

Abstract
Celiac disease (CD) is an intolerance to dietary proteins of wheat, barley, and rye. CD may have substantial morbidity, yet it is quite common with a prevalence of 1%–2% in Western populations. It is not clear why the CD phenotype is so prevalent despite its negative effects on human health, especially because appropriate treatment in the form of a gluten-free diet has only been available since the 1950s, when dietary gluten was discovered to be the triggering factor. The high prevalence of CD might suggest that genes underlying this disease may have been favored by the process of natural selection. We assessed signatures of selection for ten confirmed CD-associated loci in several genome-wide data sets, comprising 8154 controls from four European populations and 195 individuals from a North African population, by studying haplotype lengths via the integrated haplotype score (iHS) method. Consistent signs of positive selection for CD-associated derived alleles were observed in three loci: IL12A, IL18RAP, and SH2B3. For the SH2B3 risk allele, we also show a difference in allele frequency distribution (Fst) between HapMap phase II populations. Functional investigation of the effect of the SH2B3 genotype in response to lipopolysaccharide and muramyl dipeptide revealed that carriers of the SH2B3 rs3184504∗A risk allele showed stronger activation of the NOD2 recognition pathway. This suggests that SH2B3 plays a role in protection against bacteria infection, and it provides a possible explanation for the selective sweep on SH2B3, which occurred sometime between 1200 and 1700 years ago.
Main Text
Celiac disease (CD; MIM 212750) is a common intestinal inflammatory disorder resulting from intolerance to gluten, a major dietary protein of wheat, and related proteins from barley and rye. CD is the most common food intolerance in the Western world, where it affects 1%–2% of the population.1 It is also common in North Africa, India, and the Middle East.1,2 The highest prevalence of CD has been observed in the Saharawi from North Africa, where it affects 5.6% of the population. The clinical presentation of CD can vary from a classical gastrointestinal form, characterized by diarrhea, anemia, and weight loss, to a more systemic form, presenting with osteoporosis, autoimmune disease, and low fertility. Mortality in both pediatric and adult CD patients is significantly increased, especially in undiagnosed and untreated individuals.3–6
Susceptibility to CD has a strong genetic basis. The recurrence risk for siblings of CD patients to develop the disease is about 20 times higher than in the general population, and concordance between monozygotic twins is more than 80%.7 The strongest genetic risk factors are the HLA-DQ2 or HLA-DQ8 haplotypes.8 Genome-wide association studies (GWASs) and their replications recently led to the discovery of some 40 non-HLA loci.9–12 To date, CD is among the best-elucidated complex diseases; approximately 50% of its genetic susceptibility has been determined and can now be explained by association to this set of common HLA and non-HLA genetic variants.
The CD phenotype clearly could have had negative effects on fitness, given that appropriate treatment in the form of a gluten-free diet has only been available since the 1950s, when dietary gluten was discovered to be the triggering factor. Despite its negative effects on human health, the CD phenotype is quite common. Evolutionary processes such as mutations, migration, genetic drift, and natural selection have shaped the pattern of genetic variation in Homo sapiens. Most of the genetic variation is generally argued to have evolved largely under neutrality.13–15 The high prevalence of CD could therefore be the result of drift and purifying selection on its underlying genes. Alternatively, the process of natural selection may have favored genes underlying this disease given that CD is quite common, not just in a single population where it might have resulted from a bottleneck and genetic drift, but also in populations from different continents.
When a genetic variant is under positive selection, it increases in prevalence in a population and this leaves a “signature,” or pattern, in the human genome. These signatures can be identified by comparing them with the background distribution of genetic variation in humans. The recently identified CD susceptibility variants, in combination with the available genome-wide SNP data, provide the opportunity to study whether genetic variants underlying this disease have been favored by positive natural selection. We used existing data from a recently performed GWAS in five different populations, comprising 8154 controls from four European populations (UK, Dutch, Italian, and Finnish) and 195 founder individuals from Saharawi (N. Africa) CD families, to examine whether CD susceptibility loci show signs of recent positive selection by studying haplotype lengths with the Integrated Haplotype Score (iHS) method.15 To provide insight into the genetic structure and evolutionary dynamics between populations, we used the fixation index (Fst) to investigate the variance in allele frequency among populations.16 For our analysis, we selected the SNPs that were most strongly associated with the disease from each of the first published ten non-HLA loci that have been shown to be reproducibly associated with CD in several independent studies.9–11,17–20 We included a single SNP for nine of the loci (Table 1) and two independently associated SNPs (r2 = 0.101 in CEU [Utah residents with ancestry from northern and western Europe]) for the IL12A (MIM 161560) locus (Table 1). European samples were genotyped on Custom Illumina Human 670-Quad slides, which included all SNPs present on Hap550 plus 120k CNV probes (detailed quality control steps described elsewhere12). The Saharawi families were genotyped on an Illumina Human 610-Quad platform, which includes the same 550,000 probes as the Illumina Human 670-Quad slides. Only founder individuals from the Saharawi families were included in the analysis. The studies were approved by the medical-ethics committees of participating universities. The number of genotyped individuals from the five populations included in the analysis is indicated in Table S1, available online.
