18 | 5′-TCA TXG AAT CCT TCC CCC-3′ X = 1a | 5440.5 | 5440.7 |
| 19 | 5′-TCA TXG AAT CCT TCC CCC-3′ X = 3a | 5440.5 | 5440.8 |
Synthesis of DNA oligomers containing site-specific compounds 1a, 1b, 2a, 2b, 3a, and 3b (Table 1)
Method 1 (oligomers containing site specific adducts 2a and 2b) (Scheme 2, bottom)
Scheme 2.

Synthetic scheme for the preparation of oligodeoxynucleotides containing site and stereospecific adduct R,R-N6,N6-DHB-dA (Compound 2a), and R,S-1,N6-γ-HMHP-dA (compound 3a). The same methodology using different stereosisomers of 1-amino-3,4-epoxybutan-2-ol and 2,3-pyroolidinediol was used for the sythesis of oligomers containing adduct 2b and 3b.
3S,4S or 3R,4R pyrrolidine-3,4-diol (10 mg, 100 μmol) was dissolved in a mixture of DMSO (200 μL) and DIPEA (150 μL), followed by the addition of dry, 6-chloropurine containing DNA oligodeoxynucleotides (50.0 nmol). Following incubation at 37 °C for 72 h, the reaction mixtures were separated using HPLC system 1. Under these conditions, oligomers 8, 9, 14 and 15 (Table 1) eluted at 14.9, 15.0, 15.5, and 15.5 min, respectively. The final yields of the oligomers containing optically pure adducts 2a or 2b were 90 - 95 %. The identity and purity of each oligonucleotide product were confirmed by ESI- -MS (LC-MS system 1), capillary HPLC-ESI+ -MS of enzymatic digests, and exonuclease ladder sequencing as described below.
Method 2 (oligomers containing adducts 3a and 3b) (Scheme 2, top)
Our approach for synthesizing DNA oligomers containing DEB-dA exocycles 3a and 3b (10, 11, 16, and 19 in Table 1) was to generate the corresponding DNA strands containing N6-(2-hydroxy-3,4-epoxybut-1-yl)adenine isomers 1a and 1b (6, 7, 13, and 18 in Table 1), followed by their spontaneous cyclization in an aqueous solution (Scheme 2, top). To generate DNA strands containing 1a and 1b, optically pure R,R or R,S N-Fmoc-1-amino-2-hydroxy-3,4-epoxybutane (1 mg) was dissolved in a mixture of DMSO (100 μL) and DIPEA (20 μL) and added to the corresponding 6-chloropurine containing DNA oligodeoxynucleotides (45.0 nmol). Following incubation at 37 °C for 24 h, aliquots of the reaction mixture (20 μL) were diluted with water (280 μL) and filtered through Spin X centrifuge tube filters. The reaction mixtures were separated by reverse phase HPLC. N6-(2-hydroxy-3,4-epoxybut-1-yl)-adenine containing synthetic oligomers 6, 7 (HPLC system 2) and 13 (HPLC system 3) eluted at 24.7, 24.6, and 26.2 min, respectively. HPLC system 4 was used for isolation of oligomer 18, which eluted at 31.9 min. To induce spontaneous cyclization, HPLC pure oligomers containing monoepoxide adducts 1a or 1b were incubated in water for 3 hours at 25 °C. Under these conditions, adducts 1 quantitatively cyclized to form 1,N6-adenine exocycles 3 (reaction c in Scheme 2) (19), The resulting synthetic oligomers containing adducts 3a and 3b (10, 11, 16, and 19 in Table 1) were isolated using the same HPLC methods (final yield ∼30.0 %). The identity and purity of each oligonucleotide product were confirmed by ESI- -MS (LC-MS system 1), capillary HPLC-ESI+-MS of enzymatic digests, and exonuclease ladder sequencing as described below.
Enzymatic hydrolysis of synthetic oligodeoxynucleotides containing exocyclic DEB-dA lesions
HPLC pure synthetic oligodeoxynucleotides containing site- and stereospecific adducts 2 and 3 (1 nmol) were digested to 2′-deoxynuclosides in the presence of nuclease P1 (5 U) and alkaline phosphatase (40 U) in 25 mM NH4OAc/ 25mM ZnCl2, pH 7.0 (10 μL total volume) at 37 °C for 20 hours. Coformycin (1 μL, 90 ng/μL) was added to inhibit adenosine deaminase, which is present as a contaminant in commercial preparations of alkaline phosphatase. The digests were analyzed by HPLC-ESI+-MS as described below, and structurally modified nucleosides were identified from their UV spectra, MSn fragmentation patterns, and HPLC co-elution with synthetic standards.
Preparation of DNA duplexes containing site- and stereospecific DEB-dA adducts
Synthetic DNA oligomers containing site- and stereospecific compounds 2a, 2b, 3a, or unmodified dA (3.0 nmol) were annealed to the complementary strands containing either T or A opposite the modified base (3.0 nmol), in 50 μL of 0.01 M sodium phosphate buffer containing 0.05 mM NaCl (pH 7.0). The solutions were heated to 90 °C for 5 minutes and allowed to slowly cool down to room temperature without removing the sample from the heating block. The resulting DNA duplexes were diluted with 0.01 M sodium phosphate buffer containing 0.05 mM NaCl (pH 7.0) (260 μL) to bring the final volume to 310 μL.
Circular Dichroism Analyses
DNA duplexes containing compounds 2a, 2b, or 3a (3 nmol) were transferred to a 1 mm UV cell, and CD spectra were recorded with a JASCO J-815 spectropolarimeter at 25 °C.
UV Melting Temperature Determination
Synthetic DNA duplexes containing site specific adducts 2a, 2b, or 3a (3 nmol) were transferred to UV microcuvettes (2 mm light path length) (Varian Inc., Palo Alto, CA). UV melting temperatures were determined with a Varian Cary 100 Bio UV-Vis spectrophotometer (Varian Inc., Palo Alto, CA). The temperature was ramped from 30-90 °C and 90-30 °C at a rate of 0.5 °C/min (data were collected at 0.25 °C intervals). The melting temperature of each DNA duplex was calculated from the maximum of the first derivative curve from the smoothed absorption versus temperature profiles using the Cary WinUV Thermal software. Each Tm value listed in Table 2 is an average of two triplicate experiments.
Table 2.
