

Abstract
Lymphedema is the clinical manifestation of defects in lymphatic structure or function. Mutations identified in genes regulating lymphatic development result in inherited lymphedema. No mutations have yet been identified in genes mediating lymphatic function that result in inherited lymphedema. Survey microarray studies comparing lymphatic and blood endothelial cells identified expression of several connexins in lymphatic endothelial cells. Additionally, gap junctions are implicated in maintaining lymphatic flow. By sequencing GJA1, GJA4, and GJC2 in a group of families with dominantly inherited lymphedema, we identified six probands with unique missense mutations in GJC2 (encoding connexin [Cx] 47). Two larger families cosegregate lymphedema and GJC2 mutation (LOD score = 6.5). We hypothesize that missense mutations in GJC2 alter gap junction function and disrupt lymphatic flow. Until now, GJC2 mutations were only thought to cause dysmyelination, with primary expression of Cx47 limited to the central nervous system. The identification of GJC2 mutations as a cause of primary lymphedema raises the possibility of novel gap-junction-modifying agents as potential therapy for some forms of lymphedema.
Main Text
Lymphedema is the abnormal accumulation of lymphatic fluid in interstitial space. Patients with lymphedema suffer from recurrent local infections, physical impairment, and cosmetic and psychosocial stigmatization and may be at increased risk for developing lymphangiosarcoma.1 The population prevalence of lymphedema is estimated in the range of 1.3–1.4 per 1000.2 Primary (inherited) lymphedema is less common than secondary lymphedema, which is associated with conditions such as filariasis, trauma, and cancer therapy. Recent studies in families with inherited forms of lymphedema have identified six genes, FLT43,4 (encoding VEGFR3) (MIM 153100), FOXC25,6 (MIM 153400), SOX187 (MIM 607823), HGF8 (MIM 142409), MET8 (MIM 164860), and CCBE19,10 (MIM 235510), causing lymphedema.
To identify other causal genes for lymphedema, we reviewed differential gene expression in lymphatic endothelial cells (LECs) versus blood endothelial cells (BECs) and noted that GJA1 (encoding connexin [Cx] 43) (MIM 121014) is expressed in BECs and LECs whereas GJC2 (encoding Cx47) (MIM 608803) is expressed only in LECs.11 Gap junctions are intercellular channels formed by hexamers of connexin proteins on adjoining cells that facilitate the electrical and metabolic coupling of cells within a tissue via a variety of mechanisms. Rhodin first suggested a role for gap junctions on lymphatic vessels,12 but there has been limited characterization of gap junction intercellular communication (GJIC) in lymphatic vessels or LECs.13,14
We investigated the connexins as potential genes for causal lymphedema mutations in the families ascertained through the University of Pittsburgh Lymphedema Family Study (UPLFS). This study was approved by the Institutional Review Board of the University of Pittsburgh, and informed consent was obtained from all subjects. Initially, families were ascertained by a physician's diagnosis of lymphedema in the proband (confirmed by medical records) and a lymphedema occurrence in a first-degree relative. We screened 150 probands from the UPLFS for mutations in GJA1 (chromosome 6q22-q23), GJA4 (chromosome 1p35.1) (MIM 121012), and GJC2 (chromosome 1q41-q42). Sequences were aligned and curated with Sequencher v4.7 (Gene Codes Corp.). Mutations in FLT4, FOXC2, and SOX18, known lymphedema genes, were previously excluded in these probands by bidirectional sequence analysis. The sequences of GJA4 (NM002060), GJA1 (NM000165), and GJC2 (NM020435) were downloaded from Entrez Nucleotide. Unique sequence amplification and sequencing primers (see Table S1 available online) were designed to amplify genes in overlapping fragments. These fragments were then sequenced in both directions with ABI BigDye v3.1 chemistry, and the products were resolved on an ABI 3730 DNA sequencer in the Genomics and Proteomics Core Laboratory of the University of Pittsburgh. Six lymphedema families of mixed European ancestry were identified with heterozygous dominant causal GJC2 mutations (Table 1).
Table 1.