Table 1.
Characteristics of the Alleles Associated with Celiac Disease
| Chromosome | SNP ID | Gene | Associated Allele | Ancestral or Derived | Derived-Allele Frequency | _iHScorr (UK) | p Value iHS in European (UK) Population | Average Age of the Sweep (Europeans) | Derived-Allele Frequency Saharawi | _iHScorr Saharawi | p Value iHS Saharawi |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | rs6441961 | CCR2_3 | A | derived | 0.297 | −0.728 | 0.467 | n/a | 0.249 | −0.422 | 0.673 |
| 3 | rs9811792 | IL12A | G | derived | 0.46 | −1.157 | 0.247 | n/a | 0.344 | −1.174 | 0.240 |
| 3 | rs17810546 | IL12A, SCHIP1 | G | derived | 0.128 | −3.321 | 0.0009 | ∼2500 yr | 0.06 | n/a | n/a |
| 2 | rs917997 | IL18RAP | A | derived | 0.238 | −2.036 | 0.042 | ∼6500 yr | 0.154 | −1.703 | 0.089 |
| 4 | rs13151961 | IL2, IL21 | A | ancestral | 0.167 | −0.521 | 0.603 | n/a | 0.02 | n/a | n/a |
| 3 | rs1464510 | LPP | A | derived | 0.435 | 0.668 | 0.504 | n/a | 0.313 | 1.104 | 0.270 |
| 2 | rs842647 | REL | A | ancestral | 0.348 | −1.022 | 0.307 | n/a | 0.136 | −0.512 | 0.608 |
| 1 | rs2816316 | RGS1 | A | derived | 0.822 | 0.042 | 0.966 | n/a | 0.756 | 0.106 | 0.915 |
| 12 | rs3184504 | SH2B3 | A | derived | 0.468 | −2.214 | 0.027 | ∼1500 yr | 0.151 | −1.224 | 0.221 |
| 6 | rs1738074 | TAGAP | A | ancestral | 0.577 | −1.499 | 0.134 | n/a | 0.531 | −1.425 | 0.154 |
| 6 | rs2327832 | TNFAIP3 | G | derived | 0.23 | −1.531 | 0.126 | n/a | 0.249 | −0.616 | 0.538 |
p values calculated from iHS scores in the UK and the Saharawi populations, and a crude estimation of the average age of the selective sweep for alleles that show signs of selection in European populations. The OMIM information for genes is as follows: CCR2_3 (MIM 601267 and 601268), SCHIP1 (MIM 611622), LPP (MIM 600700), REL (MIM 164910), RGS1 (MIM 600323), TAGAP (MIM 609667), and TNFAIP3 (MIM 191163).
When an allele is under positive selection, its frequency rises rapidly in the population over a short time span and the haplotype carrying the advantageous allele will be longer relative to haplotypes around equally frequent alleles that have become common purely by random genetic drift. To study whether the CD loci are located in a genomic region with longer-than-expected haplotype lengths, we used the iHS statistic.15 Genotype data for all SNPs within a 4 Mb region around the CD susceptibility alleles were extracted from the genotyped control GWAS data sets. The Beagle software program was used to phase haplotypes from genotypes.21 On the basis of chimpanzee alignment, we assigned an ancestral state to all the SNPs in the data files; all the CD susceptibility alleles had known ancestral states. We used the iHS software (available online) to calculate extended haploblocks around the CD susceptibility loci in the genome-wide SNP data sets of all control samples (representing the general population). A positive iHS score means that haplotypes on the ancestral allele background are longer than the derived-allele background, whereas a negative iHS score means that the haplotypes on the derived-allele background are longer than the haplotypes associated with the ancestral allele. We standardized the iHS values by using derived frequency bins in a set of ∼5,000 randomly chosen SNPs surrounding the CD susceptibility regions but located at least 500 kb away from the associated SNP (the 500 kb distance was selected to make sure that all SNPs used for standardization were not in linkage disequilibrium with CD-associated SNPs). The standardization was performed separately in each population with control data sets for the European populations and all the founder samples for the Saharawi population. After standardization, the iHS distribution was normal with a mean of 0.00015 and a standard deviation of 0.9996. We calculated the p value with a two-sided test based on the normal distribution of the iHS values. To study signs of recent selection around the CD susceptibility loci in HapMap phase II, we used the web-based tool Haplotter.15
The haplotype structures and iHS values were similar for the four European populations. Table 1 presents the iHS values for the UK controls (which form the largest European population) and for the Saharawi. The results of each separate group are presented in Table S1. In the Saharawi population, the iHS score could be calculated for nine of the 11 SNPs. The iHS values could not be calculated for both rs13151961 (IL2/IL21 locus [MIM 147680/MIM 605348]) and rs17810546 (IL12A locus) because of a low minor-allele frequency.
