

Abstract
Purpose of review
Despite the remarkable success of intensive antiretroviral drug therapy in blocking the HIV replication, the virus persists in a small number of cells where HIV has been transcriptionally silenced. This review will focus on recent insights into the HIV transcriptional control mechanisms that provide the biochemical basis for understanding latency.
Recent findings
Latency arises when the regulatory feedback mechanism driven by HIV Tat expression is disrupted. Small changes in transcriptional initiation, induced by epigenetic silencing, can lead to restrictions in Tat levels and entry of proviruses into latency. In resting memory T-cells, which carry the bulk of the latent viral pool, additional restrictions limiting cellular levels of the essential Tat cofactor P-TEFb and the transcription initiation factors NF-κB and NFAT ensure that the provirus remains silenced unless the host cell is activated.
Summary
Strategies to purge the latent proviral pool require non-toxic activator molecules. The multiple restrictions imposed on latent proviruses that need to be overcome suggest that proviral reactivation will not be achieved when only a single reactivation step is targeted, but will require both removal of epigenetic blocks and the activation of P-TEFb. Alternatively, new inhibitors that block proviral reactivation could be developed.
Keywords: HIV Tat, HIV initiation, HIV elongation, HIV chromatin, P-TEFb
Introduction
Highly active antiretroviral therapy (HAART) for HIV infections uses a potent cocktail of antiviral drugs to stably reduce virus replication below detectable levels. Unfortunately, even after decades of treatment, the virus is able to persist and high levels of virus replication invariably resume when treatment is interrupted [1,2]. The main cause of treatment failure appears to be reactivation of virus from a pool of latently infected cells, principally residing in the pool of resting memory CD4+ T-cells [3,4].
Although it is difficult to exclude the possibility that slowly replicating virus persists in sanctuary sites which are poorly accessed by the antiviral drugs, genetic evidence strongly suggests that this is not the case. Both the residual virus recovered from treated patients [5], and the rebounding virus recovered during the short treatment interruptions [6], have much greater sequence homogeneity than would be expected for a viral population replicating at low levels, implying that both sets of viruses were derived from the reactivation of clones of long-lived latently infected cells.
During latent infections transcription from HIV proviruses is effectively suppressed. This review will describe how recent insights into the molecular mechanisms controlling HIV transcription have defined how HIV becomes initially silenced and then subsequently re-emerges from latency. These molecular studies provide the framework for developing new therapeutics that target the latent viral pool.
Transcriptional feedback: The HIV promoter is bipolar
HIV transcription is tightly controlled both at the level of transcription initiation and elongation. There is no specific host repressor that directs a provirus to become latent. Instead, the switch between productive transcription and latency is due to the manipulation of a powerful feedback mechanism fueled by the viral trans-activator protein, Tat.
The HIV LTR which includes multiple upstream DNA regulatory elements that serve as binding sites for cellular transcription initiation factors. The core promoter, which includes all the elements required for transcription in productive infections, is a powerful and highly optimized promoter comprised of three tandem SP1 binding sites [7], an efficient TATA element [8] and a highly active initiator sequence [9]. SP1 functions as an essential upstream activator; deletion of one or more of the SP1 sites effectively blocks both Tat-dependent and basal transcription [10]. In addition, the HIV LTR carries a unique “enhancer” sequence which contains two tandem NF-κB binding motifs [11]. Members of both the NF-κB family and NFAT can bind to the HIV NF-κB motifs and is stimulated by cooperative interactions with Sp1 [12]. In contrast to the elements of the core promoter, mutation of the NF-κB sites results in only a modest inhibition of virus growth in most cell lines [13], however, signaling through the viral enhancer is essential in order to re-activate latent proviruses.
The key feature that distinguishes the HIV LTR from cellular promoters is that is auto-regulated by Tat. Detailed biochemical investigations over the last 25 years have shown that Tat exclusively stimulates transcription elongation. In the absence of Tat, transcription initiation is normal but only short abortive transcripts are produced. Tat directs the cellular transcriptional elongation factor P-TEFb [14] to nascent RNA polymerases by binding to the HIV TAR element, an RNA stem-loop structure found at the 5′ end of all viral transcripts (Figure 1). P-TEFb is a protein kinase complex comprised of a regulatory Cyclin T1 (CycT1) subunit and the catalytic CDK-9 subunit. This enzyme potently stimulates HIV elongation by targeting both positive and negative elongation factors. Phosphorylation of the negative elongation factor NELF removes a powerful block to elongation [15], while phosphorylation of the C-terminal domains (CTD) of RNAP II [16] and Spt5 [17,18] results in the direct activation of polymerase processivity.
Figure 1. Reactivation of latent proviruses.

(A) Latent HIV provirus. In latent proviruses transcription elongation is very inefficient due to absence of the transcription elongation factor NF-κB as well as chromatin restrictions (not shown for simplicity). The small number of transcription complexes that are able initiate and elongate through TAR are subject to additional elongation restrictions by NELF. NELF forces premature termination leading to an exceptionally low level of transcription from the latent provirus. (B) NF-κB and Tat-activated transcription. Initiation is strongly induced by NF-κB, which acts primarily to remove chromatin restrictions near the promoter through recruitment of histone acetyltransferases. Under these circumstances promoter clearance is also much more efficient and relatively few of the elongation complexes pause near the promoter. After the transcription through the TAR element, both NELF and the Tat/P-TEFb complex (including CDK9 and CycT1 and the accessory elongation ELL2) are recruited to the elongation complex via binding interactions with TAR RNA. This activates the CDK9 kinase and leads to hyperphosphorylation of the CTD of RNA polymerase II, Spt5 and NELF-E. The phosphorylation of NELF-E leads to its release. The presence of hyperphosphorylated RNAP II and Spt5 allows enhanced transcription of the full HIV genome.
