

Abstract
Congenital heart defects (CHDs) are the most common major developmental anomalies and the most frequent cause for perinatal mortality, but their etiology remains often obscure. We identified a locus for CHDs on 6q24-q25. Genotype-phenotype correlations in 12 patients carrying a chromosomal deletion on 6q delineated a critical 850 kb region on 6q25.1 harboring five genes. Bioinformatics prioritization of candidate genes in this locus for a role in CHDs identified the TGF-β-activated kinase 1/MAP3K7 binding protein 2 gene (TAB2) as the top-ranking candidate gene. A role for this candidate gene in cardiac development was further supported by its conserved expression in the developing human and zebrafish heart. Moreover, a critical, dosage-sensitive role during development was demonstrated by the cardiac defects observed upon titrated knockdown of tab2 expression in zebrafish embryos. To definitively confirm the role of this candidate gene in CHDs, we performed mutation analysis of TAB2 in 402 patients with a CHD, which revealed two evolutionarily conserved missense mutations. Finally, a balanced translocation was identified, cosegregating with familial CHD. Mapping of the breakpoints demonstrated that this translocation disrupts TAB2. Taken together, these data clearly demonstrate a role for TAB2 in human cardiac development.
Introduction
Congenital heart defects (CHDs) are the single most important congenital cause for perinatal mortality and morbidity,1,2 but despite this manifest importance, their etiology remains largely obscure. Although epidemiological studies demonstrated that certain environmental factors are contributory,3 family and twin studies suggest a major genetic component.4,5 Indeed, mutations in several genes were associated with monogenic CHDs, mainly through linkage analysis in large families in which a CHD segregates as an autosomal-dominant trait.6 Given the mortality associated with CHDs, such large families are rare. Although family studies suggest that partially penetrant causes of CHDs are much more common than purely monogenic causes,7,8 such loci have remained unidentified in linkage studies. As a result, only few causative genes have been identified, and mutation analyses have shown that they account for only a very small fraction (< 1%) of CHD cases,9 representing a serious limitation in the genetic counseling of CHD patients and their families and in the elucidation of the pathogenesis of CHD.
To accommodate these limitations, we attempted an alternative approach by identifying loci associated with CHDs through chromosomal rearrangements. Such a strategy enables the identification of regions harboring genes involved in heart development in a dosage-sensitive manner and the construction of a human morbidity map for CHDs. We previously reported the identification of several candidate loci for CHD through the screening of patients with a CHD by means of array comparative genome hybridization (aCGH).10,11 We describe here the delineation and characterization of one of these loci, located on chromosome 6q24-q25 and containing the MAP3K7IP2 (TAB2 [MIM 605101]) gene. TAB2 maps to the critical region deleted in patients with a CHD, is dosage-sensitive in zebrafish development, and is specifically expressed in the human and zebrafish cardiovascular system. We moreover show that it is disrupted by a balanced translocation in three family members with a CHD and is mutated in two out of 402 patients with CHDs, providing strong evidence that TAB2 has a major role in cardiac development.
Subjects and Methods
Patients
Informed consent was obtained from all patients or their legal guardians for investigations on patient material and for anonymous publication. Syndromic CHD patients (Table S1, available online) were followed by the Pediatric Cardiology Unit and the Clinical Genetics Unit of the University Hospitals Leuven (patient A), by the Clinical Genetics Unit of the Ghent University Hospital (patient F), or as described (patients B, C, and I).12 Patients with isolated CHDs (Table S2) were followed by the Pediatric Cardiology Unit of the University Hospitals Leuven or collected from the CONCOR national registry database and DNA bank.13 Patients with CHD and a translocation were identified by systematic reexamination of carriers of balanced translocations.14 Patients with isolated CHDs were not investigated by aCGH. These studies were approved by the local ethics committees.
Tissue Samples and Immunohistochemistry Analysis
Human embryonic tissues were collected from legal abortions. Informed consent was obtained according to the Helsinki Declaration II. Embryonic age was based on crown-rump length measurement. Tissue samples were dissected into appropriate tissue blocks and fixed for 12–24 hr at 4°C in 10% neutral buffered formalin, 4% Formol-Calcium, or Lillie's or Bouin's fixatives. The specimens were dehydrated with graded alcohols, cleared in xylene, and embedded in paraffin. Serial sections, 3–5 μm thick, were cut in transverse, sagittal, or horizontal planes and placed on silanized slides.