In the four European populations, we observed consistent and significant signs of positive selection for three of the CD-associated alleles: rs17810546∗G (IL12A locus), rs917997∗A (IL18RAP locus [MIM 604509]), and rs3184504∗A (SH2B3 locus [MIM 605093]). For all three loci, the derived allele showed a signature of positive selection and this allele was also the CD susceptible allele (i.e., risk allele) (Table 1, Table S1). In the Saharawi population, we observed similar signs of positive selection for rs3184504∗A (SH2B3 locus) and rs917997∗A (IL18RAP locus).
The strongest signatures of selection were observed for rs17810546∗G from the IL12A locus (iHS between −2.923 and −3.434; p values between 0.0035 and 0.0006). For estimation of the age of the selective sweep (a crude estimate of the age of expansion of the derived variant), we first calculated the extended haplotype homozygosity (EHH)13 for the subset of chromosomes carrying the CD risk allele. To estimate the age, we assumed a star phylogeny of the haplotypes. The recombination distance r is the distance in cM between the points where EHH = x to the left and to the right of the core SNP. For a chosen x, r can be obtained from the data. When both x and r are then known, the generation time g can be calculated as g = (ln x / –r)∗100. Assuming an average generation length of 25 years, the age of the selective sweep equals 25g. For this study, we calculated r for the point where EHH has dropped to 0.30 (support interval EHH = 0.25 – EHH = 0.35). We estimated the age of selective sweep for the IL12A rs17810546∗G to be in the range of 2000–2500 years ago for all four European populations (Table S2A, Figure S1A).
The associated variant from the IL18RAP locus, rs917997∗A, showed a borderline-significant signature of selection in the European populations (iHS between −1.383 and −2.036; p values between 0.17 and 0.04) and in the Saharawi population (iHSSaharawi = −1.703; p = 0.089) (Table 1, Table S1). The frequency of the rs917997∗A risk allele varied from 15% in the Saharawi population to 19%–24% in the four European populations. Signs of selection for rs917997∗A were also observed in Asian HapMap samples (iHSAZN_HapMap = −2.115) (Table S1). The age of a selective sweep of rs917997∗A in the European populations was estimated to be around 6000 years ago (Table S2B, Figure S1B). The rs917997 genotype is strongly correlated with IL18RAP expression and has the lowest level of expression for carriers homozygous for the risk allele (Figure 1A). Such a cis-regulatory variant may lead to individuals having different IL18-mediated innate immune responses to infection. Interestingly, IL18RAP also confers susceptibility for Crohn's disease.22
Figure 1.
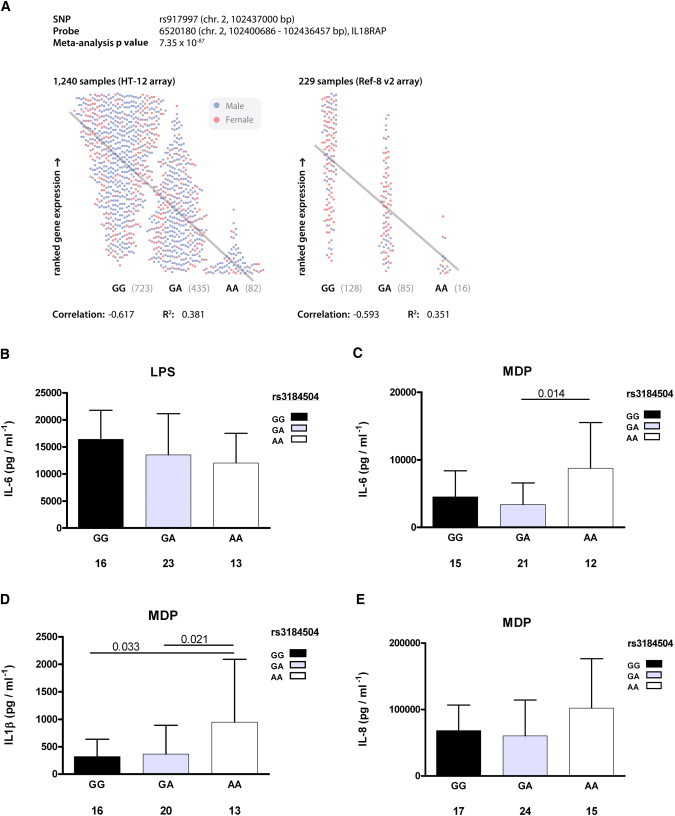
Functional Consequences of IL18RAP and SH2B3 Genotypes
(A) Individual gene expression correlation of IL18RAP with rs917997 from 1469 PAXgene samples (1240 hybridized to Illumina HT-12 arrays; 229 hybridized to Illumina Ref-8 v2 arrays). Spearman correlation coefficients are shown for HT-12 and Ref-8 v2 data, and meta-analysis p value results are shown (L.F., unpublished data).
