

Abstract
Objective
To study the role of genetic variation in the HMG-CoA reductase (HMGCR) gene in PCOS.
Design
Women with and without PCOS were genotyped for seven single nucleotide polymorphisms (SNPs) in HMGCR. SNPs and haplotypes were determined and tested for association with PCOS and its component traits.
Setting
Subjects were recruited from the reproductive endocrinology clinic at the University of Alabama at Birmingham; controls were recruited from the surrounding community. Genotyping took place at Cedars-Sinai Medical Center in Los Angeles.
Patient(s)
A total of 287 white PCOS women and 187 controls were studied.
Intervention(s)
Phenotypic and genotypic assessment.
Main outcome measure(s)
HMGCR genotype, PCOS diagnosis, androgen levels, metabolic traits.
Result(s)
No association with PCOS was observed. SNP rs4629571 was associated with increased HOMA-IR. Haplotype 3 was associated with increased HOMA-IR; Haplotype 5 was associated with higher sex hormone binding globulin (SHBG) and lower free testosterone.
Conclusion
Variation in the HMGCR gene may influence component features of PCOS including insulin resistance, SHBG, and free testosterone. HMGCR may thus act as a modifier gene in PCOS.
Keywords: polycystic ovary syndrome, HMGCR, insulin resistance, SHBG, androgens
Introduction
Polycystic ovary syndrome (PCOS) is the most common endocrinopathy of reproductive-aged women, and is in part determined by inherited factors as a complex trait. PCOS is characterized by hyperandrogenism, ovulatory dysfunction and polycystic ovarian morphology. Patients with PCOS may also present with infertility, obesity, insulin resistance, and may have an increased risk of cardiovascular disease (1–3).
Cholesterol is a precursor of steroid hormones, and mevalonate is a key compound in the cholesterol biosynthetic pathway. The rate-limiting step in this pathway is conversion of HMG-CoA (3-hydroxy-3-methylglutaryl-Coenzyme A) to mevalonate by the enzyme HMG-CoA reductase (HMGCR). HMGCR can be reversibly blocked by inhibitors such as statins. Studies in rodent ovarian theca-interstitial cells have shown that statins may reduce ovarian androgen production (4). Furthermore, a clinical trial showed that the addition of simvastatin to oral contraceptives resulted in additional lowering of circulating testosterone (T) levels in PCOS women (5, 6). In another clinical trial of statins and metformin, statins used alone improved insulin sensitivity and markers of inflammation in PCOS women (7). This was confirmed by recent studies of atorvastatin in PCOS patients, showing significant reductions in insulin resistance, hyperandrogenemia, C-reactive protein, and homocysteine (8, 9).
Given the role of HMGCR in cholesterol synthesis and the in vitro and in vivo data that statins can ameliorate hyperandrogenism and improve insulin sensitivity, we decided to investigate the relationship of genetic variation in the HMGCR gene with PCOS. We hypothesized that variants in the HMGCR gene would be associated with PCOS and insulin- and androgen- related traits in affected women.
Materials and Methods
Patients
A total of 287 white patients with PCOS, and 187 healthy white control women, were recruited from the Birmingham, Alabama area. All subjects were unrelated. PCOS subjects were recruited consecutively from the reproductive endocrine practice of one of the investigators (RA) at the University of Alabama at Birmingham (UAB). Participation in research studies was offered to patients meeting inclusion criteria (premenopausal, non-pregnant, on no hormonal therapy, including oral contraceptives, for at least three months, and meeting diagnostic criteria for PCOS). In order to ensure the inclusion of women with the classic disorder, the presence of PCOS was defined by the 1990 NIH consensus criteria (10), including: (i) clinical hyperandrogenism and/or hyperandrogenemia, (ii) oligo-ovulation, and (iii) the exclusion of related disorders, including androgen-producing tumors, nonclassic 21-hydroxylase-deficient adrenal hyperplasia (NCAH), hyperprolactinemia, active thyroid disease, or Cushing's syndrome. The specific parameters for defining hirsutism, hyperandrogenemia, ovulatory dysfunction, and exclusion of related disorders were previously reported (11).