In the culmination of over 2 decades of research on P-TEFb by David Price and his colleagues, the crystal structure of a Tat:pTEFb complex was determined earlier this year [19••]. The structure shows that Tat forms extensive contacts both with the CycT1 subunit of P-TEFb and also with the T-loop of the Cdk9 subunit. Importantly, Tat induces significant conformational changes in CDK9 providing an explanation for how it is able to constitutively activate the enzyme [19••].
The strong amplification of transcription stimulated by Tat, coupled with the disproportionate decline in transcription that ensues when Tat levels become restricted, gives the HIV promoter a “bipolar” character (Figure 2). Insightful studies by Weinberger et al. [20,21] and Burnett et al. [22•] have emphasized how stochastic fluctuations in Tat gene expression can act as a molecular switch. Small changes in initiation rates, which can be experimentally mimicked by introducing mutations into the NF-κB and Sp1 binding sites, are sufficient to restrict Tat production and lead to enhanced rates of viral entry into latency [22•]. This switching mechanism crucially depends on the auto-regulation of Tat. When Tat is expressed in trans from an ectopic promoter, HIV proviruses become constitutively active and are unable to enter latency [23].
Figure 2. Autoregulation of HIV transcription by Tat.
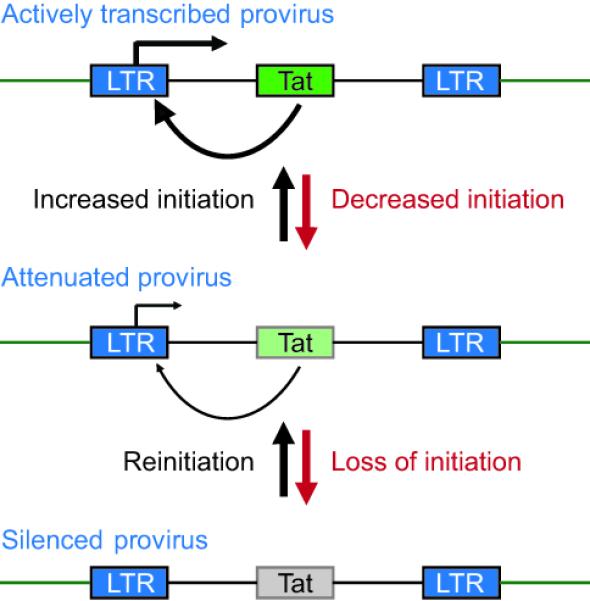
Small in initiation efficiency, due to transcriptional interference or epigenetic silencing, reduce Tat levels in the cell and disproportionately inhibit transcription, driving the HIV provirus into latency. Reinitiation stimulates Tat production and restores full transcription efficiency.
The idea that the switch between active transcription and latency is regulated by Tat expression levels is also supported by the observation that introduction of mutations that attenuate Tat activity leads to an increased frequency of viruses entering latency [23]. Similarly, viruses recovered from the latently-infected CD4+ T cells of patients are enriched for HIV-1 Tat variants with impaired transactivation activity [24].
In summary, changes in the cellular environment that restrict transcription initiation are able to reduce Tat availability and force the virus into latency, but the virus remains poised to resume its replication in response to triggers that stimulate transcription initiation and restore Tat levels.
How is this subtle balance achieved?
Epigenetic silencing and promoter occlusion: Triggering the depression
When HIV infects cells it preferentially integrates into active transcription units that provide a favorable environment for viral transcription [25]. An early hypothesis to explain latency was that latently infected cells were generated by rare integrations into heterochromatic regions [26]. However extensive sequencing studies have shown that latent proviruses are almost invariably found integrated into actively transcribed genes [27-29]. This implied that following the initial integration and expression of the proviruses epigenetic silencing events restricted HIV expression. In support of this hypothesis, numerous studies have shown that the LTRs of latent proviruses acquire heterochromatic structures (Figure 3). Typically latent proviruses display a fixed nucleosomal structure that blocks the transcription start site [30], high levels of histone deacetylases (HDACs), deactylated histones [31-33] and methylated histones [23,34•, 35-36]. The functional importance of these modifications is clearly demonstrated by the observation that drugs that inhibit histone deacetylation or methylation are potent inducers of latent proviruses [37,38].
Figure 3. Stages in epigenetic silencing.
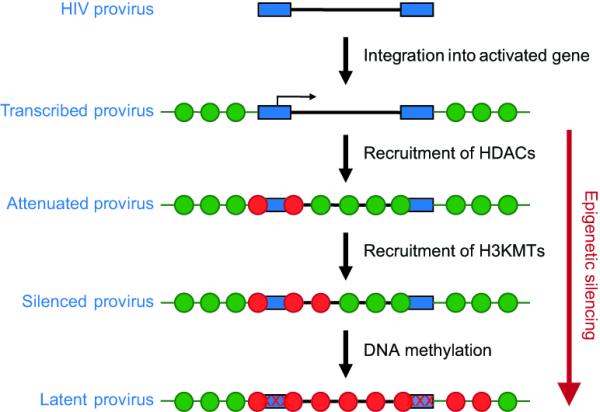
Latent HIV proviruses almost invariably have deacteylated histones, but clones show heterogenous levels of methylated histones and methylated DNA. This suggests that silencing is an ordered and progressive process, with DNA methylation being a sign of the most repressed state.
Two recent reports have demonstrated that the acquisition of hypermethylated CpG islands near the HIV promoter correlates with the silencing of HIV transcription in both Jurkat T-cells and primary isolates aviremic patients [39•,40•]. Treatment of latently infected cells with the DNA methylation inhibitor 5-aza-deoxycytidine (5-aza-CdR) potentiates the reactivation and outgrowth of silenced proviruses [39•,40•]. Typical clonal cell lines carrying a single latent proviruses show heterogenous levels of methylated histones and methylated DNA. This suggests that silencing is an ordered and progressive process, with DNA methylation repressing one of the most repressed states.