(B) Differences between the SH2B3 genotype after LPS stimulation show a moderately reduced IL-6 cytokine production in PBMCs isolated from individuals heterozygous for the celiac risk allele.
(C and D) After MDP stimulation, a specific ligand of the NOD2 innate immune receptor, homozygous individuals carrying the risk allele showed an increased production of the IL-6 and IL1-β cytokines.
(E) IL-8 production in these individuals also showed an elevated production of the cytokine.
All data in panels (B)–(E) are presented as means and standard deviation (SD). The total number of individuals for each SH2B3 genotype is given under each bar and only the significant differences are included (Mann-Whitney U test).
The haplotype containing the SH2B3 rs3184504∗A allele showed consistent signs of positive selection in all European populations (maximum IHSIT = −2.597, p = 0.009) (Table 1, Table S1, Figure 2, Figure S1C). In the Saharawi population, the rs3184504∗A was also located on an extremely extended haplotype (Figure S1C); however, because of the lower allele frequency of this allele (MAF 0.15 in Saharawi versus MAF 0.40–0.49 in European populations), the iHS p value was not significant after correction for allele frequency. The age of a selective sweep in SH2B3 was estimated to be in the range of 1200–1700 years ago in the European populations (Table S2C). The haplotype containing the SH2B3∗A allele is associated with many diseases, including several immune-related diseases (CD, type 1 diabetes, and rheumatoid arthritis) and metabolic disorders (hypertension and myocardial infarction).23–27
Figure 2.
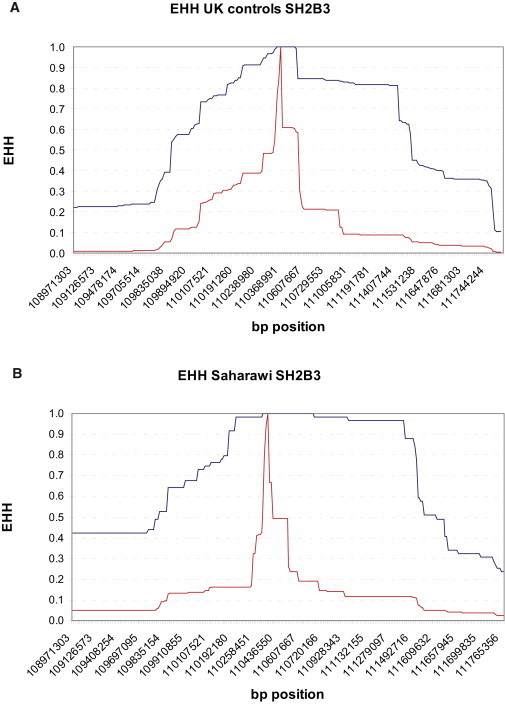
Example of EHH Plot of SH2B3 rs3184504 SNP in European and Saharawi Populations
(A) European (UK controls) population.
(B) Saharawi population.
Blue shows the derived haplotype; red shows the ancestral haplotype. Included are 1.4 Mb left and right from rs3184504. EHH plots for all the selected SNPs and all the studied populations are presented in Figure S1A–S1C.
When a genetic variation is under positive selection, it increases in prevalence in a population. Because diet, climate, and pathogen load vary across the world, there are population differences in selective pressure resulting in global allele frequency variations. Therefore, allele frequency differences between populations could indicate that the alleles show signs of selection in a certain population (although it could also point toward a population bottleneck). The Fst is a measure of population differentiation based on data of genetic variation, and the statistic compares the genetic variability within and between populations.28 Without selection, allele frequency differences between populations are the result of random genetic drift, which affects all SNPs in the population in a similar way. We studied the Fst values of the CD susceptibility loci in the HapMap II populations and compared these values with an empirical genome-wide distribution.29 Fst is directly related to the variance in allele frequency among populations and, conversely, to the degree of resemblance among individuals within populations. If Fst is small, it means that the allele frequencies within each population are similar; if it is large, it means that the allele frequencies are different.16 The SH2B3 risk allele had an Fst value of 0.61, which was a significant outlier compared to a genome-wide distribution (p value < 0.05). This allele shows relatively large between-population frequency differences, which could be a sign of differential selection in HapMap II populations (Table 2). The worldwide allele frequency distribution of rs3184504 in SH2B3 in the Human Diversity Project Data is shown in Figure 3.
Table 2.