UV melting temperatures (Tm) of DNA duplexes containing unmodified dA and exocyclic DEB-dA adducts:
- 5′- GAA GAC CTX GGC GTC C -3′
- 3′- CTT CTG GAY CCG CAG G -5′
where X is dA or DEB-dA adduct 2 or 3, and Y is either T or A.
| X | Y | Tm (calculated), °C | ΔTm, (Vs unmodified dA) | |
|---|---|---|---|---|
| Y = dT | ||||
| 20 | dA | dT | 60.6 (60.0) | 0.0 |
| 21 | 2a | dT | 52.1 | -8.5 |
| 22 | 2b | dT | 51.4 | -9.2 |
| 23 | 3a ⇄ 4a | dT | 51.5 | -9.1 |
| Y = dA | ||||
| 24 | dA | dA | 52.5 | 0.0 |
| 25 | 2b | dA | 52.6 | + 0.1 |
| 26 | 3a ⇄ 4a | dA | 55.5 | + 3.0 |
Exonuclease Ladder Sequencing of Site-Specifically Modified DNA
DNA oligodeoxynucleotides containing site specific adducts 2a, 2b, 3a, or 3b (400 pmol, in 8 μL of water) were dissolved in 10 mM Tris-HCl (pH 7.0, 6 μL) and 15 mM MgCl2 (6 μL) and incubated with phosphodiesterase I (1 mU, 12 μL) at 37 °C. Aliquots (6 μL) were taken following digestion for 1, 8, 18, 28, and 38 min, along with a 2 μL aliquot taken prior to digestion, and immediately frozen. The aliquots were combined prior to MALDI-MS analysis. In a separate experiment, site specifically modified oligodeoxynucleotides were dissolved in 10 mM Tris-HCl (pH 7.0, 7.5 μL) and 15 mM MgCl2 (7.5 μL) and subjected to partial digestion with phosphodiesterase II (PDE II, 1 mU, 9 μL). Exonuclease digests containing oligodeoxynucleotide ladders were desalted with Micro Bio spin Bio-gel P-6 polyacrylamide gel size exclusion cartridges (which were pre-rinsed with deionized water) and dried under reduced pressure prior to MALDI-MS analyses.
MALDI-TOF MS analysis
A Bruker Reflex III MALDI-TOF Mass Spectrometer (Bruker Daltonics) was equipped with a 337 nm N2 laser set to a repetition rate of 3 Hz at an energy of 175 microjoule/pulse. The instrument was operated in the linear mode at a laser attenuation of 60 - 70 and a pulse delay time of 200 ns. Positive ions were detected. The mass range was m/z 1000 - 10000. Spectra were averaged over 200 laser shots. The instrument was internally calibrated using synthetic oligodeoxynucleotides of known molecular weight.
MALDI matrix solution was prepared by combining 50 μL of saturated aqueous solution of 3-HPA with 6.67 μL of 133 mM ammonium citrate, 150 μL of methanol, and a few Dowex beads. Small pieces of Parafilm were stretched into a thin film over a 10-position Multi PROBE target (Bruker Daltonics) to create a smooth lining over the disc. MALDI matrix solution (1μL) was applied to the Parafilm surface and allowed to crystallize for ∼ 1 h at room temperature in the dark. The desalted DNA oligonucleotides were dissolved in 3 μL of deionized water and incubated with Dowex ion exchange beads for 2 h at 4°C. For sample loading, 1 μL of the aqueous sample solution was placed on the top of a crystallized matrix spot and allowed to dry.
Liquid chromatography-mass spectrometry
The majority of HPLC-ESI-MS experiments were performed with an Agilent 1100 capillary HPLC-ion trap mass spectrometer (Agilent Technologies, Inc.; Wilmington, DE). The instrument was operated in the ESI+ mode for the analyses of modified nucleosides and in the ESI- mode for analyses of synthetic oligodeoxynucleotides. Target ion abundance value was set to 30,000, and the maximum accumulation time was 300 milliseconds. In a typical experiment, 12 scans were taken per average, and a typical mass range was 200 to 2200 m/z. Nitrogen was used as a nebulizing (15 psi) and a drying gas (5 L/min, 200 °C). Electrospray ionization was achieved at a spray voltage of 3-3.5 kV.
LC-MS System 1
For analyses of DNA oligodeoxynucleotides, a Zorbax SB-C18 column (150 × 0.5 mm, 5 μm, Agilent Technologies, Inc.; Wilmington, DE) was eluted at a flow rate of 15 μL/min with a gradient of 15 mM ammonium acetate (pH 6.6) (A) and 100% acetonitrile (B). The column was maintained at 25 °C. Solvent composition was changed linearly from 5% B to 45% B over 15 min.
LC-MS System 2
For analyses of the enzymatic digests containing adducts 2a and 2b, a Zorbax Extend-C18 column (150 × 0.5 mm, 5 μm, Agilent Technologies, Inc.; Wilmington, DE) was eluted at a flow rate of 15 μL/min with a gradient of 15 mM ammonium acetate (pH 6.6) (A) and 100% acetonitrile (B). The column was maintained at 25 °C. Solvent composition was kept at 0% B for 2.5 minutes and then changed linearly from 0% B to 2% B over 19 min and to 8% B in 11.5 minutes, and finally kept at 8 % B for 10 min.
To analyze enzymatic digests of synthetic oligodeoxynucleotides containing adducts 3a and 3b, the same column was eluted at a flow rate of 15 μL/min with a gradient of 15 mM ammonium acetate (pH 7.5) (A) and 100% acetonitrile (B). After holding at 0% B for 5 minutes, the solvent composition was changed linearly from 0% B to 2% B over 17 minutes and further to 7.7% B in 11 min. Column was eluted with 7.7% B for another 8 min, followed by a linear increase to 50% B in 5 min.
LC-MS System 3
To enable separate detection of regioisomeric nucleosides 3 and 4, a capillary Thermo Hypercarb Hypersil column (150 × 0.5 mm, 5 μm, Thermo Fisher Scientific, Inc. Waltham, MA) was eluted at a flow rate of 14 μL/min with a gradient of 0.05% acetic acid (pH 4.0) (A) and 100% acetonitrile (B). The column was maintained at 25 °C. Solvent composition was changed linearly from 0% B to 20% B over 30 min. Under these conditions, compound 3 eluted at 11.7 min, and compound 4 eluted at 13.2 min.
Results
Site- and stereospecific synthesis of oligonucleotides containing lesions 1, 2, and 3
Three DNA sequences were selected for the present study. DNA 11-mer, 5′-CGGACXAGAAG-3′, was derived from the human N-ras protooncogene, with exocyclic DEB-dA adduct 2 or 3 (X) inserted at the second position of the N-ras codon 61 (24-26). The sequence of DNA 16 mers, 5′-GAAGACCTXGGCGTCC-3′, where X = DEB-dA adduct 2 or 3, was selected to enable their incorporation into ss-M13 viral DNA genomes for the REAP site specific mutagenesis assay (27). The DNA 18 mer 5′-TCA TXG AAT CCT TCC CCC-3′ X = DEB-dA adduct 3, was engineered to be used for in vitro polymerase bypass assay and steady-state kinetics studies (28,29).
Synthetic DNA oligomers containing site- and stereospecific, structurally defined adducts N6,N6-DHB-dA and 1,N6-HMHP-dA (2a, 2b, 3a, and 3b in Chart 1) were prepared by a postoligomerization approach starting with the corresponding 6-chloropurine containing DNA oligomers (Scheme 2). DNA oligomers containing 6-chloropurine at specific positions were generated by solid phase synthesis using commercial 6-chloropurine-2′-deoxyribose phosphoramidite (20). Phenoxyacetyl (PAC) protected phosphoramidites (p-iPr-PAC-dG-CE and PAC-dA-CE) were used for the assembly of the modified oligomer. 6-Chloropurine containing oligomers were cleaved from the solid support in the presence of 0.1 M NaOH at room temperature for 72 h in the dark and immediately neutralized using 0.1 M acetic acid to avoid hydrolytic decomposition, following desalting by size exclusion on NAP 25 cartridges and HPLC. The desalted oligonucleotides were kept frozen at -20 °C to prevent hydrolysis.