The observation that HIV can integrate within active transcription units prompted investigations into whether latency arises because of transcriptional interference between the host promoter and the viral LTR. Lenasi et al. [41] showed that in latently infected Jurkat T-cell lines, host-initiated transcripts terminated at the polyadenylation site in the 5’ LTR of the integrated proviruses. Similarly, Han et al. [42] showed in cell lines that were engineered to insert HIV proviruses in either orientation downstream of the HPRT gene, readthrough transcription inhibited HIV-1 gene expression for convergently orientated provirus but enhanced HIV-1 gene expression when HIV-1 was in the same orientation as the host gene.
Although transcriptional interference was clearly documented in both these studies, it remains unclear whether it is a primary cause of latency or a consequence of the insertion of a repressed provirus into an active gene. Duvinger et al. [43•] have argued that transcriptional interference leads to the “silent integration” of proviruses in the majority of latently infected cells. However, these experiments were designed to select for population of viruses that were subjected to immediate silencing events. By contrast, studies from our laboratory have shown that after the selection of cells that carry highly expressed viruses there is progressive silencing due to the imposition of epigenetic restrictions [23,34•].
In the natural setting, epigenetic silencing is imposed when HIV infects T-cells just prior to their natural reversion to a quiescent state during the differentiation of both naïve and memory T-cells [3,44,45]. Importantly, these silencing events can also be recapitulated in vitro using primary cells that are induced to enter quiescence [34•,40,46-48]. It therefore seems likely that both transcriptional interference and epigenetic silencing can contribute to the development of latency in vivo. The net effect of both mechanisms is to restrict proviral transcription initiation and induce a decline in Tat levels.
Active restriction of HIV transcription elongation: Deepening the depression
In contrast to transformed cells, resting CD4+ T-cells further ensure that latent proviruses remain transcriptionally inactive by imposing additional powerful blocks that restrict P-TEFb levels [49]. In these cells, the majority of the essential elongation factor P-TEFb is sequestered into a large RNP complex comprising 7SK RNA and a series of RNA binding proteins (7SK snRNP) [50,51]. Essential components of the 7SK snRNP complex include HEXIM1 or HEXIM2, which inhibit the CDK9 kinase in a 7SK-dependent manner [52,53].
The inactivation of P-TEFb effectively prevents any basal transcriptional activation by Tat-independent recruitment of P-TEFb to the provirus. For example, we have observed that induction of latently infected primary CD4+ T-cells requires T-cell receptor mediated induction of P-TEFb [34•]. Although the mechanisms leading to P-TEFb release from the 7SK snRNP during T-cell activation are not yet fully defined, it seems likely that post-translational modifications of the complex are required. For example, the HIV activator HMBA is able to induce dephosphorylation of the T-loop of P-TEFb by PP1a and PP2B resulting in its release from the 7SK snRNP [54]. An important recent study suggests that cyclin T1 acetylation also triggers dissociation of HEXIM1 and 7SK RNA from the inactive 7SK snRNP complex and activates the transcriptional activity of P-TEFb [55•].
Unpublished studies from our laboratory have shown that the restriction of elongation from the HIV LTR from latent proviruses is much stronger than for cellular promoters. This is due to powerful additional restrictions on HIV elongation which are imposed by NELF [15,56,57]. Furthermore, we have shown that depletion of NELF provides a potent signal to reactivate latent proviruses.
Transcription initiation and chromatin remodeling: Dispelling the lassitude
In addition to restricting P-TEFb, quiescent T-cells also sequester the cellular transcription initiation factors NF-κB and NFAT in the cytoplasm [11,58]. TCR stimulation induces a complex cascade of pathways leading to the activation of NF-κB through a protein kinase C-mediated pathway and NFAT through the Ca+2-calcineurine pathway. Recent structural studies have shown that both NF-κB [59] and NFAT [60] assume unique, mutually exclusive, conformations upon binding the HIV LTR.
NF-κB binding to the HIV LTR triggers proviral reactivation by directing recruitment of the histone acetyltransferases (HATs) to the HIV LTR [61-64]. The acetylation of histones near the HIV promoter in turn provides a signal for the recruitment of the chromatin remodeling complex BAF which activates transcription by displacing the restrictive nucleosome-1 (Nuc-1) positioned immediately downstream from the transcriptional start site [65]. The recruitment of HATs may also help to stabilize NF-κB on the viral promoter, since acetylation [66] and methylation [67] of the p65 subunit enhances its affinity its DNA binding affinity. NFAT also interacts with the HIV LTR via the NF-κB binding sites, it seems likely that members of the NFAT family also promote chromatin remodeling of the HIV-1 since they are known to recruit the coactivators p300 and CBP to cellular genes [68].
There has been a long standing controversy about whether NFAT [46,69,70] or NF-κB [34•,71] is the dominant factor mediating proviral reactivation in CD4+ T-cells. This seems to be an unnecessary distinction because it is now evident that multiple cellular pathways are able to independently reactivate latent HIV expression. In latently infected primary T-cells derived from thymocytes both the PKC pathway leading to NF-κB activation and the NFAT are able to stimulate virus expression [71]. TLR5 stimulation induces activation of NF-κB and can reactivate latent HIV-1 in quiescent central memory CD4+ T cells [72]. Similarly, inducers of NF-κB such as prostratin and HIV-1-reactivating protein factor (HRF) [73] are potent inducers of latent HIV proviruses in resting memory T-cells. By contrast, in polarized T-cells generated in vitro, NFAT is clearly the exclusive activator of latent proviruses [46].