GJC2 Mutations Observed in Primary Lymphedema Families
| Family | Sequence Substitution | Amino Acid Change | Predicted Domain |
|---|---|---|---|
| 337 | AGATCCACAACC(A>C)CTCCACCTTCGT | H19P | N-terminal |
| 135 | GAGGCCATCTACT(C>T)GGCGGAGCAGGCC | S48L | Extracellular loop 1 |
| 251 | CCACGCCGCGCGCCCC(G>A)GCGCACCTGCCG | R125Q | Intracellular loop |
| 104 | GAGGAGCCCATGCTG(G>A)GCCTGGGCGAGGAG | G149S | Intracellular loop |
| 168 | TGCTTCGTGTCG(C>T)GCCCTACTGAAAAG | R260C | Extracellular loop 2 |
| 151 | CCCGCGCCGCCCC(C>T)G CCCTGCGCCTTC | P316L | C-terminal |
We identified two GJC2 mutations in families suitable for linkage analysis: one cosegregating lymphedema and a C>T transition at nucleotide 143 leading to an S48L (family 135) substitution in extracellular loop 1 of Cx47, and another cosegregating lymphedema and a C>T transition at nucleotide 778 resulting in an R260C (family 168) substitution in extracellular loop 2 (Figure 1). Linkage analysis in these two families yielded a LOD score of 6.5 under a model of disease frequency = 0.0001, penetrance = 0.9, phenocopy rate = 0.0, assuming no recombination. The R260C mutation is located within the conserved SRPTEK motif, important for connexon docking. This motif is a target of peptide mimetic inhibitors of GJIC for Cx43 and Cx32.15,16 Four additional unique GJC2 mutations were observed in other, smaller families: H19P in the N-terminal domain, R125Q in the intracellular loop, G149S in the intracellular loop, and P316L in the C-terminal domain were transmitted from an affected parent to an affected child. Samples were not available from other family members, and these cases are consistent with, but not informative for, linkage. (Figure 2; Table 1).
Figure 1.
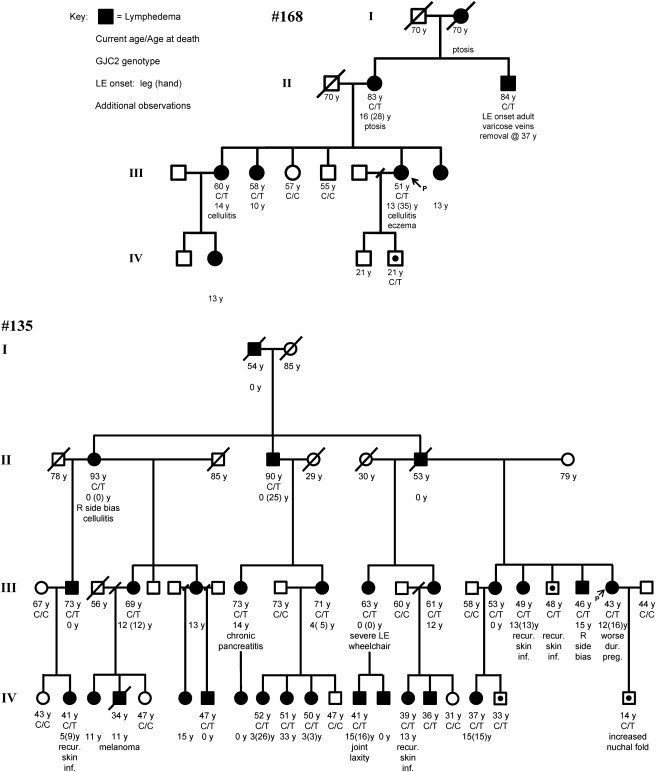
Pedigrees of the Two Linked Families
Pedigrees of the two linked families showing current age or age at death, cosegregation of GJC2 missense mutation with lymphedema, age at onset of lymphedema of the leg and/or hand, and other phenotypic features. Family 168, R260C, and family 135, S48L, are shown. Filled shapes indicate affected individuals with lymphedema. LOD = 6.5. Arrows indicates the probands.
Figure 2.