DNA strands containing site specific and optically pure 1,N6-HMHP-dA adducts 3a and 3b were prepared by a two step process (Scheme 2, top). First, 6-chloropurine containing oligomers were incubated with an excess of S,S or R,R 1-amino-3,4-epoxybutan-2-ol in the presence of DIPEA to obtain the corresponding strands containing site and stereospecific N6-(2-hydroxy-3,4-epoxybut-1-yl)-2′-deoxyadenosine adducts 1. The incubation time was limited to 24 hours in order to minimize side reactions, such as the formation of N6,N6-DHB-dA (19). A two-step desalting procedure (NAP-5 size exclusion columns followed by desalting by HPLC) was required to ensure good coupling yields.
Following coupling, the reaction mixtures were diluted with water, filtered, and purified by reversed phase HPLC using triethylammonium acetate: acetonitrile buffer system (LC systems 2, 3 and 4). Under our HPLC conditions, DNA 11-mers containing exocyclic DEB adducts 1, 2 and 3 (6, 7, 8, 9, 10, and 11 in Table 1 and Figure 1) eluted before 6-chloropurine containing DNA 5. The same elution order was maintained for oligonucleotide 18-mers (17, 18 and 19 in Table 1, Figure S-2). In contrast, N6-HEB-dA containing 16-mer 13 had a slightly longer retention time than the starting material (oligomer 12, Table 1), while the corresponding DEB-dA containing oligonucleotides 14, 15 and 16 eluted before 6-chloropurine containing DNA (Figure S-1). This HPLC system could not resolve oligomers containing N6,N6-DHB-dA and 1,N6-γ-HMHP-dA adducts.
Figure 1.
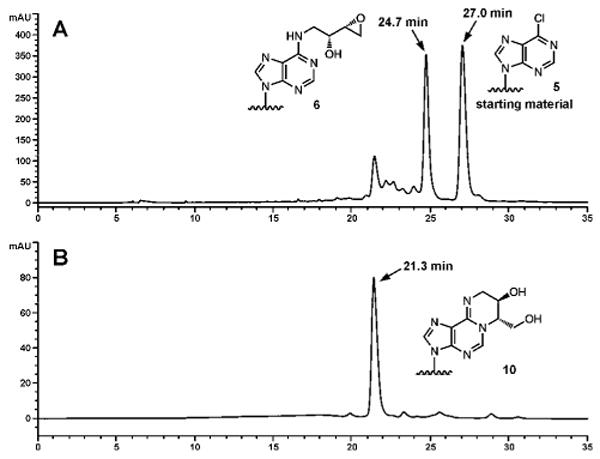
HPLC separation of the reaction mixtures following incubation of synthetic DNA 11-mer, 5′-CGG ACX AGA AG -3′ (X = 6-chloropurine), with R,R N-Fmoc-1-amino-2-hydroxy-3,4-epoxybutane (A) and HPLC purity check of the resulting purified 11 mer containing R,S-1,N6-γ-HMHP-dA (10) at position X (B).
In order to obtain optically pure oligomers containing 1,N6-γ-HMHP-dA adducts, the corresponding N6-HEB-dA oligomers were isolated by HPLC and allowed to cyclize under aqueous conditions (Scheme 2). HPLC pure oligomers 6, 7, 13 and 18 (Table 1) were incubated in water for 3 h at room temperature to convert N6–HEB-dA to 1,N6-γ-HMHP-dA (Scheme 2). The overall yield of target oligomers containing adducts 3a and 3b was 30 % (Figures 1, S-1 and S-2). When the incubation time was increased to more than 3 hours, the formation of 1,N6-α-HMHP-dA 4 was also observed via base-catalyzed rearrangement (Scheme 1). However, we found that a 3 hour cyclization reaction produced DNA oligomers containing adduct 3 with no evidence for rearrangement of 3 to 4 as indicated by HPLC-ESI+-MS/MS of enzymatic digests (see below and Figure 4C).
Figure 4.
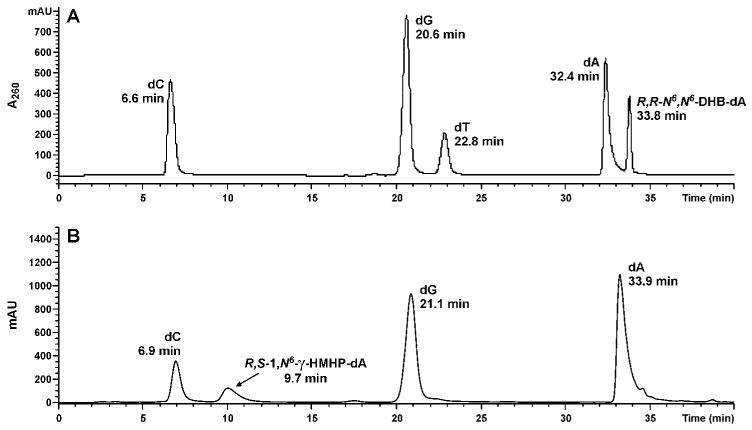
Capillary HPLC-ESI+-MS3 analyses of nucleosides generated from enzymatic digestion of synthetic DNA 16-mer 5′-GAAGACCTXGGCGTCC-3′, where X = R,R-N6,N6-DHB-dA (A) and synthetic DNA 11-mer, 5′-CGG ACX AGA AG -3′, where X = R,S-1,N6-γ-HMHP-dA (compound 3) (B).
N6,N6-DHB-dA containing oligomers were prepared analogously by reacting the corresponding 6-chloropurine containing strands (5 and 12, in Table 1) with enantiomerically pure pyrrolidine-3,4-diols (Scheme 2, top) and purified by HPLC to give oligomers 8, 9, 14 and 15 in an excellent yield (90-95%, see Table 1 and Supplement S-2).
Mass spectrometric characterization of DNA oligomers
Synthetic DNA oligomers containing site specific DEB-dA lesions 2a, 2b, 3a and 3b were initially characterized by electrospray ionization mass spectrometry (LC-MS system 1). The molecular weights observed experimentally were consistent with the calculated values (Table 1, Figures 3 and S-3).
Figure 3.
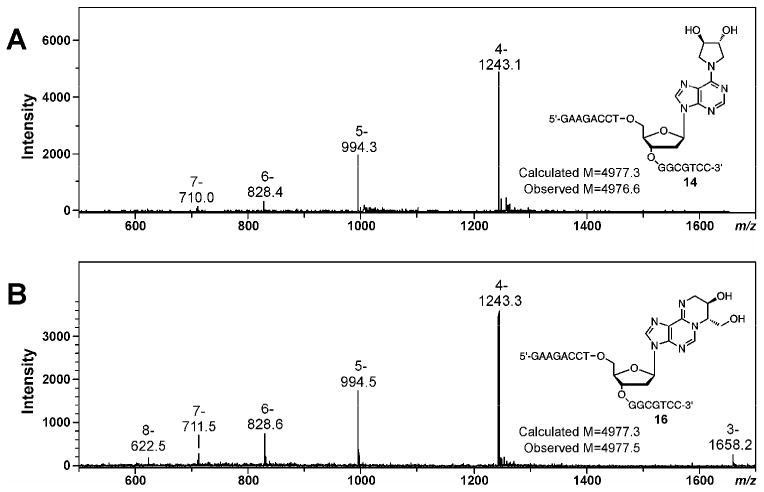
ESI- mass spectra of synthtic DNA 16 mers 5′-GAAGACCTXGGCGTCC-3′, containing exocyclic DEB-dA adducts, X = R,R-N6,N6-DHB-dA (oligonuclotide 14 in Table 1) (A) and X = R,S-1,N6-γ-HMHP-dA (oligonuclotide 16 in Table 1) (B).