Tat, P-TEFb and the positive control of HIV elongation: Mania
When proviruses emerge from latency, the initial rounds of HIV transcription are restricted until the resumption of new Tat synthesis after about 2 hrs [23,74]. Our picture of how Tat and P-TEFb stimulate HIV elongation has been radically altered by two recent papers which used modern proteomic methods to discover two distinct and stable complexes containing Tat and P-TEFb [75••,76••] (Figure 4). The most abundant complex contains active P-TEFb and the human transcription factors/coactivators AFF4, ENL, AF9, and ELL2. He et al. [75••] showed that ELL2, an elongation factor previously shown to enhance transcription elongation by preventing RNAP II backtracking, is critical both for basal HIV transcription and Tat-mediated transactivation. Thus, any model for the stimulatory effects of P-TEFb on HIV-1 transcription has to take into account the role of ELL2 and possibly several additional elongation factors.
Figure 4. Control of P-TEFb by Tat.
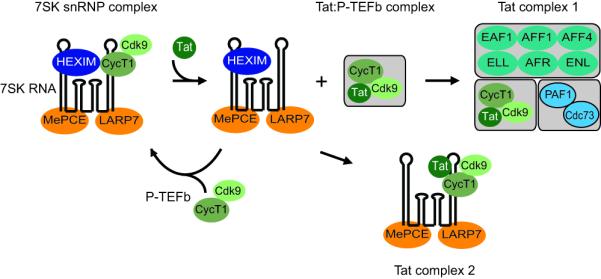
In resting CD4+ T-cells the majority of the P-TEFb in cells is found in a transcriptionally inactive snRNP complex containing 7SK RNA, HEXIM and the RNA binding proteins MePCE and LARP7. Tat disrupts this complex by displacing HEXIM and forming a stable complex with P-TEFb. Prior to recruitment to the transcription complex a larger complex is formed between P-TEFb and transcription elongation factors from the mixed lineage leukemia (MLL) family, including ELL2. A small fraction of the Tat in cell is also able to form a complex with 7SK RNA in the absence of HEXIM1.
A second completely unexpected finding to emerge from these studies is that Tat forms a stable complex with 7SK snRNP lacking HEXIM1 [76••]. Formation of Tat 7SK snRNP is likely mediated through direct interaction between Tat and 7SK RNA and is possibly plays an important role in HIV transcription by providing a pool of activated P-TEFb.
Conclusions
Devising strategies to eliminate this latent reservoir presents formidable challenges since the reservoir is established early during infection [77], is extremely stable, with an estimated half-life of 44 months [78], and can be replenished during episodes of viremia [79,80] or by homeostatic replacement of latently infected cells [81]. Since intensification of antiviral regimens has essentially no impact on eradicating the latent pool from the infected host [82], there have been recent calls to develop entirely novel forms of therapy to purge the pool of latent proviruses [83,84].
In the “shock and kill” strategy for HIV eradication [85,86], a “shock” phase is used to reactivate latent proviruses, and a “kill” phase is used to eliminate the induced cells through immune responses, viral cytopathogenicity or cytotoxic drugs. The detailed molecular studies described above strongly imply that effective activation of the entire latent viral pool may ultimately require a cocktail of drugs that stimulate both transcription initiation and P-TEFb mobilization.
The molecular studies reviewed here also emphasize that HIV has a plethora of potential drug vulnerabilities that could be exploited to prevent its re-emergence from latency. Although historically little progress has been made in finding selective inhibitors of Tat-activation of transcription the recent molecular and structural work on the Tat:P-TEFb complex suggest that the time is ripe to look for inhibitors of this critical interaction. Additionally there may be important opportunities to target the unique initiation complexes created by NF-κB and NFAT on the viral LTR.
It is a tribute to the success of molecular investigations into HIV transcription control and latency that the dream of achieving a functional cure for HIV infections, based devising new approaches to purge the latent reservoir, has now become a well-defined and realistic long-term research goal.
Keypoints.
HIV latency arises when levels of the regulatory protein Tat fall to below threshold levels.
Transcriptional blocks restricting Tat levels are the result of epigenetic silencing, and in some cases, transcriptional interference by host promoters.
Resting T-cells maintain latent proviruses by sequestering the Tat cofactor P-TEFb and the initiation factors NF-κB and NFAT in the cytoplasm.
It is unlikely that purging of the latent viral pool can be achieved without activating HIV transcriptional initiation and elongation.
Acknowledgments
This work was supported by grants from the National Institutes of Health, R01-AI067093 and DP1-DA028869 to JK.