Fst Measure of Population Differentiation
| Chromosome | SNP ID | Gene | Associated Allele | Ancestral or Derived | Fst HapMapII |
|---|---|---|---|---|---|
| 3 | rs6441961 | CCR2_3 | A | derived | 0.19 |
| 3 | rs9811792 | IL12A | G | derived | 0.18 |
| 3 | rs17810546 | IL12A, SCHIP1 | G | derived | 0.20 |
| 2 | rs917997 | IL18RAP | A | derived | 0.27 |
| 4 | rs13151961 | IL2, IL21 | A | ancestral | 0.31 |
| 3 | rs1464510 | LPP | A | derived | 0.21 |
| 2 | rs842647 | REL | A | ancestral | 0.46 |
| 1 | rs2816316 | RGS1 | A | derived | 0.00 |
| 12 | rs3184504 | SH2B3 | A | derived | 0.61a |
| 6 | rs1738074 | TAGAP | A | ancestral | 0.09 |
| 6 | rs2327832 | TNFAIP3 | G | derived | 0.18 |
Significant outlier (p < 0.05) compared with an empirical genome-wide distribution.
Figure 3.
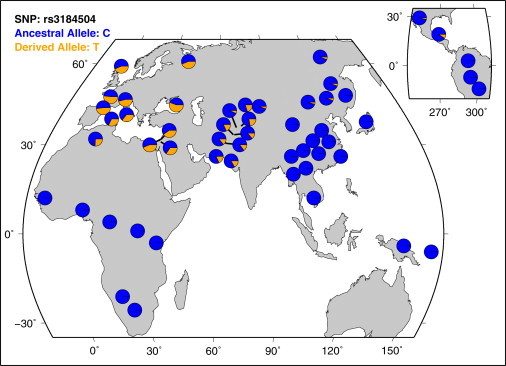
Worldwide Allele Frequency Distribution of rs3184504 in SH2B3
The derived allele is the CD risk allele that shows a signature of recent selection in our European study populations. In this figure, the rs3184504 alleles are annotated on the reverse strand.
An important question concerns the mechanism that underlies the signatures of selection of these gene variants. It is tempting to speculate that the same alleles that predispose to autoimmune diseases might be protective against infections—the major cause of mortality in the past. An example of the interplay between predisposition to autoimmunity and infections has recently been shown for the FCGRIIb (MIM 604590) gene polymorphism rs1050501, which is associated to susceptibility to systemic lupus erythematosus (SLE, [MIM 152700]) and protection against malaria.30 It is interesting to note that two of the genes identified in our study (IL12A and IL18RAP) are involved in the activation of proinflammatory cytokine pathways, and the phenotypic effect of the selected variants most likely involves modulation of cytokine responses. Cytokine responses are one of the main host defense mechanisms during infections, which exert a major selective pressure on the genes of the immune system during history. SH2B3, the third gene identified to show signs of recent positive selection, contains an SH2 domain, which is common to master regulatory genes of innate immunity (such as SOCS genes).31 Given that an SH2B3 variant is associated with several autoimmune and metabolic disorders, we hypothesized that it also might play a central role in the cytokine responses. To test this hypothesis, we investigated genotype differences in inflammatory cytokine responses (IL-6, IL-8, and IL1-β). Venous blood was drawn from 56 European individuals from the Netherlands from whom we obtained informed consent. Peripheral blood mononuclear cells (PBMCs) were isolated and resuspended in RPMI-1640 medium and adjusted to 5 × 106 cells/ml. A volume of 100 μl was added to round-bottom 96-well plates (Greiner) and incubated with 100 μl of culture medium (negative control) or various stimuli. Stimuli added to the PBMCs were lipopolysaccharide (LPS, 10 ng/ml), muramyl dipeptide (MDP, 10 μg/ml), or Pam3Cys (10 μg/ml). IL-6, IL-8, and IL1-β were measured by commercial ELISA kits (Sanquin, Amsterdam, The Netherlands). The genotype frequencies were tested for Hardy-Weinberg equilibrium with a χ2 test for goodness of fit. Association between genotypes (as the independent variable) and IL-6, IL-8, and IL1β production (as dependent variables) was determined with the Mann-Whitney U test. We also performed trend analyses to test for a dose-response effect for the CD risk alleles. For this analysis, we used log-transformed data, because the cytokine production levels were not normally distributed among the tested individuals.
LPS stimulation revealed a moderately (albeit nonsignificantly) decreased cytokine production in heterozygous rs3184504 individuals compared to individuals homozygous for the nonrisk allele G (Figure 1B). A much more striking difference in cytokine production was obtained after cell stimulation with MDP, a component of the peptidoglycans present in all bacteria cell walls: the production of the proinflammatory cytokines and IL1-β was 3- to 5-fold higher in homozygous AA individuals, i.e., individuals homozygous for the CD risk allele, compared to individuals homozygous for the nonrisk G allele (Figure 1D). A similar trend was observed for IL-6 and IL-8 (Figures 1C and 1E). We observed a dose-response relationship of the risk-allele A with IL1β production (p = 0.034 for trend), meaning that IL1β production was lowest in individuals carrying two nonrisk G alleles and that it increased with each extra risk allele (Figure 1D).