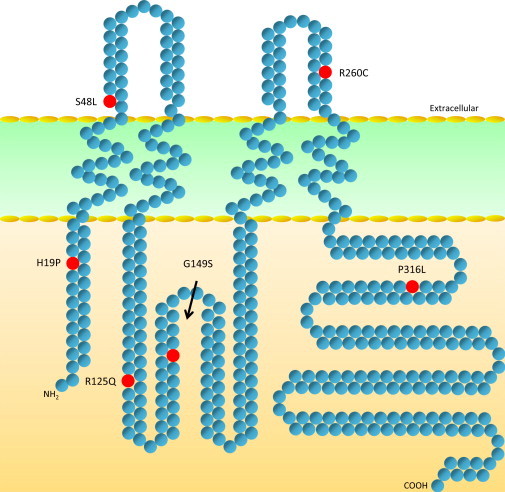
Schematic Drawing of Connexin 47 Protein Locating Lymphedema-Associated Amino Acid Substitutions
Domain break points and sequence are as described by Uhlenberg et al.22 and http://www.uniprot.org/uniprot/Q5T442.
GJC2 mutations occur only in affected or at-risk individuals, cause a change in a conserved amino acid of Cx47, and were not present in 250 sequenced, ethnically matched controls (0 of 500 alleles). These missense mutations affect amino acids highly conserved in mammalian evolution, showing only one variation of glycine to alanine in the case of the G149S mutation (Figure 3). Non-lymphedema-associated sequence variants were also identified (Table S2).
Figure 3.

Amino Acid Alignment of Cx47 from Different Species
Tan indicates intracellular domains; green indicates transmembrane domains; white indicates extracellular domains. Red dots represent the positions of amino acids altered in lymphedema families.
The current age or age at death, genotype with respect to GJC2, age at onset of lymphedema of the leg and/or hand, and other phenotypic features in the families demonstrating linkage are shown in Figure 1. Uncomplicated lymphedema of the leg or hand was the only constant feature reported in the affected individuals. Individual IV-20, family 135, was reported to have a nuchal fold at birth but was nonpenetrant for lymphedema. Many affected individuals had onset of lymphedema in childhood or adolescence. Individuals IV-4, family 168, and III-18, IV-19, and IV-20, family 135, were nonpenetrant males, showing reduced penetrance of GJC2 mutations in these families. Generally, males showed a later age at onset than females. Other features reported in some lymphedema pedigrees (ptosis, cellulitis, venous insufficiency, etc.) appeared sporadically in these families. Four individuals in family 135 reported recurrent skin infections. In the four smaller families with mutations, the clinical phenotypes were similar to the families demonstrating linkage, including a later age at onset.
Of note, two additional rare mutations, one leading to a truncated Cx47 protein (E44ter) and a 22 bp deletion leading to a truncation of the GJC2 protein at residue 30, were identified. These changes were not present in 500 control alleles but failed to segregate with disease in pedigrees. These early nonsense changes are predicted to code for a prematurely truncated polypeptide, leading to a null allele. The carriers of these truncation mutations showed no discernable phenotype, consistent with the Cx47-deficient mouse, in which heterozygous or homozygous null animals have no gross phenotype and no Cx47-specific developmental or functional abnormality.17,18
We show here that mutations in GJC2 cause primary lymphedema, through linkage in two families and significant genetic evidence from four independent families. We hypothesize that coordinated gap junction function is needed to optimize the conduction of lymph from the periphery to the thoracic duct and is compromised in individuals with GJC2 missense mutations. In vivo evidence in rat mesenteric lymphatics shows significant impairment of contraction propagation upon treatment with nonspecific gap junction inhibitors.13,14 The GJC2 mutations are notable because they support an abnormality in lymphatic function rather than the previously identified mutations in genes causing abnormal lymphatic development. Such functional abnormalities could potentially benefit from the current development of gap-junction-modifying drugs,19,20 offering a novel medical treatment for lymphedema.