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Chun TW, Davey RT, Jr., Engel D, Lane HC, Fauci AS. Re-emergence of HIV after stopping therapy. Nature. 1999;401:874–875. doi: 10.1038/44755. [DOI] [PubMed] [Google Scholar]
- 2.Davey RT, Jr., Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R, Natarajan V, Lempicki RA, Adelsberger JW, Miller KD, et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. U S A. 1999;96:15109–15114. doi: 10.1073/pnas.96.26.15109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen A, Zink MC, Mankowski JL, Chadwick K, Margolick JB, Carruth LM, Li M, Clements JE, Siliciano RF. Resting CD4+ T lymphocytes but not thymocytes provide a latent viral reservoir in a simian immunodeficiency virus-Macaca nemestrina model of human immunodeficiency virus type 1-infected patients on highly active antiretroviral therapy. J. Virol. 2003;77:4938–4949. doi: 10.1128/JVI.77.8.4938-4949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 5.Brennan TP, Woods JO, Sedaghat AR, Siliciano JD, Siliciano RF, Wilke CO. Analysis of HIV-1 Viremia and Provirus in Resting CD4+ T Cells Reveals a Novel Source of Residual Viremia in Patients on Antiretroviral Therapy. J. Virol. 2009;83:8470–8481. doi: 10.1128/JVI.02568-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joos B, Fischer M, Kuster H, Pillai SK, Wong JK, Boni J, Hirschel B, Weber R, Trkola A, Gunthard HF. HIV rebounds from latently infected cells, rather than from continuing low-level replication. Proc. Natl. Acad. Sci. U S A. 2008;105:16725–16730. doi: 10.1073/pnas.0804192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones K, Kadonaga J, Luciw P, Tjian R. Activation of the AIDS retrovirus promoter by the cellular transcription factor, Sp1. Science. 1986;232:755–759. doi: 10.1126/science.3008338. [DOI] [PubMed] [Google Scholar]
- 8.Olsen HS, Rosen CA. Contribution of the TATA motif to Tat-mediated transcriptional activation of the human immunodeficiency virus gene expression. J. Virol. 1992;66:5594–5597. doi: 10.1128/jvi.66.9.5594-5597.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rittner K, Churcher MJ, Gait MJ, Karn J. The human immunodeficiency virus long terminal repeat includes a specialised initiator element which is required for Tat-responsive transcription. J. Mol. Biol. 1995;248:562–580. doi: 10.1006/jmbi.1995.0243. [DOI] [PubMed] [Google Scholar]
- 10.Ross EK, Buckler-White AJ, Rabson AB, Englund G, Martin MA. Contribution of NF-kB and Sp1 binding motifs to the replicative capacity of human immunodeficiency virus type 1: Distinct patterns of viral growth are determined by T-cell types. J. Virol. 1991;65:4350–4358. doi: 10.1128/jvi.65.8.4350-4358.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nabel G, Baltimore DA. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature. 1987;326:711–713. doi: 10.1038/326711a0. [DOI] [PubMed] [Google Scholar]
- 12.Perkins ND, Edwards NL, Duckett CS, Agranoff AB, Schmid RM, Nabel GJ. A cooperative interaction between NF-κB and Sp1 is required for HIV-1 enhancer activation. EMBO J. 1993;12:3551–3558. doi: 10.1002/j.1460-2075.1993.tb06029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen BK, Feinberg MB, Baltimore D. The κB sites in the human immunodeficiency virus type 1 long terminal repeat enhance virus replication yet are not absolutely required for viral growth. J. Virol. 1997;71:5495–5504. doi: 10.1128/jvi.71.7.5495-5504.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei P, Garber ME, Fang S-M, Fischer WH, Jones KA. A novel cdk9-associated c-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop specific binding to TAR RNA. Cell. 1998;92:451–462. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- 15.Fujinaga K, Irwin D, Huang Y, Taube R, Kurosu T, Peterlin BM. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol. Cell. Biol. 2004;24:787–795. doi: 10.1128/MCB.24.2.787-795.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim YK, Bourgeois CF, Isel C, Churcher MJ, Karn J. Phosphorylation of the RNA polymerase II carboxyl-terminal domain by CDK9 is directly responsible for human immunodeficiency virus type 1 Tat-activated transcriptional elongation. Mol. Cell. Biol. 2002;22:4622–4637. doi: 10.1128/MCB.22.13.4622-4637.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bourgeois CF, Kim YK, Churcher MJ, West MJ, Karn J. Spt5 cooperates with Tat by preventing premature RNA release at terminator sequences. Mol. Cell. Biol. 2002;22:1079–1093. doi: 10.1128/MCB.22.4.1079-1093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ivanov D, Kwak YT, Guo J, Gaynor RB. Domains in the SPT5 protein that modulate its transcriptional regulatory properties. Mol. Cell. Biol. 2000;20:2970–2983. doi: 10.1128/mcb.20.9.2970-2983.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •• 19.Tahirov TH, Babayeva ND, Varzavand K, Cooper JJ, Sedore SC, Price DH. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature. 2010;465:747–751. doi: 10.1038/nature09131. This paper presents the first structure of a complex between HIV-1 Tat protein and its cofactor P-TEFb. The structure demonstrates how interactions with P-TEFb direct the folding of the Tat protein and how Tat, in turn, alters the conformation of P-TEFb to activate its kinase activity. Prior to this work structural studies on Tat were intractable.
- 20.Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, Schaffer DV. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. 2005;122:169–182. doi: 10.1016/j.cell.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 21.Weinberger LS, Dar RD, Simpson ML. Transient-mediated fate determination in a transcriptional circuit of HIV. Nat. Genet. 2008;40:466–470. doi: 10.1038/ng.116. [DOI] [PubMed] [Google Scholar]
- • 22.Burnett JC, Miller-Jensen K, Shah PS, Arkin AP, Schaffer DV. Control of stochastic gene expression by host factors at the HIV promoter. PLoS Pathog. 2009;5:e1000260. doi: 10.1371/journal.ppat.1000260. This paper describes the impact of mutations in the viral LTR on the behavior of the Tat feedback mechanism. The results demonstrate how even a slight attenuation of transcription initiation can have a profound impact on the stochastic induction of HIV transcription by Tat.