Sections were deparaffinized and rehydrated in xylene followed by a series of graded alcohols in accordance with established procedures. The sections were treated with a fresh 0.5% solution of hydrogen peroxide in methanol for 15 min for quenching of endogenous peroxidase and were then rinsed in TRIS buffered saline (TBS, 5 mM Tris-HCl, 146 mM NaCl, pH 7.6). Nonspecific binding was inhibited by incubation for 30 min with blocking buffer (ChemMate antibody diluent S2022, DakoCytomation, Glostrup, Denmark) at room temperature. The sections were then incubated overnight at 4°C with a polyclonal rabbit antibody which specifically recognizes human TAB2 (MAP3K7IP2), by immunoblotting and immunohistochemistry (ARP32402, Aviva Systems Biology, 1:1000) in blocking buffer (ChemMate antibody diluent S2022, DakoCytomation).
The sections were washed with TBS and then incubated for 30 min with a peroxidase-labeled anti-rabbit polymer (DAKO EnVision + System/HRP K4011, DakoCytomation). The sections were washed with TBS, followed by incubation for 10 min with 3,3′-diamino-benzidine chromogen solution. Positive staining was recognized as a brown color. The sections were dehydrated in graded alcohols followed by xylene and coverslipped with DPX mounting media. Nonimmune rabbit IgG1 (X0936) and staining without primary antibody were used as negative controls. Sections from human embryos containing osteoclasts served as positive controls. Control sections stained without antibody or with a nonimmune rabbit IgG1 were blank, whereas stained osteoclasts were always positive.
Molecular Cytogenetics
aCGH was performed as described15 on in-house-created microarray slides, constructed with bacterial artificial chromosome (BAC) or P1-derived artificial chromosome (PAC) probes chosen in a genome-wide manner with 1 Mb spacing (set donated by the Sanger Institute) or chosen from chromosomes 6 and 2 with tiling resolution (set obtained from BACPAC Chori, Oakland, CA, USA). 244K arrays were obtained from Agilent and hybridized according to the manufacturer's protocol. Genomic DNA was labeled by the BioPrime Array CGH Genomic Labeling System (Invitrogen, Carlsbad, CA, USA) with the use of Cy3- and Cy5-labeled dCTPs (2′-deoxycytidine 5′-triphosphate) (Amersham Biosciences, Boston, MA, USA) as recommended by the manufacturer with minor modifications.15
Translocation breakpoints were mapped by fluorescence in situ hybridization (FISH) analysis with the use of 200 ng BAC DNA according to standard procedures. The FISH signals were visualized with an avidin-FITC detection system. Chromosomes were counterstained with DAPI (4,6-diamino-2-phenylindole), and the signals were investigated with a Leica DMRB epifluorescence microscope equipped with a Sensys 1400 CCD camera (Photometrics, Tucson, AZ, USA) and IPLab Spectrum imaging software (Abbott Laboratories, Abbott Park, IL, USA).
Candidate-Gene Prioritization
Candidate-gene prioritization was performed with the use of an adapted version of Endeavour.16 In brief, two data sources for prioritization were added to the regular set of data sources: an expression microarray data set of murine heart development (GEO accession number GSE1479—transposed to human gene identifiers with the use of BioMart) and a set representing gene homology, extracted from HomoloGene, BioMart, and Inparanoid. These data were summarized by vector representations, and scoring was done with the use of Pearson correlation. Additionally, prioritizations that were based on data sources displaying strong Spearman rank correlation (> 0.3) were fused prior to fusing with results from prioritizations based on more independent data sources (Figure S1). Data sources were validated by leave-one-out cross-validation (LOOCV) as described previously,16 and sources with an area under the curve below 0.6 were omitted (Figure S2). Genes were prioritized on the basis of seven training sets (Table S5), representing discrete aspects of cardiac development and genetics, and results from the seven sets were fused with the use of rank-order statistics.16 In silico testing by LOOCV demonstrated that this adapted algorithm readily ranks genes with an established involvement in heart development, on average in the top 5%. All 105 annotated protein-coding genes from 6q24-q25 were prioritized. Gene identification and prioritization was based on Ensembl Release 49 and Endeavour databases of May 2008.