The role of GJC2/Cx47 in lymphatic function is unexpected because it has a demonstrated primary role in the central nervous system (CNS), with expression reportedly limited to oligodendrocytes.17,21 Homozygous loss-of-function mutations in GJC2 cause Pelizaeus-Merzbacher-like disease (PMLD; MIM 608804), characterized by severe CNS dysmyelination.22,23 Neither individuals affected with PMLD nor their obligate heterozygous carriers of GJC2 mutations are reported to have a lymphatic phenotype, although the clinical phenotype of lymphedema is often subtle. Likewise, the clinical information available on our lymphedema patients and families would be insensitive to a mild clinical neurological abnormality. We observed no mutations in the transmembrane domains where many of the PMLD mutations are found.23
The GJC2 lymphedema mutations are distributed throughout the protein, with no geographical clustering. However, the two mutations located in the extracellular loop domains (i.e., S48L and R260C) are predicted to interfere with connexon (i.e., hemichannel) assembly into functional channels. The linked R260C mutation is located in a conserved SRPTEK motif important for connexon docking; the importance of this motif is further underscored by a homologous autosomal-dominant GJA1 mutation (R202H) identified in families with oculodentodigital dysplasia (MIM 164200), with functional characteristics of poor plaque formation and impaired dye transfer and electrical coupling.24,25 Similarly, we expect these two extracellular mutations to result in impaired channel activity and propose that this might result in impaired coordination of pulsatile lymphatic flow.14 The mechanism through which the identified intracellular mutations mediate their effects is not clear, especially in light of the more recent recognition that connexin function is not limited only to their well-recognized channel activity but may involve hemichannel function or changes in cell adhesion or motility.26–29 Further characterization of the mutations reported here, especially with regard to their predicted dominant-negative effect with wild-type Cx47 or transdominant effect with other endogenous connexins expressed in LECs, will contribute to our understanding of the role of connexins in lymphatic function.
Although we are approaching the 400th anniversary of the first description of the lymphatic system in 1627,30 lymphatics remain a poorly understood and largely neglected biological system. Despite an essential and active role in the maintenance of fluid homeostasis, trafficking of antigen-presenting and other immune cells, and pathological processes like chronic inflammation and cancer, the lymphatic vascular system is often viewed as a passive conduit rather than an interactive participant in these processes. The identification of mutations in the Cx gene family in primary lymphedema justifies further investigation into the manipulation of gap-junction-coordinated lymphatic flow and function as a possible therapeutic option for primary lymphedema.
Acknowledgments
We acknowledge the partial support of institutional funds from the Office of the Dean of the Graduate School of Public Health and the Office of the Senior Vice Chancellor of the University of Pittsburgh, the Competitive Medical Research Fund of the UPMC Health System, and National Institutes of Health grants R01 HL092866 and HD37243. We gratefully acknowledge the cooperation and generous assistance of the patients and families participating in the University of Pittsburgh Lymphedema Family Study.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
University of Pittsburgh Lymphedema Family Study, http://www.hgen.pitt.edu/projects/lymph/
Entrez Nucleotide, http://www.ncbi.nlm.nih.gov/nuccore/
Genomics and Proteomics Core Laboratory of the University of Pittsburgh, http://www.genetics.pitt.edu/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Kobayashi M.R., Miller T.A. Lymphedema. Clin. Plast. Surg. 1987;14:303–313. [PubMed] [Google Scholar]