To confirm the presence of specific DEB-dA adducts in oligonucleotide structure, they were enzymatically digested to 2′-deoxynuclosides, followed by HPLC-ESI+-MS/MS analyses in parallel with authentic standards of R,R -N6,N6-DHB-dA (2a), S,S - N6,N6-DHB-dA (2b), R,S -1,N6-γ-HMHP-dA (3a), R,R -1,N6-γ-HMHP-dA (3b), S,R-1,N6-α-HMHP-dA (4a) and R,R-1,N6-α-HMHP-dA (4b) and using LC-MS system 2 (19). HPLC-ESI+-MS/MS analyses of the enzymatic digests of N6,N6-DHB-dA-containing oligomers (8, 9, 14 and 15) revealed a peak eluting at 33.8 min (Figure 4A). The HPLC retention time, UV spectrum (λmax = 274 nm), and MS/MS fragmentation patterns of this compound (MS2 m/z 338.3 → m/z 222.2 [M+2H-dR]+, MS3 m/z 222.2 → m/z 148.0 [Ade-CH2+H]+, 186.0 [M+2H-dR-2H2O]+, 204.0 [M+2H-dR-H2O]+) were identical to those of synthetic N6,N6-DHB-dA (compounds 2a and 2b) prepared independently (Figures 4A, 5A, and S-4A). Furthermore, nucleoside molar ratios obtained from HPLC-UV analysis were consistent with DNA sequence (not shown).
Figure 5.
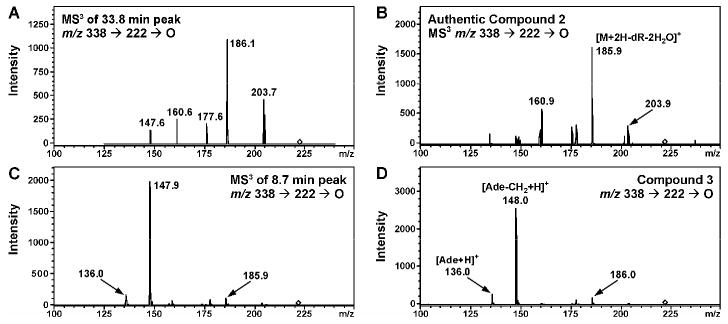
MS3 spectra (m/z 338 → 222 →) of nucleosides 2a and 3a detected in enzymatic digests of oligonucleotides 14 (A) and 10 (C) and corresponding synthetic standards (B and D).
HPLC-ESI+-MS/MS analyses of enzymatic digests of synthetic DNA oligomers containing 1,N6-γ-HMHP-dA adducts (3a) revealed a peak with a retention time matching that of synthetic 1,N6-γ-HMHP-dA (8.9-10.0 min, depending on HPLC conditions, Figure 4B and S-4C). To confirm that the HMHP-dA adduct present in synthetic oligomer was 1,N6-γ-HMHP-dA (3a) and not its Dimroth rearrangement product 1,N6-α-HMHP-dA (4a) (Scheme 1), enzymatic digests were subjected to solid phase extraction and reanalyzed using a Hypercarb Hypersil column capable of resolving regioisomeric adducts 3 and 4 (LC-MS system 3) (19). HPLC retention time of the main species present in enzymatic digests (11.7 min) matched that of authentic 3a, confirming that synthetic oligomers contained 1,N6-γ-HMHP-dA isomer (not shown). The HPLC retention time, UV spectrum (λmax = 264 nm), and MS/MS fragmentation of this compound (ESI+ MS m/z 338.3 [M+H]+, MS2 m/z 338.3 → m/z 222.2 [M+2H-dR]+, MS3 m/z 222.2 → m/z 136.0 [Ade+H]+, 148.0 [Ade-CH2+H]+, 186.0 [M+2H-dR-2H2O]+) were identical to those of synthetic 1,N6-γ-HMHP-dA standard (compound 3a) prepared independently (Figure 4C, 5C, S-4B and S-4C) (19). A small amount of 4a (< 10%) was also observed, probably due to spontaneous rearrangement of 3a to 4a during enzymatic digestion to deoxynucleosides (Scheme 1).
In order to confirm the exact location of site-specific DEB-dA adducts within DNA sequence, structurally modified oligodeoxynucleotides were subjected to controlled exonuclease digestion, followed by MALDI-TOF MS analysis of the resulting DNA ladders (30,31). Figure 6 demonstrates exonuclease ladder sequencing results for DNA 11 mer oligomer 8 containing adduct 2a at the 6th position within sequence (Table 1). MALDI-TOF analyses of time-controlled phosphodiesterase I digest revealed eight peaks, corresponding to the intact oligomer 8, [m/z = 3485.0 Da] and DNA fragments resulting from sequential removal of the first seven residues from the 3′-end of the sequence (Panel A in Figure 6). MALDI-TOF spectra of time-controlled phosphodiesterase II digest contained eight peaks corresponding to the sequential removal of seven nucleotides from 5′-terminus of the oligomer (Panel B in Figure 6). By considering the mass ladders generated from 3′- and 5′- exonuclease digests, complete sequence of the oligomer 8, CGGACXAGAAG, was established. The mass difference of 399.1 Da observed in both exonuclease digestions corresponds to N6,N6-DHB-dA nucleotide [m/z = 399.0] at the 6th position within the synthetic oligonucleotide (X). Similarly, MALDI-TOF MS of exonuclease ladders was employed to obtain the full sequence of DNA oligomers containing 1,N6-HMHP-dA adduct 3a (Figure 7). Interestingly, we found that adduct 3a blocks exonuclease digestion (Figure 7B,D), requiring longer incubation time to obtain the full set of exonuclease ladders.
Figure 6.
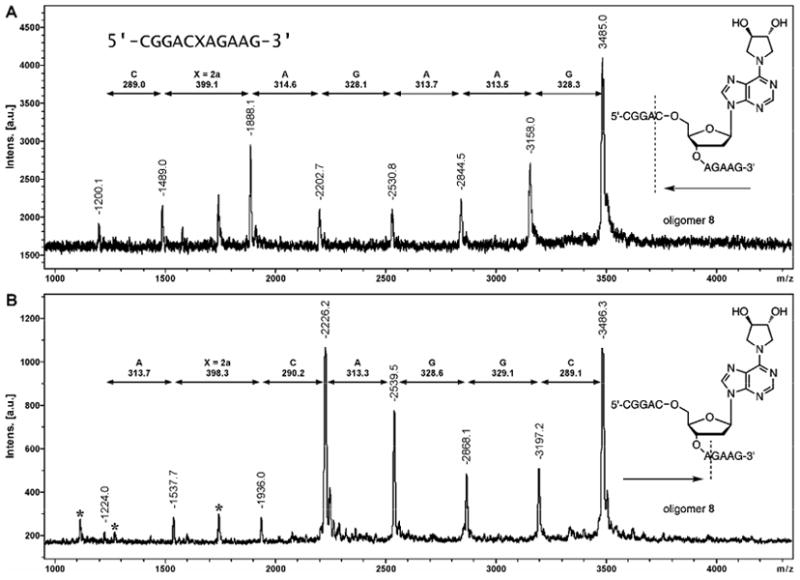
MALDI-TOF mass spectra of controlled exonuclease digests of DNA 11 mer 5′-CGG ACX AGA AG -3′ where X = 2: 3′→5′ digest with PDE I (A); 5′→3′ digest with PDE II (B).