Zebrafish Assays
Wild-type (AB) or flk-green fluorescent protein (flk-GFP)17 zebrafish (Danio rerio) stocks were maintained in accordance with standard aquaculture guidelines. Eggs were collected after natural mating within 30 min after being laid so that timed development was assured. RNA was extracted with TRIzol, and cDNA was synthesized with random primers and the Superscript III kit (all from Invitrogen, Merelbeke, Belgium).
Whole-mount in situ hybridizations (WISH) on wild-type and flk-GFP zebrafish embryos was performed as described previously18 with the use of a sequence-verified probe amplified by PCR from the synthesized cDNA. The resulting embryos were imaged or embedded in acrylamide-bisacrylamide gel, cryosectioned to 20 μm, and mounted on vectabond-pretreated microscope slides. Sections were rehydrated in phosphate-buffered saline with 0.1% Tween 20 (PBT) and incubated for 4 hr with PBT-1% preimmune donkey serum (PDS) at room temperature and then overnight at 4°C with Alexa-647-labeled GFP antibody (sc-9996 AF647 from Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1/200 in PBT-1% PDS). Slides were washed four times in PBT, mounted on vectashield-DAPI, and imaged on an Olympus FV1000 for confocal imaging and an Olympus AX51 for bright-field imaging.
Morpholinos (MOs) were designed against the intron 2-exon 3 splice site (5′-ATC ACT CTT GTT CTG AGG AAA GAA G-3, splice-site-blocking MO [sbMO]) and the translation initiation site (5′-ATC TGC TGG TTT CCC TGT GCC ATT C-3′, translation-blocking MO [tbMO]) of tab2. Both MOs and a control MO (cMO) (5′-CCT CTT ACC TCA GTT ACA ATT TAT A-3′) were ordered from Gene Tools (Philomath, OR, USA). Fidelity of annotated nucleotide sequences of MO binding sites was confirmed by direct sequencing. MOs were diluted in a 1/50 Rhodamine solution, and an estimated 0.5 nL was injected at the one- or two-cell stage at specified doses. tab2 mRNA concentrations were measured by real-time quantitative (rtq) PCR as described previously15 and normalized to β-actin.
Mutation Analysis
DNA was collected from patients with an outflow tract defect (tetralogy of Fallot [ToF], pulmonic stenosis [PS], aortic stenosis [AS]). Exons and exon-intron boundaries of TAB2 were amplified by PCR with the use of oligonucleotide primers described in Table S3. PCR products were sized by DNA gel electrophoresis and sequenced with the BigDye DNA sequencing kit (Applied Biosystems, Foster City, CA, USA).
Results
We present the identification and delineation of a locus for CHDs, and we describe how a candidate gene was selected from this locus. We investigated the role of this gene in heart development through expression analyses and functional studies, and we present the results of mutation analysis.
Molecular Characterization of Deletions in 6q24-q25
We observed deletions of 6q24-q25 in multiple syndromic CHD patients. In the framework of an aCGH screening study,4 aCGH on 1Mb arrays revealed the presence of a 6q24.2-q25.1 deletion in patient A, shown to have occurred de novo by rtqPCR analysis of parental DNA. Patient A had an aortic coarctation, a hypoplastic aortic arch, and a ventricular septal defect, as well as additional problems. During a routine analysis of patients with an unexplained syndromic disorder, a second deletion was detected and delineated in patient Fp with the use of Agilent 44K arrays. It was shown by rtqPCR to be inherited from his mother (Fm). This mother (Fm) was diagnosed in her childhood with aortic and mitral valve stenosis and had episodes of sinus tachycardia. Our studies prompted us to perform an echocardiographic evaluation of patient Fp, which revealed mild centrovalvular insufficiency of the aortic and pulmonary valves. Previously reported interstitial 6q deletions (patients B, C, and I)12 were further delineated by aCGH on 244K arrays. Genotype data on other patients (D, E, G, H, J, and K) were collected from literature reports12,19–23 or from an online database on cardiogenetics, CHDWiki.24 The extent of all deletions and the presence or absence of CHDs in deletion carriers is depicted in Figure 1A, and more detailed phenotype and genotype data are provided in Table S1.
Figure 1.