- 2.Rockson S.G., Rivera K.K. Estimating the population burden of lymphedema. Ann. N Y Acad. Sci. 2008;1131:147–154. doi: 10.1196/annals.1413.014. [DOI] [PubMed] [Google Scholar]
- 3.Ferrell R.E., Levinson K.L., Esman J.H., Kimak M.A., Lawrence E.C., Barmada M.M., Finegold D.N. Hereditary lymphedema: evidence for linkage and genetic heterogeneity. Hum. Mol. Genet. 1998;7:2073–2078. doi: 10.1093/hmg/7.13.2073. [DOI] [PubMed] [Google Scholar]
- 4.Karkkainen M.J., Ferrell R.E., Lawrence E.C., Kimak M.A., Levinson K.L., McTigue M.A., Alitalo K., Finegold D.N. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat. Genet. 2000;25:153–159. doi: 10.1038/75997. [DOI] [PubMed] [Google Scholar]
- 5.Fang J., Dagenais S.L., Erickson R.P., Arlt M.F., Glynn M.W., Gorski J.L., Seaver L.H., Glover T.W. Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am. J. Hum. Genet. 2000;67:1382–1388. doi: 10.1086/316915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finegold D.N., Kimak M.A., Lawrence E.C., Levinson K.L., Cherniske E.M., Pober B.R., Dunlap J.W., Ferrell R.E. Truncating mutations in FOXC2 cause multiple lymphedema syndromes. Hum. Mol. Genet. 2001;10:1185–1189. doi: 10.1093/hmg/10.11.1185. [DOI] [PubMed] [Google Scholar]
- 7.Irrthum A., Devriendt K., Chitayat D., Matthijs G., Glade C., Steijlen P.M., Fryns J.P., Van Steensel M.A., Vikkula M. Mutations in the transcription factor gene SOX18 underlie recessive and dominant forms of hypotrichosis-lymphedema-telangiectasia. Am. J. Hum. Genet. 2003;72:1470–1478. doi: 10.1086/375614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finegold D.N., Schacht V., Kimak M.A., Lawrence E.C., Foeldi E., Karlsson J.M., Baty C.J., Ferrell R.E. HGF and MET mutations in primary and secondary lymphedema. Lymphat. Res. Biol. 2008;6:65–68. doi: 10.1089/lrb.2008.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alders M., Hogan B.M., Gjini E., Salehi F., Al-Gazali L., Hennekam E.A., Holmberg E.E., Mannens M.M., Mulder M.F., Offerhaus G.J. Mutations in CCBE1 cause generalized lymph vessel dysplasia in humans. Nat. Genet. 2009;41:1272–1274. doi: 10.1038/ng.484. [DOI] [PubMed] [Google Scholar]
- 10.Connell F., Kalidas K., Ostergaard P., Brice G., Homfray T., Roberts L., Bunyan D.J., Mitton S., Mansour S., Mortimer P., Jeffery S., Lymphoedema Consortium Linkage and sequence analysis indicate that CCBE1 is mutated in recessively inherited generalised lymphatic dysplasia. Hum. Genet. 2010;127:231–241. doi: 10.1007/s00439-009-0766-y. [DOI] [PubMed] [Google Scholar]
- 11.Wick N., Saharinen P., Saharinen J., Gurnhofer E., Steiner C.W., Raab I., Stokic D., Giovanoli P., Buchsbaum S., Burchard A. Transcriptomal comparison of human dermal lymphatic endothelial cells ex vivo and in vitro. Physiol. Genomics. 2007;28:179–192. doi: 10.1152/physiolgenomics.00037.2006. [DOI] [PubMed] [Google Scholar]
- 12.Rhodin J.A. Microscopic anatomy of the pulmonary vascular bed in the cat lung. Microvasc. Res. 1978;15:169–193. doi: 10.1016/0026-2862(78)90017-1. [DOI] [PubMed] [Google Scholar]
- 13.Zawieja D.C., Davis K.L., Schuster R., Hinds W.M., Granger H.J. Distribution, propagation, and coordination of contractile activity in lymphatics. Am. J. Physiol. 1993;264:H1283–H1291. doi: 10.1152/ajpheart.1993.264.4.H1283. [DOI] [PubMed] [Google Scholar]
- 14.McHale N.G., Meharg M.K. Co-ordination of pumping in isolated bovine lymphatic vessels. J. Physiol. 1992;450:503–512. doi: 10.1113/jphysiol.1992.sp019139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warner A., Clements D.K., Parikh S., Evans W.H., DeHaan R.L. Specific motifs in the external loops of connexin proteins can determine gap junction formation between chick heart myocytes. J. Physiol. 1995;488:721–728. doi: 10.1113/jphysiol.1995.sp021003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berthoud V.M., Beyer E.C., Seul K.H. Peptide inhibitors of intercellular communication. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000;279:L619–L622. doi: 10.1152/ajplung.2000.279.4.L619. [DOI] [PubMed] [Google Scholar]