- 23.Pearson R, Kim YK, Hokello J, Lassen K, Friedman J, Tyagi M, Karn J. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J. Virol. 2008;82:12291–12303. doi: 10.1128/JVI.01383-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yukl S, Pillai S, Li P, Chang K, Pasutti W, Ahlgren C, Havlir D, Strain M, Gunthard H, Richman D, et al. Latently-infected CD4+ T cells are enriched for HIV-1 Tat variants with impaired transactivation activity. Virology. 2009;387:98–108. doi: 10.1016/j.virol.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lewinski MK, Yamashita M, Emerman M, Ciuffi A, Marshall H, Crawford G, Collins F, Shinn P, Leipzig J, Hannenhalli S, et al. Retroviral DNA integration: viral and cellular determinants of target-site selection. PLoS Pathog. 2006;2:e60. doi: 10.1371/journal.ppat.0020060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jordan A, Bisgrove D, Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003;22:1868–1877. doi: 10.1093/emboj/cdg188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lewinski MK, Bisgrove D, Shinn P, Chen H, Hoffmann C, Hannenhalli S, Verdin E, Berry CC, Ecker JR, Bushman FD. Genome-wide analysis of chromosomal features repressing human immunodeficiency virus transcription. J. Virol. 2005;79:6610–6619. doi: 10.1128/JVI.79.11.6610-6619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han Y, Lassen K, Monie D, Sedaghat AR, Shimoji S, Liu X, Pierson TC, Margolick JB, Siliciano RF, Siliciano JD. Resting CD4+ T cells from human immunodeficiency virus type 1 (HIV-1)-infected individuals carry integrated HIV-1 genomes within actively transcribed host genes. J. Virol. 2004;78:6122–6133. doi: 10.1128/JVI.78.12.6122-6133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vatakis DN, Kim S, Kim N, Chow SA, Zack JA. Human immunodeficiency virus integration efficiency and site selection in quiescent CD4+ T cells. J. Virol. 2009;83:6222–6233. doi: 10.1128/JVI.00356-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verdin E, Paras PJ, Van Lint C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993;12:3249–3259. doi: 10.1002/j.1460-2075.1993.tb05994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keedy KS, Archin NM, Gates AT, Espeseth A, Hazuda DJ, Margolis DM. A limited group of class I histone deacetylases act to repress human immunodeficiency virus type-1 expression. J. Virol. 2009;83:4749–4756. doi: 10.1128/JVI.02585-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. NF-κB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006;25:139–149. doi: 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tyagi M, Karn J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007;26:4985–4995. doi: 10.1038/sj.emboj.7601928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- • 34.Tyagi M, Pearson RJ, Karn J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J. Virol. 2010;84:6425–6437. doi: 10.1128/JVI.01519-09. In this paper we show that transcriptionally active proviruses are subject to epigenetic silencing in primary CD4+ T-cells. This experiment demonstrates that latency can be acquired and is not necessarily the result of integration into a restrictive site on the genome. Once silenced, the proviruses can be reactivated by TCR signaling which activates both NF-κB and P-TEFb, but not by agents that induce only NF-κB.
- 35.du Chene I, Basyuk E, Lin YL, Triboulet R, Knezevich A, Chable-Bessia C, Mettling C, Baillat V, Reynes J, Corbeau P, et al. Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 2007;26:424–435. doi: 10.1038/sj.emboj.7601517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marban C, Suzanne S, Dequiedt F, de Walque S, Redel L, Van Lint C, Aunis D, Rohr O. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007;26:412–423. doi: 10.1038/sj.emboj.7601516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Archin NM, Keedy KS, Espeseth A, Dang H, Hazuda DJ, Margolis DM. Expression of latent human immunodeficiency type 1 is induced by novel and selective histone deacetylase inhibitors. AIDS. 2009;23:1799–1806. doi: 10.1097/QAD.0b013e32832ec1dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imai K, Togami H, Okamoto T. Involvement of Histone H3 Lysine 9 (H3K9) Methyltransferase G9a in the Maintenance of HIV-1 Latency and Its Reactivation by BIX01294. J. Biol. Chem. 2010;285:16538–16545. doi: 10.1074/jbc.M110.103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- • 39.Blazkova J, Trejbalova K, Gondois-Rey F, Halfon P, Philibert P, Guiguen A, Verdin E, Olive D, Van Lint C, Hejnar J, et al. CpG methylation controls reactivation of HIV from latency. PLoS Pathog. 2009;5:e1000554. doi: 10.1371/journal.ppat.1000554. DNA methylation is a key epigenetic modification that is associated with the terminal silencing of cellular genes during differentiation. Earlier experiments had suggested that DNA methylation does not play a role in silencing HIV. However this study demonstrates that even with clones carrying single proviral integrants DNA methylation of latent HIV provirals is highly variable. The viruses that are most heavily methylated are the most resistant to reactivation.
- • 40.Kauder SE, Bosque A, Lindqvist A, Planelles V, Verdin E. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 2009;5:e1000495. doi: 10.1371/journal.ppat.1000495. This paper extends the study of DNA methylation of latent HIV proviruses to a new primary cell model for HIV latency developed by Bosque and Planelles. Consistent with the results of Blazkova et al., DNA methylation is associated with the most effectively silenced HIV proviruses in the population.
- 41.Lenasi T, Contreras X, Peterlin BM. Transcriptional interference antagonizes proviral gene expression to promote HIV latency. Cell Host Microbe. 2008;4:123–133. doi: 10.1016/j.chom.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han Y, Lin YB, An W, Xu J, Yang HC, O’Connell K, Dordai D, Boeke JD, Siliciano JD, Siliciano RF. Orientation-dependent regulation of integrated HIV-1 expression by host gene transcriptional readthrough. Cell Host Microbe. 2008;4:134–146. doi: 10.1016/j.chom.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- • 43.Duverger A, Jones J, May J, Bibollet-Ruche F, Wagner FA, Cron RQ, Kutsch O. Determinants of the establishment of human immunodeficiency virus type 1 latency. J. Virol. 2009;83:3078–3093. doi: 10.1128/JVI.02058-08. This provocative paper suggests that transcriptional interference is the primary mechanism for establishing latency in cells. While the authors make a strong that “silent integrations” are possible other studies have shown that epigenetic silencing can also play an important role in establishing latency.