Figure 7.
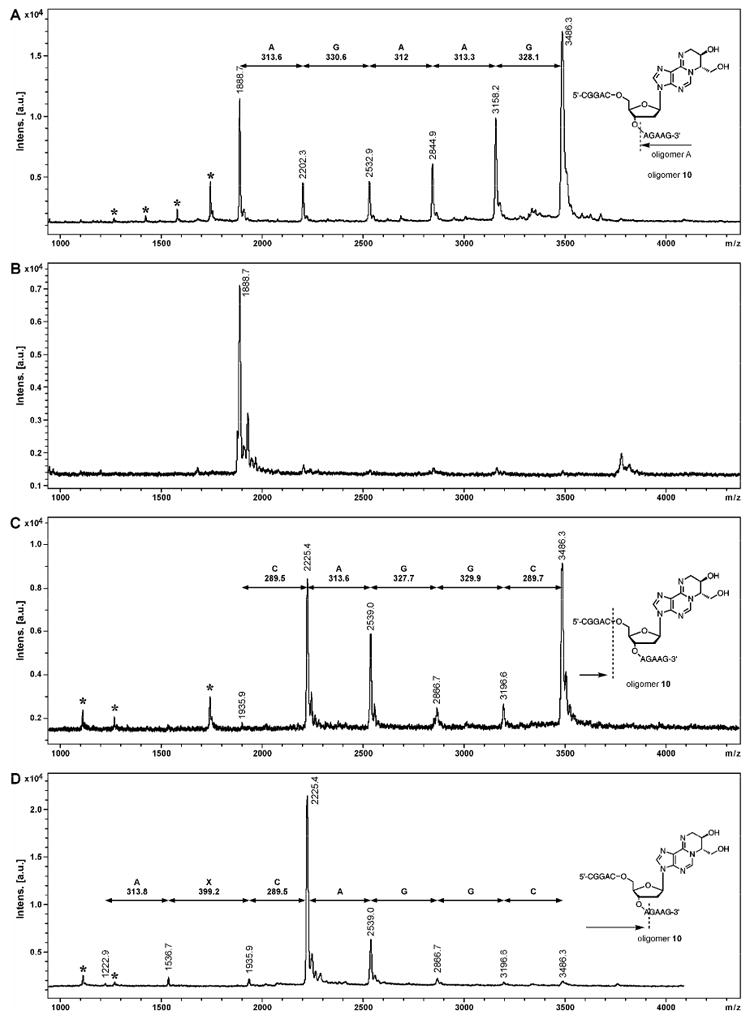
MALDI-TOF mass spectra of controlled exonuclease digests of DNA 11 mer 5′-CGG ACX AGA AG -3′ where X = 3: 3′→5′ digest with PDE 1 (A), digest with PDE 1 for extended time (B); 5′→3′ digest with PDE II (C) and digest with PDE II for extended time (D). The signals corresponding to doubly charged ions are marked with an asterisk (*).
CD Profiles
DNA duplexes containing site specific N6,N6-DHB-dA and 1,N6-HMHP-dA adducts (2 and 3) were obtained by annealing structurally modified 16-mers to the complementary strands containing either adenine or thymine opposite the adducts (duplexes 21, 22, 23, 25, 26 in Table 2 and in Figure S-7), and the effects of exocyclic DEB-dA adducts on the overall duplex structure was studied by CD spectroscopy. No major changes in CD spectra of DNA duplexes was observed when N6,N6-DHB-dA or 1,N6-HMHP-dA were placed in duplex DNA in place of dA (Figure S-7).
Thermal Melting Studies
The thermodynamic stability of structurally modified duplexes was investigated by UV spctroscopy. UV melting temperatures of DNA duplexes 21, 22 and 23 containing exocyclic DEB-dA adducts opposite to thymine residue were 8-9 °C lower than those of the corresponding native DNA 20 (51.4 °C vs 60.6 °C, Table 2 and Figure S-6). These results suggest that the presence of DEB-derived exocyclic dA adducts prevents hydrogen bonding between dT and modified adenine. Interestingly, introduction of adenine opposite the 1,N6-HMHP-dA lesion (duplex 26) resulted in a greater duplex stability (55.5 °C vs 51.5 °C), suggesting that adduct 3 can pair with adenine via an alternative base pairing mode (see below and Scheme 3). In contrast, no increased stability was observed for duplex 25 containing N6,N6-DHB-dA: dA mismatch (Table 2).
Scheme 3.

Proposed Watson-Crick and Hoogsteen base pairing of DEB-dA adduct 3 with thymine and protonated adenine.
Discussion
The ability of DEB to alkylate two sites within DNA by consecutive reactions of the two epoxide rings is critical for its powerful mutagenic and cytotoxic effects (8). DEB analogs possessing a single oxirane functionality, e.g. EB and EBD, are much less potent than DEB (4). Although DEB preferentially targets guanine nucleobases within DNA, the number of AT base pair substitutions equals or exceeds the number of mutations at GC base pairs (32), suggesting that strongly mispairing DEB-dA lesions are formed (8,32-34). However, none of the previously characterized DEB-dA adducts are capable of inducing A to T transversions known to be associated with exposure to DEB (12,26,33).
We hypothesized that DEB produces highly mispairing exocyclic lesions at adenine nucleobases. This hypothesis was supported by our previous studies that identified three types of fused ring DEB-dA lesions: N6,N6-(2,3-dihydroxybutan-1,4-diyl)-2′-deoxyadenosine 2, 1,N6-(2-hydroxy-3-hydroxymethylpropan-1,3-diyl)-2′-deoxyadenosine 3, and 1,N6-(1-hydroxymethyl-2-hydroxypropan-1,3-diyl)-2′-deoxyadenosine 4 (Chart 1) (19). Adducts 3 and 4 are in equilibrium with each other, slowly inter-converting under physiological conditions (Scheme 1). On the nucleoside level, this equilibrium lies towards the more thermodynamically stable isomer 4 (3:7 molar ratio at equilibrium). Among exocyclic DEB-dA adducts, 4 is the most abundant adduct observed in DEB treated DNA, and also in DNA extracted from the liver tissues of laboratory mice exposed to BD by inhalation (19).