Controls were healthy women, with regular menstrual cycles and without family history of hirsutism. These women had no evidence of hirsutism, acne, alopecia, or endocrine dysfunction and had not taken hormonal therapy (including oral contraceptives) for at least three months prior to testing. Controls were recruited by word of mouth and advertisements in the Birmingham, Alabama area, through a call for “healthy women” without detailing further the nature of the studies.
All subjects gave written informed consent, and the study was performed according to the guidelines of the Institutional Review Boards of UAB and Cedars-Sinai Medical Center.
Phenotyping
Subjects underwent a brief physical examination, hirsutism scoring using a modification of the Ferriman-Gallwey method (mFG) (12), and underwent blood sampling. Hormonal measures, including total and free T, dehydroepiandrosterone sulfate (DHEAS), 17α-hydroxyprogesterone (17-HP), and SHBG, were obtained between days 3 and 8 (follicular phase) following a spontaneous menstrual cycle or progesterone-induced withdrawal bleed, per a previously described protocol (11). Total T was measured after serum extraction by an in-house RIA method, SHBG activity was measured by competitive binding analysis, using Sephadex G-25 (Sigma-Aldrich Corp., St. Louis, MO) and [3H]T as the ligand, and the free T was calculated as previously described (13, 14). The SHBG method gives values of approximately 100–300 nmol/L in normal adult women. DHEAS and 17-HP were measured by direct RIA using commercially available kits (from Diagnostic Products Corp., Los Angeles, CA). The intra- and interassay variations for the hormonal assays have been previously reported (15). The same laboratory assays were employed for all subjects. For these androgen-related traits measured in the women with PCOS, completeness of data was over 98%. The total and free T values of three cases were statistical outliers; therefore, these values were deleted from analysis.
Fasting glucose and insulin were also obtained in a subset of the cohort (~70%). The computer-based homeostasis model assessment (HOMA, www.dtu.ox.ac.uk/homa) utilizes fasting glucose and insulin to calculate indices of insulin resistance (HOMA-IR) and insulin secretion (HOMA-%B) (16, 17). An ideal, normal-weight person less than 35 yr of age has a HOMA-IR=1 and HOMA-%B=100% (18). For the insulin-related traits only, subjects with diabetes (n=6) were excluded because the hyperglycemia of diabetes may induce secondary changes in insulin-related traits that reduce their utility for genetic analyses. The resulting subset of subjects with fasting glucose and insulin did not differ demographically or hormonally from the study subjects overall.
Genotyping and haplotype determination
We genotyped 7 single nucleotide polymorphisms (SNPs) in HMGCR, rs10038095, rs17244841, rs6453131, rs3846662, rs17228540, rs3846663, and rs4629571, which span the 24.8 kb genomic length of HMGCR. The SNPs were selected from the Pharmacogenetics and Risk of Cardiovascular Disease (PARC) database (http://droog.gs.washington.edu/parc/data/hmgcr/welcome.html). Two of these SNPs (rs17244841 and rs17238540) were reported to reduce cholesterol response to statin therapy (19). The 7 SNPs were genotyped using the 5’-exonuclease assay (TaqMan MGB, Applied Biosystems, Foster City, CA) described previously (20, 21); duplicate genotyping of 96 samples for one SNP yielded 100% concordance. The genotyping success rate was 94.4%.
Haploview (22) was used to determine haplotypes as well as haplotype blocks. Haploview constructs haplotypes using an accelerated expectation maximization algorithm similar to the partition/ligation method (23). Haploview was used to calculate linkage disequilibrium (LD, the D’ statistic) between each pairwise combination of SNPs. Haplotype blocks were determined using the solid spine of LD algorithm in Haploview (22). Haplotypes were assigned to individual subjects only when the assignment could be made with a greater than 95% certainty.
Statistical Analysis
For all analyses, quantitative trait values were log- or square root-transformed as appropriate to reduce non-normality. Unpaired T-tests and chi-square tests were used to compare clinical characteristics between women with and without PCOS. Quantitative data are presented as median (interquartile range).