- 44.Brooks DG, Kitchen SG, Kitchen CM, Scripture-Adams DD, Zack JA. Generation of HIV latency during thymopoiesis. Nat. Med. 2001;7:459–464. doi: 10.1038/86531. [DOI] [PubMed] [Google Scholar]
- 45.Brenchley JM, Hill BJ, Ambrozak DR, Price DA, Guenaga FJ, Casazza JP, Kuruppu J, Yazdani J, Migueles SA, Connors M, et al. T-cell subsets that harbor human immunodeficiency virus (HIV) in vivo: implications for HIV pathogenesis. J. Virol. 2004;78:1160–1168. doi: 10.1128/JVI.78.3.1160-1168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bosque A, Planelles V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood. 2008;113:58–65. doi: 10.1182/blood-2008-07-168393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang HC, Xing S, Shan L, O’Connell K, Dinoso J, Shen A, Zhou Y, Shrum CK, Han Y, Liu JO, et al. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J. Clin. Invest. 2009;119:3473–3486. doi: 10.1172/JCI39199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burke B, Brown HJ, Marsden MD, Bristol G, Vatakis DN, Zack JA. Primary cell model for activation-inducible human immunodeficiency virus. J. Virol. 2007;81:7424–7434. doi: 10.1128/JVI.02838-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adams M, Sharmeen L, Kimpton J, Romeo JM, Garcia JV, Peterlin BM, Groudine M, Emerman M. Cellular latency in human immunodeficiency virus-infected individuals with high CD4 levels can be detected by the presence of promoter-proximal transcripts. Proc. Natl. Acad. Sci. U S A. 1994;91:3862–3866. doi: 10.1073/pnas.91.9.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nguyen VT, Kiss T, Michels AA, Bensaude O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature. 2001;414:322–325. doi: 10.1038/35104581. [DOI] [PubMed] [Google Scholar]
- 51.Yang Z, Zhu Q, Luo K, Zhou Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature. 2001;414:317–322. doi: 10.1038/35104575. [DOI] [PubMed] [Google Scholar]
- 52.Yik JH, Chen R, Nishimura R, Jennings JL, Link AJ, Zhou Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell. 2003;12:971–982. doi: 10.1016/s1097-2765(03)00388-5. [DOI] [PubMed] [Google Scholar]
- 53.Michels AA, Fraldi A, Li Q, Adamson TE, Bonnet F, Nguyen VT, Sedore SC, Price JP, Price DH, Lania L, et al. Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J. 2004;23:2608–2619. doi: 10.1038/sj.emboj.7600275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen R, Liu M, Li H, Xue Y, Ramey WN, He N, Ai N, Luo H, Zhu Y, Zhou N, et al. PP2B and PP1alpha cooperatively disrupt 7SK snRNP to release P-TEFb for transcription in response to Ca2+ signaling. Genes Dev. 2008;22:1356–1368. doi: 10.1101/gad.1636008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- • 55.Cho S, Schroeder S, Kaehlcke K, Kwon HS, Pedal A, Herker E, Schnoelzer M, Ott M. Acetylation of cyclin T1 regulates the equilibrium between active and inactive P-TEFb in cells. EMBO J. 2009;28:1407–1417. doi: 10.1038/emboj.2009.99. This paper provides the first evidence that the acetylation of cyclin T1 may provide an important signal regulating the stability of the large inactive 7SK snRNP complex which is used to store P-TEFb in an inactive form in cells. Since P-TEFb levels are highly restricted in resting memory CD4+ T-cells learning how P-TEFb is regulated is an an essential step in developing activators of latent proviruses.
- 56.Zhang Z, Klatt A, Gilmour DS, Henderson AJ. Negative elongation factor NELF represses human immunodeficiency virus transcription by pausing the RNA polymerase II complex. J. Biol. Chem. 2007;282:16981–16988. doi: 10.1074/jbc.M610688200. [DOI] [PubMed] [Google Scholar]
- 57.Ping Y-H, Rana TM. DSIF and NELF interact with RNA polymerase II elongation complex and HIV-1 Tat stimulates P-TEFb-mediated phosphorylation of RNA polymerase II and DSIF during transcription elongation. J. Biol. Chem. 2001;276:12951–12958. doi: 10.1074/jbc.M006130200. [DOI] [PubMed] [Google Scholar]
- 58.Kinoshita S, Su L, Amano M, Timmerman LA, Kaneshima H, Nolan GP. The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity. 1997;6:235–244. doi: 10.1016/s1074-7613(00)80326-x. [DOI] [PubMed] [Google Scholar]
- 59.Stroud JC, Oltman A, Han A, Bates DL, Chen L. Structural basis of HIV-1 activation by NF-κB - a higher order complex of p50:RelA bound to the HIV-1 LTR. J. Mol. Biol. 2009;393:98–112. doi: 10.1016/j.jmb.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Giffin MJ, Stroud JC, Bates DL, von Koenig KD, Hardin J, Chen L. Structure of NFAT1 bound as a dimer to the HIV-1 LTR κB element. Nature Struct. Biol. 2003;10:800–806. doi: 10.1038/nsb981. [DOI] [PubMed] [Google Scholar]
- 61.Lusic M, Marcello A, Cereseto A, Giacca M. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 2003;22:6550–6561. doi: 10.1093/emboj/cdg631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Benkirane M, Chun RF, Xiao H, Ogryzko VV, Howard BH, Nakatani Y, Jeang K-T. Activation of integrated provirus requires histone acetyltransferase: p300 and p/CAF are coactivators for HIV-1 Tat. J. Biol. Chem. 1998;273:24989–24905. doi: 10.1074/jbc.273.38.24898. [DOI] [PubMed] [Google Scholar]
- 63.Hottiger MO, Nabel GJ. Interaction of human immunodeficiency virus type 1 Tat with the transcriptional coactivators p300 and CREB binding protein. J. Virol. 1998;72:8252–8256. doi: 10.1128/jvi.72.10.8252-8256.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marzio G, Tyagi M, Gutierrez MI, Giacca M. HIV-1 Tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc. Natl. Acad. Sci. U S A. 1998;95:13519–13524. doi: 10.1073/pnas.95.23.13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mahmoudi T, Parra M, Vries RG, Kauder SE, Verrijzer CP, Ott M, Verdin E. The SWI/SNF chromatin-remodeling complex is a cofactor for tat transactivation of the HIV promoter. J. Biol. Chem. 2006;281:19960–19968. doi: 10.1074/jbc.M603336200. [DOI] [PubMed] [Google Scholar]