Because exocyclic DEB-adenine adducts 2, 3, and 4 lack the structural features required to form Watson-Crick base pair with thymine, they are likely to block DNA polymerases and to cause mispairing during DNA synthesis. The N6-amino group of N6,N6-DHB-dA is doubly alkylated and can no longer serve as H bond donor in a base pair with dT. Watson-Crick base pairing between 1,N6-HMHP-dA and dT is completely blocked by the butanediol bridge (Scheme 3). Structurally similar exocyclic nucleobase adducts 1,N6-ethenoadenine (ε-dA), 3,N4-ethenocytosine, 1,N2 – ethenoguanine, N2,3-ethenoguanine, and 1,N6-ethanoadenine, are known to be miscoding during DNA synthesis (35-38). For example, polymerase bypass of ε-dA leads to A → G and A→T mutations, while G, T or A are incorporated opposite 3,N4-ethenocytoine during DNA synthesis (39-42). Structural studies by X-ray crystallography suggest that ε-dA assumes the syn conformation around the glycosidic bond, forming a Hoogsteen base pairing with protonated dC (20). Similarly, Patel and co-workers reported the NMR evidence of syn conformation of ε-dA when placed opposite dG (23). It is possible that DEB-dA adducts 3 and 4 can similarly assume syn conformation and participate in Hoogsteen base pairing with protonated dA, leading to A to T transversions (Scheme 3).
The main goal of the present work was to generate DNA duplexes containing site- and stereospecific exocyclic DEB-dA adducts for structural and biological studies. Our strategy for preparing DNA oligomers containing adducts 2a, 2b, 3a, and 3b, is based on a post-oligomerization methodology. DNA oligomers containing site specific 6-chloropurine were coupled with optically pure 1-amino-2-hydroxy-3,4-epoxybutanes to generate oligomers containing N6-(2-hydroxy-3,4-epoxybut-1-yl)adenine adducts 1, followed by their spontaneous cyclization to the corresponding 1,N6-γ-HMHP-dA lesions 3 (Scheme 2). N6,N6-DHB-dA containing DNA strands were prepared analogously by coupling 6-chloropurine containing DNA with 3S,4S or 3R,4R pyrrolidine-3,4-diols (Scheme 2). Synthetic oligonucleotide structures were confirmed by ESI- MS, exonucleoside ladder sequencing, and HPLC-MS/MS of enzymatic digests.
The effects of exocyclic DEB-dA adducts on DNA structure and stability were evaluated by CD spectroscopy and UV melting studies. CD spectra of structurally modified duplexes are consistent with B-type DNA duplex structure (Figure S-7). However, the presence of DEB-dA adducts 2 and 3 leads to significant thermodynamic destabilization of the DNA duplexes, as revealed by the 8 - 9° decrease in UV melting temperatures as compared to unmodified DNA (Table 2). It should be noted that a small but significant shift of the UV-melting curve was observed upon heating and cooling of DNA duplex containing adduct 3 opposite adenine (Figure S-6D). This is explained by a partial conversion of adduct 3 to 4 when adduct containing duplex is heated to a high temperature conditions in water in order to obtain the UV melting curve (Scheme 1).
Interestingly, DNA duplexes containing DEB-dA lesions 3 are more stable when adenine is introduced opposite the structurally modified adenine (duplex 26 versus duplex 23 in Table 2, Figure S-6). This can be explained by the formation of a non-standard base pair between adduct 3 and dA. We hypothesize that by analogy with εA, adduct 3 assumes syn conformation about the N-glycosidic bond, and forming a Hoogsteen base pairing with incoming protonated dA (Scheme 3). Such protonation at the N-1of adenine (pKa = 3.6) is thermodynamically favorable if it allows for the formation of a stable base pair with the 1,N6-HMHP-dA. This alternative base pairing could allow for the incorporation of dATP opposite 1,N6-HMHP-dA during DNA replication, ultimately leading to A → T mutations. Our future studies will employ site specifically modified DNA strands prepared in this work for structural investigations by NMR, polymerase bypass studies, and site-specific mutagenesis experiments to establish the effects of the exocyclic DEB-dA lesions on DNA structure and DNA polymerase fidelity.
Supplementary Material
Figure 2.

HPLC separation of synthetic DNA 11-mers, 5′-CGG ACX AGA AG -3′: X = 6-Chloropurine (A), X = R,R-N6,N6-(2,3-dihydroxybutan-1,4-diyl)-2′-deoxyadenosine (R,R -N6,N6-DHB-dA) (B), X = S,S-N6,N6-(2,3-dihydroxybutan-1,4-diyl)-2′-deoxy-adenosine (R,R -N6,N6-DHB-dA) (C).
Acknowledgments
We thank Carry Thompson, Alina Trukhina, and Srikanth Kotapathi for their assistance with HPLC purification of DNA strands, Brock Matter (University of Minnesota Cancer Center) for his help with the HPLC and mass spectrometry experiments, and Bob Carlson (University of Minnesota Cancer Center) for preparing figures for this manuscript. This research was supported by a grant from the NCI (R01 CA095039).
List of abbreviations
- 1
N6-(2-hydroxy-3,4-epoxybut-1-yl)-2′-deoxyadenosine
- 2
N6,N6-(2,3-dihydroxybutan-1,4-diyl)-2′-deoxyadenosine (N6,N6-DHB-dA)
- 3
1,N6-(2-hydroxy-3-hydroxymethylpropan-1,3-diyl)-2′-deoxyadenosine (1,N6-γ-HMHP-dA)
- 4
1,N6-(1-hydroxymethyl-2-hydroxypropan-1,3-diyl)-2′-deoxyadenosine (1,N6-α-HMHP-dA)
- AMP
adenosine monophosphate
- BCNU
1,3-bis(2-chloroethyl)-1-nitroso-urea
- bis-N7G-BD
4-bis-(guan-7-yl)-2,3-butanediol
- BD
1,3-butadiene
- CD
circular dichroism
- DEB
1,2,3,4-diepoxybutane
- DIPEA
N,N-diisopropylethyl amine
- DMSO
dimethylsulphoxide
- dR
2′-deoxyribose
- ds DNA
double stranded oligodeoxynucleotide
- HEB
2-hydroxy-3,4-epoxybut-1-yl
- 3-HPA
3-hydroxypicolinic acid
- HPLC-ESI-MS
high pressure liquid chromatography-electrospray ionization mass spectrometry
- MALDI-TOF MS
Matrix Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry
- N7G-N1A-BD
1-(guan-7-yl)-4-(aden-1-yl)-2,3-butanediol
- PAC-dA-CE
N6-phenoxyacetyl protected 5′-O-(4,4′-Dimethoxytrityl)-3′-O-(2-cyanoethyl)-N,N-diisopropylphosphoramidite of 2′-deoxyadenosine
- p-iPr-PAC-dG-CE
N2-4-isopropyl phenoxyacetyl protected 5′-O-(4,4′-Dimethoxytrityl)-3′-O-(2-cyanoethyl)-N,N-diisopropylphosphoramidite of 2′-deoxyguanosine
- SPE
solid phase extraction
Footnotes
Supporting Information Available: HPLC chromatograms of the 16 mer and 18 mer oligonucleotides reaction mixture separation, capillary HPLC-ESI- -MS of oligonucleotides 8, 9, 10, 15, and 19; HPLC-ESI+-MS/MS analyses of the enzymatic digest of oligonucleotides 16 compared with the nucleoside standards, MALDI-TOF-MS spectra of the oligonucleotide containing 3a (oligomer 10), UV melting curves with their first derivatives of oligomers 20, 23, 24, and 26, and CD spectra of the adducted duplexes. This material is available free of charge via the Internet at http://pubs.acs.org.