Association of SNPs or haplotypes with presence/absence of PCOS was evaluated using logistic regression, adjusting for age and body mass index (BMI) by including them as independent variables in all analyses. Association with quantitative phenotypic variables utilized analysis of covariance (ANCOVA), again adjusting for age and BMI.
As a measure to handle multiple testing, significance was taken as P<0.017, considering that we analyzed one linkage disequilibrium group of SNPs against three families of traits (PCOS diagnosis, androgens, metabolic traits), yielding a Bonferroni correction factor of three (i.e. three independent comparisons). Analyses were carried out using Statview 5.0 (SAS Institute, Cary, NC).
Results
Clinical characteristics of the subjects are presented in Table 1. We genotyped 7 SNPs in the HMGCR gene with LD (D’) of 1 between each pair of SNPs (Figure 1 and Table 2 and Supplementary Table 1). The overall high degree of LD confirmed the possibility of constructing haplotypes across the entire gene. Table 3 displays the HMGCR haplotypes and their frequencies. The five haplotypes of frequency >1% were tested for association with PCOS and its component traits.
Table 1.
Clinical characteristics of the study group.
| Control (n=187) | PCOS (n=287) | |
|---|---|---|
| Age (yr) | 33.0 (17.0) | 27.5 (11.5)a |
| BMI (kg/m2) | 24.1 (6.4) | 34.7 (13.5)a |
| WHR | 0.78 (0.08) | 0.83 (0.10)a |
| mFG score | 0 (0) | 7.0 (5.0)a |
| Hirsute (%) | 0 | 73.9a |
| Total testosterone (ng/dl) | 41.0 (26.5) | 80.0 (31.0)a |
| Free testosterone (pg/ml) | 0.35 (0.26) | 0.84 (0.47)a |
| DHEAS (ng/ml) | 950.0 (749.0) | 2084.0 (1697.8)a |
| SHBG (nmol/l)b | 220.0 (120.0) | 150.0 (70.0)a |
| Insulin (µIU/ml) | 6.9 (6.4) | 18.0 (18.0)a |
| Glucose (mg/dl) | 86.0 (10.0) | 86.0 (13.0) |
| HOMA-IR | 0.92 (0.83) | 2.29 (1.93)a |
| HOMA-%B | 103.9 (59.5) | 175.3 (99.3)a |
Data are median (interquartile range).
P < 0.001 compared to control group, by unpaired T-tests or chi-square tests as appropriate; quantitative data were transformed to approximate normality.