- 66.Chen LF, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kB action regulated by reversible acetylation. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 67.Lu T, Jackson MW, Wang B, Yang M, Chance MR, Miyagi M, Gudkov AV, Stark GR. Regulation of NF-κB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc. Natl. Acad. Sci. U S A. 2010;107:46–51. doi: 10.1073/pnas.0912493107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.García-Rodríguez C, Rao A. Nuclear Factor of Activated T Cells (NFAT)- dependent Transactivation Regulated by the Coactivators p300/CREB-binding Protein (CBP) J. Exptl. Med. 1998;187:2031–2036. doi: 10.1084/jem.187.12.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Robichaud GA, Barbeau B, Fortin J-F, Rothstein DM, Tremblay MJ. Nuclear factor of activated T cells is a driving force for preferential productive HIV-1 infection of CD45RO-expressing CD4+ T cells. J. Biol. Chem. 2002;277:23733–23741. doi: 10.1074/jbc.M201563200. [DOI] [PubMed] [Google Scholar]
- 70.Kinoshita S, Chen BK, Kaneshima H, Nolan GP. Host control of HIV-1 parasitism in T cells by the nuclear factor of activated T cells. Cell. 1998;95:595–604. doi: 10.1016/s0092-8674(00)81630-x. [DOI] [PubMed] [Google Scholar]
- 71.Brooks DG, Arlen PA, Gao L, Kitchen CM, Zack JA. Identification of T cell-signaling pathways that stimulate latent HIV in primary cells. Proc. Natl. Acad. Sci. U S A. 2003;100:12955–12960. doi: 10.1073/pnas.2233345100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thibault S, Imbeault M, Tardif MR, Tremblay MJ. TLR5 stimulation is sufficient to trigger reactivation of latent HIV-1 provirus in T lymphoid cells and activate virus gene expression in central memory CD4+ T cells. Virology. 2009;389:20–25. doi: 10.1016/j.virol.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 73.Wolschendorf F, Duverger A, Jones J, Wagner FH, Huff J, Benjamin WH, Saag MS, Niederweis M, Kutsch O. Hit-and-Run Stimulation: A Novel Concept to Reactivate Latent HIV-1 Infection Without Cytokine Gene Induction. J. Virol. 2010;84:8712–8720. doi: 10.1128/JVI.00523-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Williams SA, Kwon H, Chen LF, Greene WC. Sustained induction of NF-κB is required for efficient expression of latent human immunodeficiency virus type 1. J. Virol. 2007;81:6043–6056. doi: 10.1128/JVI.02074-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •• 75.He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, Krogan NJ, Alber T, Zhou Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell. 2010;38:428–438. doi: 10.1016/j.molcel.2010.04.013. This paper presents detailed proteomic studies demonstrating that HIV Tat not only associates with P-TEFb but also helps to recruit the transcription elongation factor ELL. The interactions between Tat and ELL are mediated by the AFF4 protein. The recognition that Tat stimulates elongation through more than one mechanism came as a complete surprise to the field and represents a major advance in our understanding of elongation control mechanisms.
- •• 76.Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, Benkirane M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell. 2010;38:439–451. doi: 10.1016/j.molcel.2010.04.012. This paper, which appears simultaneously with the work of He et al., provides additional evidence for Tat association with large complexes containing transcription factors in addition to P-TEFb. Furthermore the paper identifies a previously undetected complex of Tat, P-TEFb and 7SK RNA that lacks HEXIM1. This complex may represent an important pool of partially activated P-TEFb in the cell.
- 77.Chun T-W, Engel D, Berrey MM, Shea T, Corey L, Fauci AS. Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV-1 infection. Proc. Natl. Acad. Sci. U S A. 1998;95:8869–8873. doi: 10.1073/pnas.95.15.8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ, Siliciano RF. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003;9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 79.Ramratnam B, Mittler JE, Zhang L, Boden D, Hurley A, Fang F, Macken CA, Perelson AS, Markowitz M, Ho DD. The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy. Nat. Med. 2000;6:82–85. doi: 10.1038/71577. [DOI] [PubMed] [Google Scholar]
- 80.Chun T-W, Nickle DC, Justement JS, Large D, Semerjian A, Curlin ME, O’Shea MA, Hallahan CW, Daucher M, Ward DJ, et al. HIV-infected individuals receiving effective antiviral therapy for extended periods of time continually replenish their viral reservoir. J. Clin. Invest. 2005;115:3250–3255. doi: 10.1172/JCI26197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel MR, Ghattas G, Brenchley JM, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009;15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dinoso JB, Kim SY, Wiegand AM, Palmer SE, Gange SJ, Cranmer L, O’Shea A, Callender M, Spivak A, Brennan T, et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc. Natl. Acad. Sci. U S A. 2009;106:9403–9408. doi: 10.1073/pnas.0903107106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Richman DD, Margolis DM, Delaney M, Greene WC, Hazuda D, Pomerantz RJ. The challenge of finding a cure for HIV infection. Science. 2009;323:1304–1307. doi: 10.1126/science.1165706. [DOI] [PubMed] [Google Scholar]
- 84.Trono D, Van Lint C, Rouzioux C, Verdin E, Barre-Sinoussi F, Chun TW, Chomont N. HIV persistence and the prospect of long-term drug-free remissions for HIV-infected individuals. Science. 2010;329:174–180. doi: 10.1126/science.1191047. [DOI] [PubMed] [Google Scholar]
- 85.Savarino A, Mai A, Norelli S, El Daker S, Valente S, Rotili D, Altucci L, Palamara AT, Garaci E. “Shock and kill” effects of class I-selective histone deacetylase inhibitors in combination with the glutathione synthesis inhibitor buthionine sulfoximine in cell line models for HIV-1 quiescence. Retrovirology. 2009;6:52. doi: 10.1186/1742-4690-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bowman MC, Archin NM, Margolis DM. Pharmaceutical approaches to eradication of persistent HIV infection. Expert Rev. Mol. Med. 2009;11:e6. doi: 10.1017/S1462399409000970. [DOI] [PubMed] [Google Scholar]