Molecules that contain an SH2 domain in their structure, like SH2B3, are known to modulate intermolecular interactions and to inhibit cytokine responses.32 Stimulation of PBMCs with MDP, a specific ligand of the pattern-recognition receptor NOD2, shows that cells isolated from individuals homozygous for the SH2B3 CD risk allele display an increased proinflammatory cytokine production. This suggests that the SH2B3 protein has an inhibiting function on the MDP-NOD2-RIP2 signaling pathway, and this inhibition is diminished in individuals carrying the SH2B3 risk allele. The increased cytokine production observed in these individuals is in line with the interaction of SH2B3 with the ERK1/2 and p38MAPK pathways33,34; this interaction in turn mediates NOD2-induced IL1-β production.35 These functional consequences of different SH2B3 gene variants suggest, on the one hand, a possible mechanism of how this polymorphism contributes to the increased risk of developing immune-related diseases and, on the other hand, that the cause of the signature of positive selection should be sought in improved host defense against infections. The improved response to bacterial ligands, followed by positive selection, is reminiscent of the similar observation reported on the selection of TIRAP/Mal (MIM 606252) variants, an important adaptor molecule for the innate immune responses.36
In summary, we have demonstrated signs of positive selection for three common loci associated with CD. Our study of SH2B3 reveals the function of the protein in the innate immune response and provides a possible explanation for its signature of positive selection. The specific pressure that influenced the selective sweep 1200–1700 years ago was most likely an infectious disease. Given that NOD2 is known to be an important receptor for bacterial pathogens,37 it is tempting to speculate that SH2B3 is protective during strong bacterial infection pressures, but more functional studies are needed to prove this relationship.
Acknowledgments
The study was supported by the Celiac Disease Consortium, an Innovative Cluster approved by the Netherlands Genomics Initiative and partially funded by the Dutch Government, the Netherlands Organization for Scientific Research, EU STREP KP6, SenterNovem (IOP genomics), and the Wellcome Trust. We acknowledge use of DNA from the British 1958 Birth Cohort collection, funded by the UK Medical Research Council and the Wellcome Trust. G.T. was awarded a Ter Meulen Fund travel grant by the Royal Netherlands Academy of Arts and Sciences (KNAW). M.G.N. was supported by a VIDI grant from the Netherlands Organization for Scientific Research (NWO). P.C.D. is an MRC Clinical Training Fellow. We thank all the clinicians and Coeliac UK for their help in recruiting individuals for this study. The Finnish Celiac Disease Study Group is represented by Katri Kaukinen, Kalle Kurppa, and Markku Mäki. This study used data generated by the Wellcome Trust Case-Control Consortium 2 and resources provided by the Type 1 Diabetes Genetics Consortium, a collaborative clinical study sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institute of Allergy and Infectious Diseases (NIAID), National Human Genome Research Institute (NHGRI), National Institute of Child Health and Human Development (NICHD), and Juvenile Diabetes Research Foundation International (JDRF) and supported by U01 DK062418. We thank all the individuals who participated in the study. A full list of personal acknowledgements is given in the Supplemental Data.
Supplemental Information
Web Resources
The URLs for data presented herein are as follows:
Haplotter, http://hg-wen.uchicago.edu/selection/haplotter.htm
Human Diversity Project Data, http://hgdp.uchicago.edu/cgi-bin/gbrowse/HGDP/
iHS software, http://hgdp.uchicago.edu/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Catassi C., Fasano A. Celiac disease. Curr. Opin. Gastroenterol. 2008;24:687–691. doi: 10.1097/MOG.0b013e32830edc1e. [DOI] [PubMed] [Google Scholar]
- 2.Abu-Zekry M., Kryszak D., Diab M., Catassi C., Fasano A. Prevalence of celiac disease in Egyptian children disputes the east-west agriculture-dependent spread of the disease. J. Pediatr. Gastroenterol. Nutr. 2008;47:136–140. doi: 10.1097/MPG.0b013e31815ce5d1. [DOI] [PubMed] [Google Scholar]