WHR, waist to hip ratio; mFG: modified Ferriman–Gnonewey; DHEAS, dehydroepiandrosterone sulfate; SHBG, sex hormone-binding globulin; HOMA-IR, homeostasis model assessment of insulin resistance; HOMA-%B, homeostasis model assessment of beta-cell function (insulin secretion).
Figure 1.
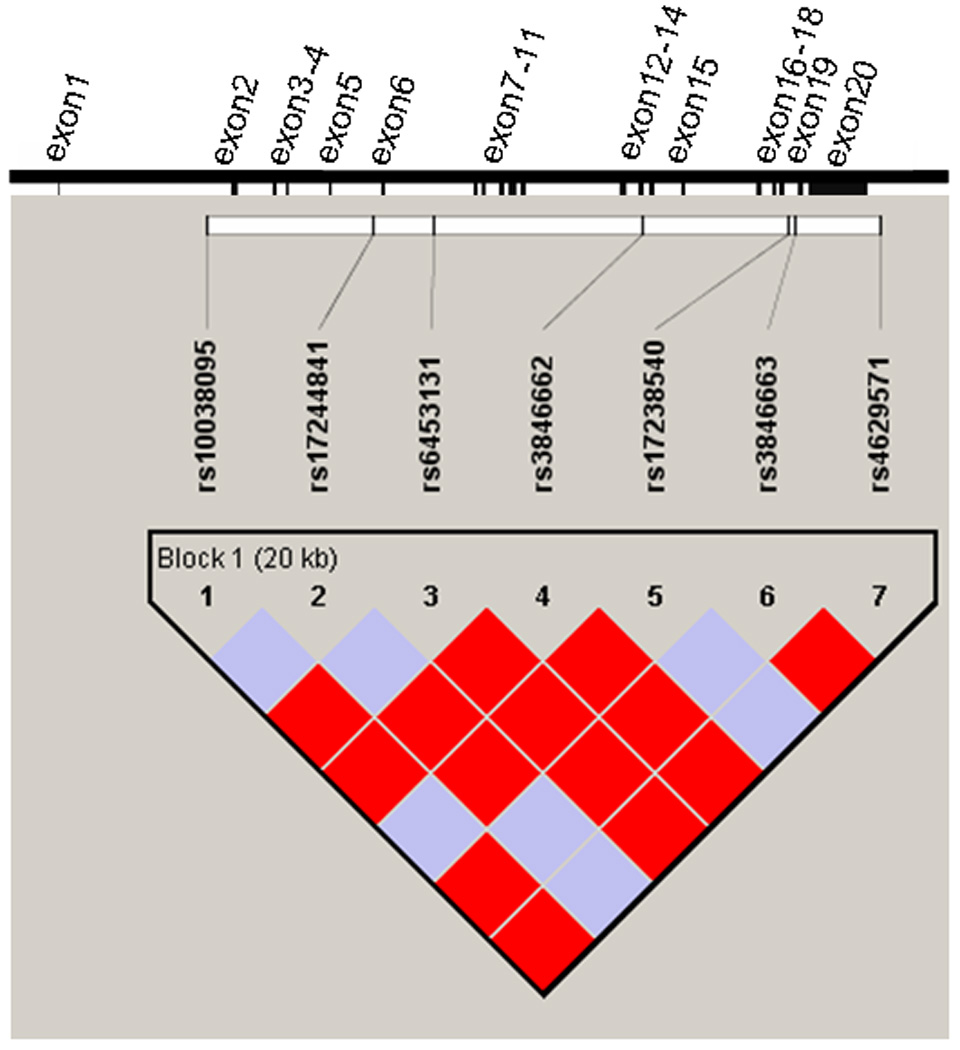
Gene structure and LD plot for the HMGCR gene. The gene structure of HMGCR is shown at top; the gene has 20 exons (represented by vertical bars) and is located on the forward strand of chromosome 5 (5q13.3-q14). The locations of the genotyped 7 SNPs relative to the exons are indicated. D’=1 for all pairs of SNPs. The darker solid blocks indicate a logarithm of the odds (LOD) score ≥2 for the corresponding pair of variants; lighter solid blocks indicate a LOD score <2. The SNPs were considered together in one haplotype block as indicated.
Table 2.
Frequency and location information on HMGCR variants.
| Variant designation |
noneeles (major/minor) |
Location | Overnone MAF |
PCOS MAF |
Control MAF |
|---|---|---|---|---|---|
| rs10038095 | A/T | Intron 1 | 0.345 | 0.343 | 0.349 |
| rs17244841 | A/T | Intron 5 | 0.033 | 0.036 | 0.029 |
| rs6453131 | T/G | Intron 6 | 0.343 | 0.337 | 0.354 |
| rs3846662 | T/C | Intron 13 | 0.412 | 0.420 | 0.401 |
| rs17238540 | T/G | Intron 18 | 0.031 | 0.032 | 0.029 |
| rs3846663 | C/T | Intron 18 | 0.342 | 0.338 | 0.349 |
| rs4629571 | A/G | 3' to gene | 0.075 | 0.078 | 0.071 |
MAF=minor noneele frequency.
Table 3.
HMGCR haplotypes and haplotype frequencies.
| Haplotype | Overnone Freq | PCOS Freq |
PCOS Counta |
Control Freq |
Control Counta |
|
|---|---|---|---|---|---|---|
| 1 | AATTTCA | 0.587 | 0.579 | 321 | 0.600 | 207 |
| 2 | TAGCTTA | 0.268 | 0.261 | 145 | 0.279 | 96 |
| 3 | TAGCTTG | 0.074 | 0.077 | 42 | 0.070 | 24 |
| 4 | AATCTCA | 0.035 | 0.044 | 24 | 0.022 | 8 |
| 5 | ATTCGCA | 0.031 | 0.032 | 18 | 0.029 | 10 |
Order of SNPs in HMGCR haplotypes is rs10038095, rs17244841, rs6453131, rs3846662, rs17228540, rs3846663, and rs4629571.