Reference List
- 1.Morrow NL. The industrial production and use of 1,3-butadiene. Environ Health Perspect. 1990;86:7–8. doi: 10.1289/ehp.90867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 3.Kligerman AD, DeMarini DM, Doerr CL, Hanley NM, Milholland VS, Tennant AH. Comparison of cytogenetic effects of 3,4-epoxy-1-butene and 1,2:3, 4- diepoxybutane in mouse, rat and human lymphocytes following in vitro G0 exposures. Mutat Res. 1999;439:13–23. doi: 10.1016/s1383-5718(98)00170-3. [DOI] [PubMed] [Google Scholar]
- 4.Cochrane JE, Skopek TR. Mutagenicity of butadiene and its epoxide metabolites: I. Mutagenic potential of 1,2-epoxybutene, 1,2,3,4-diepoxybutane and 3,4-epoxy-1,2- butanediol in cultured human lymphoblasts. Carcinogenesis. 1994;15:713–717. doi: 10.1093/carcin/15.4.713. [DOI] [PubMed] [Google Scholar]
- 5.Henderson RF, Thornton-Manning JR, Bechtold WE, Dahl AR. Metabolism of 1,3-butadiene: species differences. Toxicology. 1996;113:17–22. doi: 10.1016/0300-483x(96)03422-1. [DOI] [PubMed] [Google Scholar]
- 6.Kligerman AD, Hu Y. Some insights into the mode of action of butadiene by examining the genotoxicity of its metabolites. Chem Biol Interact. 2006 doi: 10.1016/j.cbi.2006.03.013. Epub ahead of print 4/18/2006. [DOI] [PubMed] [Google Scholar]
- 7.Millard JT, White MM. Diepoxybutane cross-links DNA at 5′-GNC sequences. Biochemistry. 1993;32:2120–2124. doi: 10.1021/bi00059a034. [DOI] [PubMed] [Google Scholar]
- 8.Lawley PD, Brookes P. Interstrand cross-linking of DNA by difunctional alkylating agents. J Mol Biol. 1967;25:143–160. doi: 10.1016/0022-2836(67)90285-9. [DOI] [PubMed] [Google Scholar]
- 9.Park S, Anderson C, Loeber R, Seetharaman M, Jones R, Tretyakova N. Interstrand and intrastrand DNA-DNA cross-linking by 1,2,3,4-diepoxybutane: role of stereochemistry. J Am Chem Soc. 2005;127:14355–14365. doi: 10.1021/ja051979x. [DOI] [PubMed] [Google Scholar]
- 10.Loeber R, Rajesh M, Fang Q, Pegg AE, Tretyakova N. Cross-linking of the human DNA repair protein O6-alkylguanine DNA alkyltransferase to DNA in the presence of 1,2,3,4-diepoxybutane. Chem Res Toxicol. 2006;19:645–654. doi: 10.1021/tx0600088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carmical JR, Kowalczyk A, Zou Y, Van Houten B, Nechev LV, Harris CM, Harris TM, Lloyd RS. Butadiene-induced intrastrand DNA cross-links: a possible role in deletion mutagenesis. J Biol Chem. 2000;275:19482–19489. doi: 10.1074/jbc.M002037200. [DOI] [PubMed] [Google Scholar]
- 12.Carmical JR, Nechev LV, Harris CM, Harris TM, Lloyd RS. Mutagenic potential of adenine N6 adducts of monoepoxide and diolepoxide derivatives of butadiene. Environ Mol Mutagen. 2000;35:48–56. [PubMed] [Google Scholar]
- 13.Kanuri M, Nechev LV, Tamura PJ, Harris CM, Harris TM, Lloyd RS. Mutagenic spectrum of butadiene-derived N1-deoxyinosine adducts and N6,N6-deoxyadenosine intrastrand cross-links in Mammalian cells. Chem Res Toxicol. 2002;15:1572–1580. doi: 10.1021/tx025591g. [DOI] [PubMed] [Google Scholar]
- 14.Zhang XY, Elfarra AA. Characterization of 1,2,3,4-diepoxybutane-2′-deoxyguanosine cross-linking products formed at physiological and nonphysiological conditions. Chem Res Toxicol. 2006;19:547–555. doi: 10.1021/tx0503395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michaelson-Richie E, Loeber R, Codreanu S, Ming X, Liebler DC, Tretyakova N. DNA-Protein Cross-Linking by 1,2,3,4-Diepoxybutane. J Proteome Res. 2010 doi: 10.1021/pr1000835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang XY, Elfarra AA. Identification and characterization of a series of nucleoside adducts formed by the reaction of 2′-deoxyguanosine and 1,2,3,4-diepoxybutane under physiological conditions. Chem Res Toxicol. 2003;16:1606–1615. doi: 10.1021/tx0341355. [DOI] [PubMed] [Google Scholar]
- 17.Zhang XY, Elfarra AA. Characterization of the reaction products of 2′-deoxyguanosine and 1,2,3,4-diepoxybutane after acid hydrolysis: formation of novel guanine and pyrimidine adducts. Chem Res Toxicol. 2004;17:521–528. doi: 10.1021/tx034243r. [DOI] [PubMed] [Google Scholar]
- 18.Zhang XY, Elfarra AA. Reaction of 1,2,3,4-diepoxybutane with 2′-deoxyguanosine: initial products and their stabilities and decomposition patterns under physiological conditions. Chem Res Toxicol. 2005;18:1316–1323. doi: 10.1021/tx0500979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seneviratne U, Antsypovich S, Goggin M, Dorr DQ, Guza R, Moser A, Thompson C, York DM, Tretyakova N. Exocyclic deoxyadenosine adducts of 1,2,3,4-diepoxybutane: synthesis, structural elucidation, and mechanistic studies. Chem Res Toxicol. 2010;23:118–133. doi: 10.1021/tx900312e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antsypovich S, Quirk Dorr D, Pitts C, Tretyakova N. Site specific N6-(2-hydroxy-3,4-epoxybuty-1-yl)adenine oligodeoxynucleotide adducts of 1,2,3,4-diepoxybutane: synthesis and stability at physiological pH. Chem Res Toxicol. 2007;20:641–649. doi: 10.1021/tx060178k. [DOI] [PubMed] [Google Scholar]
- 21.Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Hoogsteen base pair formation promotes synthesis opposite the 1,N6-ethenodeoxyadenosine lesion by human DNA polymerase iota. Nat Struct Mol Biol. 2006;13:619–625. doi: 10.1038/nsmb1118. [DOI] [PubMed] [Google Scholar]
- 22.VanderVeen LA, Hashim MF, Nechev LV, Harris TM, Harris CM, Marnett LJ. Evaluation of the mutagenic potential of the principal DNA adduct of acrolein. J Biol Chem. 2001;276:9066–9070. doi: 10.1074/jbc.M008900200. [DOI] [PubMed] [Google Scholar]
- 23.de los SC, Kouchakdjian M, Yarema K, Basu A, Essigmann J, Patel DJ. NMR studies of the exocyclic 1,N6-ethenodeoxyadenosine adduct (epsilon dA) opposite deoxyguanosine in a DNA duplex. Epsilon dA(syn).dG(anti) pairing at the lesion site. Biochemistry. 1991;30:1828–1835. doi: 10.1021/bi00221a015. [DOI] [PubMed] [Google Scholar]