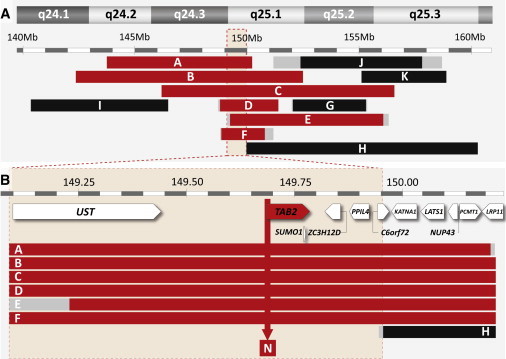
Fine Mapping of Chromosome Aberrations in 6q24-q25, and Candidate-Gene Prioritization
(A) Position of deletions found in patients with a CHD (red bars) or without a CHD (black bars). Deletion reference letters are shown on the bars. Regions containing deletion breakpoints are shown in gray. Phenotypes shown as in Table S1. Deletion F was found in a male and his mother, both affected by CHDs. The critically deleted region for CHDs is demarcated by a light red box.
(B) Position of deletions found in the region deleted in all CHD patients (commonly deleted region) with respect to the 11 annotated genes encoded in this region. The deletion found in patient H (black, no CHD) does not affect the TAB2 gene, and it demarcates the critically deleted region, which contains five genes. The arrow indicates the position of the translocation breakpoint on chromosome 6 found in family N.
Cardiac defects identified in deletion patients mostly affected the outflow tract. They included stenosis of one or more heart valves, hypoplasia of the aortic arch, and atrial or ventricular septal defects. Patients with larger deletions typically also had a mild mental retardation and additional congenital problems, whereas those with smaller deletions (patients Fp, Fm, and J) had a (low) normal mental development. Genotype comparison of CHD patients revealed a commonly deleted region of 1.2 Mb, affecting 11 genes (Figure 1B). Deletion H, found in a patient without a CHD, moreover demarcates an 850 kb region deleted in all CHD patients and unaffected in patients without a CHD. This critically deleted region extends from 149.09 to 149.96 Mb (Figure 1B) and contains five genes unaffected by deletion H.
Prioritization of Genes on 6q24-q25 for CHD
The clustering of CHD-associated deletions on 6q24-q25 suggested that haploinsufficiency of one or more genes in this locus causes CHDs. Although this putative CHD gene most likely resides in the commonly deleted region, we could not exclude the alternative hypothesis that two or more genes located elsewhere on 6q24-q25 cause CHDs with incomplete penetrance. The in silico analysis was therefore extended to all genes on 6q24-q25. In addition, this extension of our test set enabled a better assessment of the statistical power of our findings. Gene prioritization yielded a ranked list of candidate genes for CHDs, with MAP3K7IP2 (also known as TAB2) ranking first of all 105 genes from 6q24-q25 (Table S4 and Figure S3). The corresponding p value (4.17 × 10−7) was highly significant; it is located in the critically deleted region, and no significant prioritization (p < 0.01) was obtained for any other gene in the commonly deleted region. Upon genome-wide prioritization, TAB2 moreover ranks 44th among all genes of the human genome (Table S6). It thus seemed like a likely candidate gene for CHDs.
TAB2 Expression in Human Embryonic Hearts
To explore the potential role of the TAB2 candidate gene in human heart development, we analyzed its expression in human embryos. Immunohistochemistry analysis of tissue sections from 5.5 wk and 7.5 wk human embryos showed cytoplasmic expression of TAB2 in cells of the ventricular trabeculae (Figure 2A), in endothelial cells of the conotruncal cushions of the outflow tract (Figures 2A and 2B), and in the endothelial cells lining the developing aortic valves (Figures 2C and 2D). These results indicate a function of TAB2 in the developing human heart.
Figure 2.
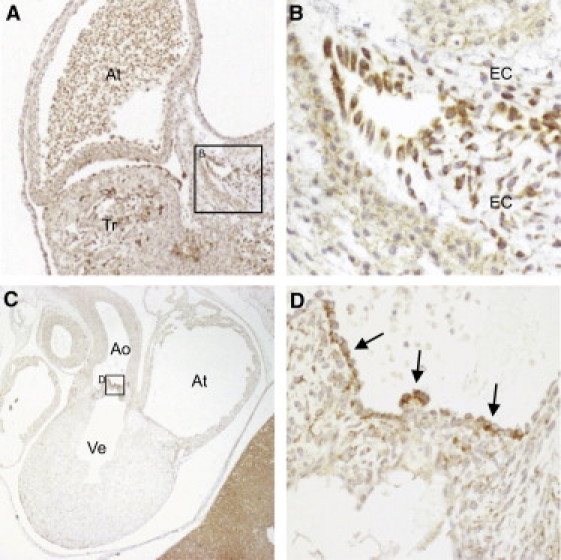
TAB2 Expression in Human Embryonic Hearts
(A) Section of a 5.5 wk embryo. TAB2 expression (brown color) is prominent in the ventricular trabeculae and in the endocardial cushions of the outflow tract.