Count represents number of chromosomes assigned a particular haplotype by the expectation maximization algorithm.
No association was observed with the risk of PCOS. Table 4 displays the association between SNPs and HOMA-IR and HOMA-%B. In women with PCOS, SNP rs4629571 was associated with increased HOMA-IR [carriers: 2.39 (1.22), non-carriers: 2.13 (2.02), P=0.011]. Carriers of 4 other SNPs exhibited associations with increased HOMA-IR (P values 0.019 to 0.055) that were not statistically significant considering for multiple testing. The variants rs4629571 and rs384662 were associated (not significant after Bonferroni correction) with increased HOMA-%B (P=0.035 and 0.037, respectively). Haplotype 3 (TAGTTCG), which is composed of minor alleles of the 5 SNPs that were associated with HOMA-IR, was also associated with increased HOMA-IR [carriers: 2.41 (1.31), non-carriers: 2.13 (1.98), P=0.0096]. Haplotype 3 was also associated with increased HOMA-%B [carriers: 186.20 (118.05), non-carriers: 173.60 (101.00), P=0.032, not significant considering multiple testing]. No significant association of HMGCR variants with HOMA-IR or HOMA-%B were observed in control women (Supplementary Table 2).
Table 4.
Associations of HMGCR SNPs with HOMA-IR and HOMA-%B in women with PCOS.
| HOMA-IR | HOMA-%B | |||||
|---|---|---|---|---|---|---|
| SNP | Carriers | Non- carriers |
P | Carriers | Non- carriers |
P |
| rs10038095 | 2.36 (1.83) | 2.08 (1.80) | 0.048 | 174.60 (91.73) | 175.30 (108.15) | 0.16 |
| rs17244841 | 2.38 (1.99) | 2.16 (1.94) | 0.80 | 169.70 (65.90) | 175.30 (103.40) | 0.93 |
| rs6453131 | 2.36 (1.89) | 2.02 (1.81) | 0.032 | 173.60 (91.10) | 175.00 (106.60) | 0.12 |
| rs3846662 | 2.37 (2.04) | 1.96 (1.71) | 0.019 | 175.90 (108.03) | 175.30 (91.40) | 0.037 |
| rs17238540 | 2.52 (2.01) | 2.21 (1.88) | 0.80 | 170.40 (74.40) | 175.90 (98.15) | 0.87 |
| rs3846663 | 2.34 (1.83) | 2.08 (1.80) | 0.055 | 171.15 (92.85) | 175.30 (108.15) | 0.16 |
| rs4629571 | 2.39 (1.22) | 2.13 (2.02) | 0.011 | 182.95 (112.40) | 173.60 (101.50) | 0.035 |
Significance is taken as P<0.017. Comparisons carried out using analysis of covariance, adjusting for age and BMI. Data are median (interquartile range).
In the analyses between HMGCR and androgen-related traits, haplotype 5 (ATTCGTA) was associated with lower free T [carriers: 0.74 (0.48) pg/mL, non-carriers: 0.85 (0.44) pg/mL, P=0.014]. Haplotype 5 was also associated with higher SHBG [carriers: 200.00 (82.50) nmol/L, non-carriers: 150.00 (60.00) nmol/L, P=0.0037]. To further determine the primary effect of haplotype 5, we reanalyzed the haplotype 5 associations with SHBG and free T with inclusion of free T and SHBG as covariates, respectively. The P value was 0.21 when the free T association was adjusted for SHBG; when adjusting the SHBG association for free T, the P value was 0.024, suggesting that HMGCR haplotype 5 may primarily increase SHBG, which then results in a decrease in free T. This would indicate that the effect of haplotype 5 on free T is indirect.