- 3.Rubio-Tapia A., Kyle R.A., Kaplan E.L., Johnson D.R., Page W., Erdtmann F., Brantner T.L., Kim W.R., Phelps T.K., Lahr B.D. Increased prevalence and mortality in undiagnosed celiac disease. Gastroenterology. 2009;137:88–93. doi: 10.1053/j.gastro.2009.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Viljamaa M., Kaukinen K., Pukkala E., Hervonen K., Reunala T., Collin P. Malignancies and mortality in patients with coeliac disease and dermatitis herpetiformis: 30-year population-based study. Dig. Liver Dis. 2006;38:374–380. doi: 10.1016/j.dld.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Metzger M.H., Heier M., Mäki M., Bravi E., Schneider A., Löwel H., Illig T., Schuppan D., Wichmann H.E. Mortality excess in individuals with elevated IgA anti-transglutaminase antibodies: The KORA/MONICA Augsburg cohort study 1989-1998. Eur. J. Epidemiol. 2006;21:359–365. doi: 10.1007/s10654-006-9002-4. [DOI] [PubMed] [Google Scholar]
- 6.Solaymani-Dodaran M., West J., Logan R.F. Long-term mortality in people with celiac disease diagnosed in childhood compared with adulthood: A population-based cohort study. Am. J. Gastroenterol. 2007;102:864–870. doi: 10.1111/j.1572-0241.2007.01111.x. [DOI] [PubMed] [Google Scholar]
- 7.Greco L., Romino R., Coto I., Di Cosmo N., Percopo S., Maglio M., Paparo F., Gasperi V., Limongelli M.G., Cotichini R. The first large population based twin study of coeliac disease. Gut. 2002;50:624–628. doi: 10.1136/gut.50.5.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karell K., Louka A.S., Moodie S.J., Ascher H., Clot F., Greco L., Ciclitira P.J., Sollid L.M., Partanen J., European Genetics Cluster on Celiac Disease HLA types in celiac disease patients not carrying the DQA1∗05-DQB1∗02 (DQ2) heterodimer: Results from the European Genetics Cluster on Celiac Disease. Hum. Immunol. 2003;64:469–477. doi: 10.1016/s0198-8859(03)00027-2. [DOI] [PubMed] [Google Scholar]
- 9.Hunt K.A., Zhernakova A., Turner G., Heap G.A., Franke L., Bruinenberg M., Romanos J., Dinesen L.C., Ryan A.W., Panesar D. Newly identified genetic risk variants for celiac disease related to the immune response. Nat. Genet. 2008;40:395–402. doi: 10.1038/ng.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trynka G., Zhernakova A., Romanos J., Franke L., Hunt K.A., Turner G., Bruinenberg M., Heap G.A., Platteel M., Ryan A.W. Coeliac disease-associated risk variants in TNFAIP3 and REL implicate altered NF-kappaB signalling. Gut. 2009;58:1078–1083. doi: 10.1136/gut.2008.169052. [DOI] [PubMed] [Google Scholar]
- 11.van Heel D.A., Franke L., Hunt K.A., Gwilliam R., Zhernakova A., Inouye M., Wapenaar M.C., Barnardo M.C., Bethel G., Holmes G.K. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat. Genet. 2007;39:827–829. doi: 10.1038/ng2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dubois P.C., Trynka G., Franke L., Hunt K.A., Romanos J., Curtotti A., Zhernakova A., Heap G.A., Adány R., Aromaa A. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 2010;42:295–302. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sabeti P.C., Reich D.E., Higgins J.M., Levine H.Z., Richter D.J., Schaffner S.F., Gabriel S.B., Platko J.V., Patterson N.J., McDonald G.J. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- 14.Sabeti P.C., Schaffner S.F., Fry B., Lohmueller J., Varilly P., Shamovsky O., Palma A., Mikkelsen T.S., Altshuler D., Lander E.S. Positive natural selection in the human lineage. Science. 2006;312:1614–1620. doi: 10.1126/science.1124309. [DOI] [PubMed] [Google Scholar]
- 15.Voight B.F., Kudaravalli S., Wen X., Pritchard J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holsinger K.E., Weir B.S. Genetics in geographically structured populations: Defining, estimating and interpreting F(ST) Nat. Rev. Genet. 2009;10:639–650. doi: 10.1038/nrg2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romanos J., Barisani D., Trynka G., Zhernakova A., Bardella M.T., Wijmenga C. Six new coeliac disease loci replicated in an Italian population confirm association with coeliac disease. J. Med. Genet. 2009;46:60–63. doi: 10.1136/jmg.2008.061457. [DOI] [PubMed] [Google Scholar]
- 18.Garner C.P., Murray J.A., Ding Y.C., Tien Z., van Heel D.A., Neuhausen S.L. Replication of celiac disease UK genome-wide association study results in a US population. Hum. Mol. Genet. 2009;18:4219–4225. doi: 10.1093/hmg/ddp364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dema B., Martínez A., Fernández-Arquero M., Maluenda C., Polanco I., de la Concha E.G., Urcelay E., Núñez C. Association of IL18RAP and CCR3 with coeliac disease in the Spanish population. J. Med. Genet. 2009;46:617–619. doi: 10.1136/jmg.2009.067041. [DOI] [PubMed] [Google Scholar]
- 20.Amundsen S.S., Rundberg J., Adamovic S., Gudjónsdóttir A.H., Ascher H., Ek J., Nilsson S., Lie B.A., Naluai A.T., Sollid L.M. Four novel coeliac disease regions replicated in an association study of a Swedish-Norwegian family cohort. Genes Immun. 2010;11:79–86. doi: 10.1038/gene.2009.67. [DOI] [PubMed] [Google Scholar]