- 17.Odermatt B., Wellershaus K., Wallraff A., Seifert G., Degen J., Euwens C., Fuss B., Büssow H., Schilling K., Steinhäuser C., Willecke K. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J. Neurosci. 2003;23:4549–4559. doi: 10.1523/JNEUROSCI.23-11-04549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Menichella D.M., Goodenough D.A., Sirkowski E., Scherer S.S., Paul D.L. Connexins are critical for normal myelination in the CNS. J. Neurosci. 2003;23:5963–5973. doi: 10.1523/JNEUROSCI.23-13-05963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verma V., Larsen B.D., Coombs W., Lin X., Spagnol G., Sorgen P.L., Taffet S.M., Delmar M. Novel pharmacophores of connexin43 based on the “RXP” series of Cx43-binding peptides. Circ. Res. 2009;105:176–184. doi: 10.1161/CIRCRESAHA.109.200576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kjølbye A.L., Dikshteyn M., Eloff B.C., Deschênes I., Rosenbaum D.S. Maintenance of intercellular coupling by the antiarrhythmic peptide rotigaptide suppresses arrhythmogenic discordant alternans. Am. J. Physiol. Heart Circ. Physiol. 2008;294:H41–H49. doi: 10.1152/ajpheart.01089.2006. [DOI] [PubMed] [Google Scholar]
- 21.Nagy J.I., Ionescu A.V., Lynn B.D., Rash J.E. Coupling of astrocyte connexins Cx26, Cx30, Cx43 to oligodendrocyte Cx29, Cx32, Cx47: Implications from normal and connexin32 knockout mice. Glia. 2003;44:205–218. doi: 10.1002/glia.10278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uhlenberg B., Schuelke M., Rüschendorf F., Ruf N., Kaindl A.M., Henneke M., Thiele H., Stoltenburg-Didinger G., Aksu F., Topaloğlu H. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am. J. Hum. Genet. 2004;75:251–260; erratum 737. doi: 10.1086/422763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orthmann-Murphy J.L., Enriquez A.D., Abrams C.K., Scherer S.S. Loss-of-function GJA12/Connexin47 mutations cause Pelizaeus-Merzbacher-like disease. Mol. Cell. Neurosci. 2007;34:629–641. doi: 10.1016/j.mcn.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shibayama J., Paznekas W., Seki A., Taffet S., Jabs E.W., Delmar M., Musa H. Functional characterization of connexin43 mutations found in patients with oculodentodigital dysplasia. Circ. Res. 2005;96:e83–e91. doi: 10.1161/01.RES.0000168369.79972.d2. [DOI] [PubMed] [Google Scholar]
- 25.McLachlan E., Manias J.L., Gong X.Q., Lounsbury C.S., Shao Q., Bernier S.M., Bai D., Laird D.W. Functional characterization of oculodentodigital dysplasia-associated Cx43 mutants. Cell Commun. Adhes. 2005;12:279–292. doi: 10.1080/15419060500514143. [DOI] [PubMed] [Google Scholar]
- 26.Goodenough D.A., Paul D.L. Gap junctions. Cold Spring Harb. Perspect. Biol. 2009;1:a002576. doi: 10.1101/cshperspect.a002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rhee D.Y., Zhao X.Q., Francis R.J., Huang G.Y., Mably J.D., Lo C.W. Connexin 43 regulates epicardial cell polarity and migration in coronary vascular development. Development. 2009;136:3185–3193. doi: 10.1242/dev.032334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei C.J., Xu X., Lo C.W. Connexins and cell signaling in development and disease. Annu. Rev. Cell Dev. Biol. 2004;20:811–838. doi: 10.1146/annurev.cellbio.19.111301.144309. [DOI] [PubMed] [Google Scholar]
- 29.Elias L.A., Wang D.D., Kriegstein A.R. Gap junction adhesion is necessary for radial migration in the neocortex. Nature. 2007;448:901–907. doi: 10.1038/nature06063. [DOI] [PubMed] [Google Scholar]
- 30.Aselli G. Mediolani; Milan: 1627. De lactibus sive Lacteis venis quarto vasorum mesaraicortan genere novo invento Gasparis Asellii Crenionensis anatomici Ticinensis dissertatio. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.