Discussion
We observed that HMGCR SNP rs4629571 and haplotype 3 were associated with increased insulin resistance and a trend to increased beta cell function. Haplotype 5 was associated with increased SHBG levels and lower free T. No association was observed between HMGCR variants and the risk of PCOS itself. The effects on free T appear to be secondary to changes in SHBG. These results provide new evidence that HMGCR may be a modifier gene in PCOS as an interactive factor connecting both insulin sensitivity and androgen bioavailability.
HMGCR was considered a logical candidate gene for PCOS because HMGCR is the rate-limiting enzyme in cholesterol synthesis. Patients with PCOS often have dyslipidemia with elevated plasma levels of total cholesterol, LDL-C, VLDL-C, triglycerides, and reduced HDL-C (24, 25). In clinical trials, statins improved the lipid profile, decreased testosterone, reduced hirsutism, raised SHBG, improved insulin sensitivity and biochemical markers of systemic inflammation and endothelial function in PCOS women (5, 8, 9).
Our genetic data and statin treatment effects suggest that HMGCR may be involved in insulin sensitivity in PCOS. Statins administered to PCOS women reduced insulin resistance (7–9). We demonstrated that HMGCR haplotype 3 and its SNPs were associated with increased insulin resistance. Genes that regulate insulin sensitivity may modulate SHBG level. For example, the Pro12Ala variant of the peroxisome proliferator-activated receptor gamma (PPARG) gene may influence SHBG level (26). The Gly972Arg variant in the insulin receptor substrate-1 gene (IRS1) was associated with decreased circulating SHBG in post-menopausal breast cancer survivors (27). In addition to PPARG and IRS1, HMGCR may belong to this cluster of genes which can provide a genetic link between insulin resistance/sensitivity and SHBG level, although the molecular mechanism of how HMGCR variants influence SHBG level is not currently known. HMGCR might be a factor that modulates the production or secretion of hepatic SHBG, given that both are synthesized in the liver.
Our most novel result is the association of HMGCR variants with SHBG. Compared to in vitro studies wherein statins directly inhibited thecal cell androgen output (4), statins may additionally influence testosterone level indirectly via increasing SHBG. Considering the role of HMGCR in cholesterol synthesis, the activity of HMGCR may be inhibited by negative feedback when androgens are increased in PCOS, resulting in reduced production of steroid hormones. As a further protective response, this HMGCR inhibition may increase SHBG production, to reduce androgen bioavailability as well. This hypothesis deserves further study.
In our cohort, lipid levels are not available, thus we were not able to evaluate association between HMGCR variants and lipid levels in PCOS. The seven variants we genotyped capture (via linkage disequilibrium) approximately half of the polymorphic variants listed in the PARC database; therefore, there may exist other variants associated with component traits of PCOS. Our findings need to be replicated in other PCOS populations before HMGCR is firmly established as a modifier gene in PCOS.
In conclusion, our genetic results have suggested that particular variants in HMGCR are associated with insulin resistance and possibly insulin secretion, and also associated with higher SHBG and consequently decreased testosterone. HMGCR may play a role mediating both insulin action and androgen bioavailability in PCOS. Statins may have novel beneficial effects on SHBG via HMGCR. Subjects with different genotypes of HMGCR may have different responses to statin treatment, which deserves further investigation in a pharmacogenetic study.
Supplementary Material
Acknowledgments
Grant Support: This study was supported in part by NIH grants R01-HD29364 and K24-HD01346 (to RA), R01-DK79888 (to M.O.G.), and M01-RR00425 (General Clinical Research Center Grant from the NCRR), and an endowment from the Helping Hand of Los Angeles, Inc.
Footnotes
Disclosure Statement: N.X., K.T. and M.O.G. have nothing to declare. R.A. has received consulting fees from Procter & Gamble, Merck & Co., and Organon.
This work was presented at the 88th Annual Meeting of the Endocrine Society, Boston, June 24–27, 2006.