(B) Magnification of endocardial cushions.
(C) Frontal section of a 7.5 wk embryo.
(D) Magnification showing cytoplasmic expression of TAB2 in the endothelial cells lining the developing aortic valves.
Abbreviations are as follows: At, atrium; Ao, aorta; EC, endocardial cushions; Tr, trabeculae; Ve, ventricle.
tab2 Zebrafish Assays
We used zebrafish as a model organism to investigate the role and dosage sensitivity of tab2 during vertebrate development. WISH on staged wild-type zebrafish embryos revealed ubiquitous tab2 expression during early development (12–18 hr postfertilization [hpf]) and a more restricted expression pattern during later development, with expression in the developing cardiac outflow tract, the dorsal aorta, and the posterior cardinal vein (Figure 3). This expression pattern is reminiscent of the expression during human heart development and thus suggests a conserved role for tab2 in the cardiac outflow tract.
Figure 3.
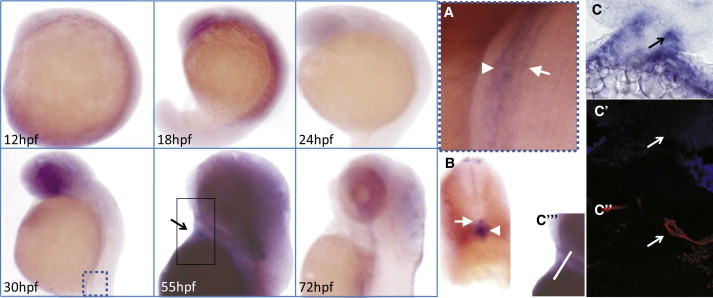
Gene Expression of tab2 in the Developing Zebrafish
mRNA expression of tab2 in the developing zebrafish at distinct developmental stages (indicated in the lower left corner). Boxed parts of embryos at 30 hpf and 55 hpf are repeated in (A) and (C″′), respectively.
(A): Magnification showing expression of tab2 in the dorsal aorta (arrow) and posterior cardinal vein (arrowhead).
(B) Transverse section through the hindbody of a zebrafish at 30 hpf depicted in (A), showing restricted expression in the dorsal aorta (arrowhead) and posterior cardinal vein (arrow).
(C): Bright-field image of the expression of tab2 in the cardiac outflow tract at 55 hpf (arrow).
(C′) DAPI staining of the section depicted in (C) (pseudocolored in blue).
(C″) The section depicted in (C), immunostained with Alexa-647-labeled GFP antibody (pseudocolored in red), displaying the position of the cardiac outflow tract (arrow).
(C″′) Image of a 55 hpf, whole-mount-stained embryo, presenting the orientation of the section shown in (C), (C′), and (C″) (white line).
To investigate the function of tab2 during zebrafish development, we knocked down its expression by using two independent MOs that were interfering with either normal splicing or translation of the tab2 RNA. Injection of either resulted in similar developmental abnormalities, whereas injection of the same amount of cMO did not result in abnormal development. Knockdown was thus demonstrated to be specific. The first embryological defects became apparent during gastrulation (Figures 4F and 4G) and included delayed epiboly progression and convergent extension defects. Later in development (36–48 hpf), severe heart failure became apparent (Figures 4D and 4E): the heart tube appeared thin and elongated, with blood pooling in the common cardinal vein before entry into the heart.
Figure 4.