- 21.Browning S.R., Browning B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhernakova A., Festen E.M., Franke L., Trynka G., van Diemen C.C., Monsuur A.J., Bevova M., Nijmeijer R.M., van 't Slot R., Heijmans R. Genetic analysis of innate immunity in Crohn's disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am. J. Hum. Genet. 2008;82:1202–1210. doi: 10.1016/j.ajhg.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coenen M.J., Trynka G., Heskamp S., Franke B., van Diemen C.C., Smolonska J., van Leeuwen M., Brouwer E., Boezen M.H., Postma D.S. Common and different genetic background for rheumatoid arthritis and coeliac disease. Hum. Mol. Genet. 2009;18:4195–4203. doi: 10.1093/hmg/ddp365. [DOI] [PubMed] [Google Scholar]
- 24.Todd J.A., Walker N.M., Cooper J.D., Smyth D.J., Downes K., Plagnol V., Bailey R., Nejentsev S., Field S.F., Payne F., Genetics of Type 1 Diabetes in Finland. Wellcome Trust Case Control Consortium Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levy D., Ehret G.B., Rice K., Verwoert G.C., Launer L.J., Dehghan A., Glazer N.L., Morrison A.C., Johnson A.D., Aspelund T. Genome-wide association study of blood pressure and hypertension. Nat. Genet. 2009;41:677–687. doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newton-Cheh C., Johnson T., Gateva V., Tobin M.D., Bochud M., Coin L., Najjar S.S., Zhao J.H., Heath S.C., Eyheramendy S. Genome-wide association study identifies eight loci associated with blood pressure. Nat. Genet. 2009;41:666–676. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhernakova A., van Diemen C.C., Wijmenga C. Detecting shared pathogenesis from the shared genetics of immune-related diseases. Nat. Rev. Genet. 2009;10:43–55. doi: 10.1038/nrg2489. [DOI] [PubMed] [Google Scholar]
- 28.Lewontin R.C., Krakauer J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics. 1973;74:175–195. doi: 10.1093/genetics/74.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng F., Chen W., Richards E., Deng L., Zeng C. SNP@Evolution: A hierarchical database of positive selection on the human genome. BMC Evol. Biol. 2009;9:221. doi: 10.1186/1471-2148-9-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Willcocks L.C., Carr E.J., Niederer H.A., Rayner T.F., Williams T.N., Yang W., Scott J.A., Urban B.C., Peshu N., Vyse T.J. A defunctioning polymorphism in FCGR2B is associated with protection against malaria but susceptibility to systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA. 2010;107:7881–7885. doi: 10.1073/pnas.0915133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hilton D.J., Richardson R.T., Alexander W.S., Viney E.M., Willson T.A., Sprigg N.S., Starr R., Nicholson S.E., Metcalf D., Nicola N.A. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc. Natl. Acad. Sci. USA. 1998;95:114–119. doi: 10.1073/pnas.95.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pawson T. Specificity in signal transduction: From phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004;116:191–203. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- 33.Fitau J., Boulday G., Coulon F., Quillard T., Charreau B. The adaptor molecule Lnk negatively regulates tumor necrosis factor-alpha-dependent VCAM-1 expression in endothelial cells through inhibition of the ERK1 and -2 pathways. J. Biol. Chem. 2006;281:20148–20159. doi: 10.1074/jbc.M510997200. [DOI] [PubMed] [Google Scholar]
- 34.Simon C., Dondi E., Chaix A., de Sepulveda P., Kubiseski T.J., Varin-Blank N., Velazquez L. Lnk adaptor protein down-regulates specific Kit-induced signaling pathways in primary mast cells. Blood. 2008;112:4039–4047. doi: 10.1182/blood-2008-05-154849. [DOI] [PubMed] [Google Scholar]
- 35.Windheim M., Lang C., Peggie M., Plater L.A., Cohen P. Molecular mechanisms involved in the regulation of cytokine production by muramyl dipeptide. Biochem. J. 2007;404:179–190. doi: 10.1042/BJ20061704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khor C.C., Chapman S.J., Vannberg F.O., Dunne A., Murphy C., Ling E.Y., Frodsham A.J., Walley A.J., Kyrieleis O., Khan A. A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat. Genet. 2007;39:523–528. doi: 10.1038/ng1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferwerda B., McCall M.B., de Vries M.C., Hopman J., Maiga B., Dolo A., Doumbo O., Daou M., de Jong D., Joosten L.A. Caspase-12 and the inflammatory response to Yersinia pestis. PLoS ONE. 2009;4:e6870. doi: 10.1371/journal.pone.0006870. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.