References
- 1.Shaw LJ, Bairey Merz CN, Azziz R, Stanczyk FZ, Sopko G, Braunstein GD, et al. Postmenopausal women with a history of irregular menses and elevated androgen measurements at high risk for worsening cardiovascular event-free survival: results from the National Institutes of Health--National Heart, Lung, and Blood Institute sponsored Women's Ischemia Syndrome Evaluation. J Clin Endocrinol Metab. 2008;93:1276–1284. doi: 10.1210/jc.2007-0425. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 2.Birdsall MA, Farquhar CM, White HD. Association between polycystic ovaries and extent of coronary artery disease in women having cardiac catheterization. Ann Intern Med. 1997;126:32–35. doi: 10.7326/0003-4819-126-1-199701010-00005. [DOI] [PubMed] [Google Scholar]
- 3.Wild RA, Grubb B, Hartz A, Van Nort JJ, Bachman W, Bartholomew M. Clinical signs of androgen excess as risk factors for coronary artery disease. Fertil Steril. 1990;54:255–259. doi: 10.1016/s0015-0282(16)53699-1. [DOI] [PubMed] [Google Scholar]
- 4.Izquierdo D, Foyouzi N, Kwintkiewicz J, Duleba AJ. Mevastatin inhibits ovarian theca-interstitial cell proliferation and steroidogenesis. Fertil Steril. 2004;82 Suppl 3:1193–1197. doi: 10.1016/j.fertnstert.2004.03.037. [DOI] [PubMed] [Google Scholar]
- 5.Duleba AJ, Banaszewska B, Spaczynski RZ, Pawelczyk L. Simvastatin improves biochemical parameters in women with polycystic ovary syndrome: results of a prospective, randomized trial. Fertil Steril. 2006;85:996–1001. doi: 10.1016/j.fertnstert.2005.09.030. [DOI] [PubMed] [Google Scholar]
- 6.Banaszewska B, Pawelczyk L, Spaczynski RZ, Dziura J, Duleba AJ. Effects of simvastatin and oral contraceptive agent on polycystic ovary syndrome: prospective, randomized, crossover trial. J Clin Endocrinol Metab. 2007;92:456–461. doi: 10.1210/jc.2006-1988. [DOI] [PubMed] [Google Scholar]
- 7.Banaszewska B, Pawelczyk L, Spaczynski RZ, Duleba AJ. Effects of simvastatin and metformin on endocrine and metabolic aspects of polycystic ovary syndrome; 90th Annual Meeting of the Endocrine Society; 2008. pp. 125–126. [Google Scholar]
- 8.Sathyapalan T, Kilpatrick ES, Coady AM, Atkin SL. The effect of atorvastatin in patients with polycystic ovary syndrome: a randomized double-blind placebo-controlled study. J Clin Endocrinol Metab. 2009;94:103–108. doi: 10.1210/jc.2008-1750. [DOI] [PubMed] [Google Scholar]
- 9.Kaya C, Cengiz SD, Berker B, Demirtas S, Cesur M, Erdogan G. Comparative effects of atorvastatin and simvastatin on the plasma total homocysteine levels in women with polycystic ovary syndrome: a prospective randomized study. Fertil Steril. 2008 Aug 8; doi: 10.1016/j.fertnstert.2008.06.006. Epub. [DOI] [PubMed] [Google Scholar]
- 10.Zawadzki JK, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: towards a rational approach. In: Dunaif A, Givens JR, Haseltine F, Merriam GR, editors. Polycystic Ovary Syndrome. Cambridge: Blackwell Scientific Publications; 1992. pp. 377–384. [Google Scholar]
- 11.Azziz R, Woods KS, Reyna R, Key TJ, Knochenhauer ES, Yildiz BO. The prevalence and features of the polycystic ovary syndrome in an unselected population. J Clin Endocrinol Metab. 2004;89:2745–2749. doi: 10.1210/jc.2003-032046. [DOI] [PubMed] [Google Scholar]
- 12.Hatch R, Rosenfield RL, Kim MH, Tredway D. Hirsutism: implications, etiology, and management. Am J Obstet Gynecol. 1981;140:815–830. doi: 10.1016/0002-9378(81)90746-8. [DOI] [PubMed] [Google Scholar]