- 24.Merritt WK, Scholdberg TA, Nechev LV, Harris TM, Harris CM, Lloyd RS, Stone MP. Stereospecific structural perturbations arising from adenine N(6) butadiene triol adducts in duplex DNA. Chem Res Toxicol. 2004;17:1007–1019. doi: 10.1021/tx049908j. [DOI] [PubMed] [Google Scholar]
- 25.Scholdberg TA, Nechev LV, Merritt WK, Harris TM, Harris CM, Lloyd RS, Stone MP. Structure of a Site Specific Major Groove (2S,3S)-N(6)-(2,3,4-Trihydroxybutyl)-2′-deoxyadenosyl DNA Adduct of Butadiene Diol Epoxide. Chem Res Toxicol. 2004;17:717–730. doi: 10.1021/tx034271+. [DOI] [PubMed] [Google Scholar]
- 26.Scholdberg TA, Nechev LV, Merritt WK, Harris TM, Harris CM, Lloyd RS, Stone MP. Mispairing of a site specific major groove (2S,3S)-N6-(2,3,4-trihydroxybutyl)-2′-deoxyadenosyl DNA Adduct of butadiene diol epoxide with deoxyguanosine: formation of a dA(anti).dG(anti) pairing interaction. Chem Res Toxicol. 2005;18:145–153. doi: 10.1021/tx049772p. [DOI] [PubMed] [Google Scholar]
- 27.Delaney JC, Essigmann JM. Assays for determining lesion bypass efficiency and mutagenicity of site-specific DNA lesions in vivo. Methods Enzymol. 2006;408:1–15. doi: 10.1016/S0076-6879(06)08001-3. [DOI] [PubMed] [Google Scholar]
- 28.Goodenough AK, Kozekov ID, Zang H, Choi JY, Guengerich FP, Harris TM, Rizzo CJ. Site specific synthesis and polymerase bypass of oligonucleotides containing a 6-hydroxy-3,5,6,7-tetrahydro-9H-imidazo[1,2-a]purin-9-one base, an intermediate in the formation of 1,N2-etheno-2′-deoxyguanosine. Chem Res Toxicol. 2005;18:1701–1714. doi: 10.1021/tx050141k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zang H, Goodenough AK, Choi JY, Irimia A, Loukachevitch LV, Kozekov ID, Angel KC, Rizzo CJ, Egli M, Guengerich FP. DNA adduct bypass polymerization by Sulfolobus solfataricus DNA polymerase Dpo4: analysis and crystal structures of multiple base pair substitution and frameshift products with the adduct 1,N2-ethenoguanine. J Biol Chem. 2005;280:29750–29764. doi: 10.1074/jbc.M504756200. [DOI] [PubMed] [Google Scholar]
- 30.Park S, Seetharaman M, Ogdie A, Ferguson D, Tretyakova N. 3′-Exonuclease resistance of DNA oligodeoxynucleotides containing O6-[4-oxo-4-(3-pyridyl)butyl]guanine. Nucleic Acids Res. 2003;31:1984–1994. doi: 10.1093/nar/gkg299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tretyakova N, Matter B, Ogdie A, Wishnok JS, Tannenbaum SR. Locating Nucleobase Lesions within DNA Sequences by MALDI-TOF Mass Spectral Analysis of Exonuclease Ladders. Chem Res Toxicol. 2001;14:1058–1070. doi: 10.1021/tx010062i. [DOI] [PubMed] [Google Scholar]
- 32.Recio L, Steen AM, Pluta LJ, Meyer KG, Saranko CJ. Mutational spectrum of 1,3-butadiene and metabolites 1,2-epoxybutene and 1,2,3,4-diepoxybutane to assess mutagenic mechanisms. Chem Biol Interact. 2001;135-136:325–341. doi: 10.1016/s0009-2797(01)00220-4. [DOI] [PubMed] [Google Scholar]
- 33.Cochrane JE, Skopek TR. Mutagenicity of butadiene and its epoxide metabolites: II. Mutational spectra of butadiene, 1,2-epoxybutene and diepoxybutane at the hprt locus in splenic T cells from exposed B6C3F1 mice. Carcinogenesis. 1994;15:719–723. doi: 10.1093/carcin/15.4.719. [DOI] [PubMed] [Google Scholar]
- 34.Steen AM, Meyer KG, Recio L. Analysis of hprt mutations occurring in human TK6 lymphoblastoid cells following exposure to 1,2,3,4-diepoxybutane. Mutagenesis. 1997;12:61–67. doi: 10.1093/mutage/12.2.61. [DOI] [PubMed] [Google Scholar]
- 35.Chung FL, Chen HJ, Nath RG. Lipid peroxidation as a potential endogenous source for the formation of exocyclic DNA adducts. Carcinogenesis. 1996;17:2105–2111. doi: 10.1093/carcin/17.10.2105. [DOI] [PubMed] [Google Scholar]
- 36.Frick LE, Delaney JC, Wong C, Drennan CL, Essigmann JM. Alleviation of 1,N6-ethanoadenine genotoxicity by the Escherichia coli adaptive response protein AlkB. Proc Natl Acad Sci U S A. 2007;104:755–760. doi: 10.1073/pnas.0607377104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nair J, Barbin A, Guichard Y, Bartsch H. 1,N6-ethenodeoxyadenosine and 3,N4-ethenodeoxycytine in liver DNA from humans and untreated rodents detected by immunoaffinity/32P-postlabeling. Carcinogenesis. 1995;16:613–617. doi: 10.1093/carcin/16.3.613. [DOI] [PubMed] [Google Scholar]
- 38.Pollack M, Oe T, Lee SH, Silva Elipe MV, Arison BH, Blair IA. Characterization of 2′-deoxycytidine adducts derived from 4-oxo-2-nonenal, a novel lipid peroxidation product. Chem Res Toxicol. 2003;16:893–900. doi: 10.1021/tx030009p. [DOI] [PubMed] [Google Scholar]
- 39.Basu AK, Wood ML, Niedernhofer LJ, Ramos LA, Essigmann JM. Mutagenic and genotoxic effects of three vinyl chloride-induced DNA lesions: 1,N6-ethenoadenine, 3,N4-ethenocytosine, and 4-amino-5-(imidazol-2-yl)imidazole. Biochemistry. 1993;32:12793–12801. doi: 10.1021/bi00210a031. [DOI] [PubMed] [Google Scholar]
- 40.Moriya M, Zhang W, Johnson F, Grollman AP. Mutagenic potency of exocyclic DNA adducts: marked differences between Escherichia coli and simian kidney cells. Proc Natl Acad Sci U S A. 1994;91:11899–11903. doi: 10.1073/pnas.91.25.11899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pandya GA, Moriya M. 1,N6-ethenodeoxyadenosine, a DNA adduct highly mutagenic in mammalian cells. Biochemistry. 1996;35:11487–11492. doi: 10.1021/bi960170h. [DOI] [PubMed] [Google Scholar]
- 42.Levine RL, Yang IY, Hossain M, Pandya GA, Grollman AP, Moriya M. Mutagenesis induced by a single 1,N6-ethenodeoxyadenosine adduct in human cells. Cancer Res. 2000;60:4098–4104. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.