- 13.Boots LR, Potter S, Potter D, Azziz R. Measurement of total serum testosterone levels using commercially available kits: high degree of between-kit variability. Fertil Steril. 1998;69:286–292. doi: 10.1016/s0015-0282(97)00464-0. [DOI] [PubMed] [Google Scholar]
- 14.Pearlman WH, Crepy O, Murphy M. Testosterone-binding levels in the serum of women during the normal menstrual cycle, pregnancy, and the post-partum period. J Clin Endocrinol Metab. 1967;27:1012–1018. doi: 10.1210/jcem-27-7-1012. [DOI] [PubMed] [Google Scholar]
- 15.Knochenhauer ES, Key TJ, Kahsar-Miller M, Waggoner W, Boots LR, Azziz R. Prevalence of the polycystic ovary syndrome in unselected black and white women of the southeastern United States: a prospective study. J Clin Endocrinol Metab. 1998;83:3078–3082. doi: 10.1210/jcem.83.9.5090. [DOI] [PubMed] [Google Scholar]
- 16.Wallace TM, Levy JC, Matthews DR. Use and abuse of HOMA modeling. Diabetes Care. 2004;27:1487–1495. doi: 10.2337/diacare.27.6.1487. [DOI] [PubMed] [Google Scholar]
- 17.Levy JC, Matthews DR, Hermans MP. Correct homeostasis model assessment (HOMA) evaluation uses the computer program. Diabetes Care. 1998;21:2191–2192. doi: 10.2337/diacare.21.12.2191. [DOI] [PubMed] [Google Scholar]
- 18.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 19.Chasman DI, Posada D, Subrahmanyan L, Cook NR, Stanton VP, Jr, Ridker PM. Pharmacogenetic study of statin therapy and cholesterol reduction. JAMA. 2004;291:2821–2827. doi: 10.1001/jama.291.23.2821. [DOI] [PubMed] [Google Scholar]
- 20.Goodarzi MO, Guo X, Taylor KD, Quiñones MJ, Samayoa C, Yang H, et al. Determination and use of haplotypes: ethnic comparison and association of the lipoprotein lipase gene and coronary artery disease in Mexican-Americans. Genet Med. 2003;5:322–327. doi: 10.1097/01.GIM.0000076971.55421.AD. [DOI] [PubMed] [Google Scholar]
- 21.Livak KJ. Allelic discrimination using fluorogenic probes and the 5' nuclease assay. Genet Anal. 1999;14:143–149. doi: 10.1016/s1050-3862(98)00019-9. [DOI] [PubMed] [Google Scholar]
- 22.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 23.Qin ZS, Niu T, Liu JS. Partition-ligation-expectation-maximization algorithm for haplotype inference with single-nucleotide polymorphisms. Am J Hum Genet. 2002;71:1242–1247. doi: 10.1086/344207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodarzi MO, Erickson S, Port SC, Jennrich RI, Korenman SG. Relative impact of insulin resistance and obesity on cardiovascular risk factors in polycystic ovary syndrome. Metabolism. 2003;52:713–719. doi: 10.1016/s0026-0495(03)00031-3. [DOI] [PubMed] [Google Scholar]
- 25.Legro RS, Kunselman AR, Dunaif A. Prevalence and predictors of dyslipidemia in women with polycystic ovary syndrome. Am J Med. 2001;111:607–613. doi: 10.1016/s0002-9343(01)00948-2. [DOI] [PubMed] [Google Scholar]
- 26.Mousavinasab F, Tahtinen T, Jokelainen J, Koskela P, Vanhala M, Oikarinen J, et al. The Pro12Ala polymorphism of the PPAR gamma 2 gene influences sex hormone-binding globulin level and its relationship to the development of the metabolic syndrome in young Finnish men. Endocrine. 2006;30:185–190. doi: 10.1385/ENDO:30:2:185. [DOI] [PubMed] [Google Scholar]
- 27.Fan J, McKean-Cowdin R, Bernstein L, Stanczyk FZ, Li AX, Ballard-Barbash R, et al. An association between a common variant (G972R) in the IRS-1 gene and sex hormone levels in post-menopausal breast cancer survivors. Breast Cancer Res Treat. 2006;99:323–331. doi: 10.1007/s10549-006-9211-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.