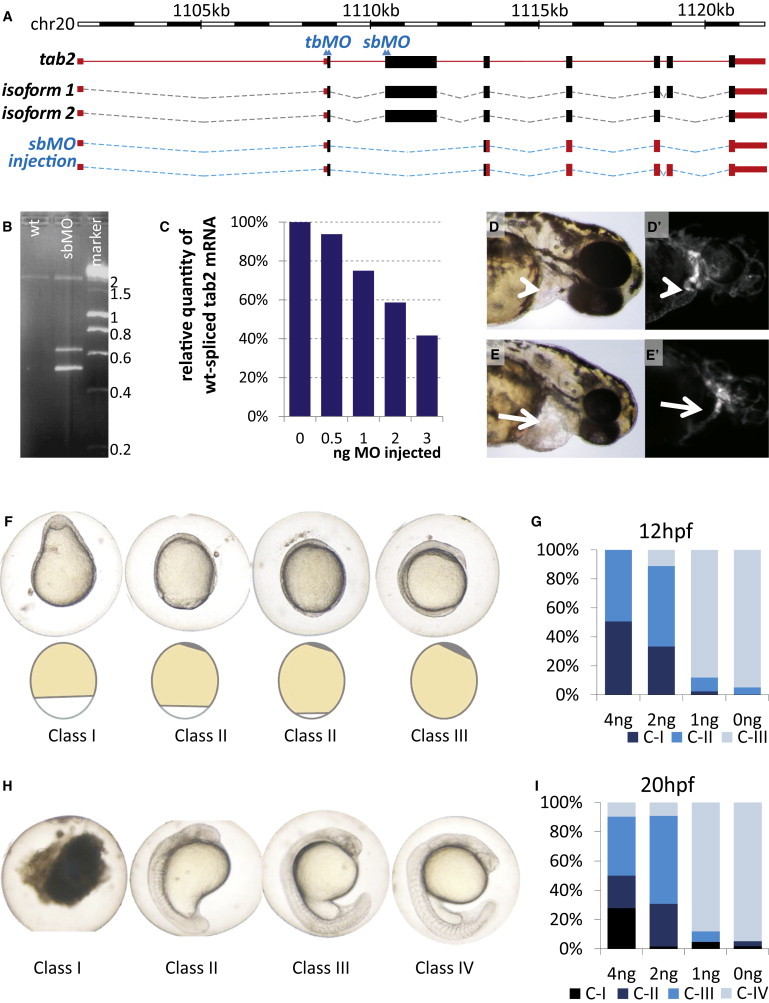
tab2 Knockdown in Zebrafish
(A) Schematic organization of the tab2 gene in the zebrafish genome. Two alternative splice isoforms as detected by PCR on the reverse-transcribed mRNA are shown. Introns are dotted lines, exons full boxes (coding sequences in black, noncoding sequences in orange). The target sites of the sbMO) and tbMO are indicated by double arrowheads. The lower schema indicates the effect of sbMO injection on the mature tab2 mRNA (isoforms 1 and 2).
(B) Gel electrophoresis of the product of PCR on cDNA extracted from nonmorphant (WT) or 2 ng sbMO-injected zebrafish embryos 24 hpf and a DNA marker for size comparison, showing one band around 2000 bp in the wild-type situation, and two additional fragments in the morphant situation.
(C) Effect of TAB2 sbMO injection on the amount of correctly spliced mRNA.
(D) Wild-type AB embryo at 48 hpf.
(D′) Noninjected embryo at 48 hpf expressing GFP in vascular endothelial cells under the influence of an flk promoter (flk-GFP).
(E) Morphant embryo at 48 hpf. Note the enlarged pericardial sac (arrow).
(E′) Morphant flk-GFP embryo at 48 hpf. Note the thin and elongated heart.
(F) Phenotypical classification of sbMO-injected zebrafish embryos at 12 hpf. Defects in epiboly progression are evident, and progression of the yolk-syncytial layer is schematically displayed below. Phenotypes are ordered from severe to normal. Class I: anterior-posterior gradient is not evident. Class II: the yolk-syncytial layer has not progresses until the vegetal pole; two different embryos are shown to illustrate the continuum in phenotypes in class II. Class III: wild-type. Left, posterior; up, dorsal.
(G) Distribution of phenotypes at 12 hpf dependent upon the sbMO dose injected. Y axis: percentage of embryos in each class as indicated by the color code. X axis: amount of TAB2 sbMO injected at the one-cell stage. At least 42 embryos were successfully injected for each dose. In total, over 230 embryos were successfully injected.
(H) Phenotypical classification of sbMO-injected zebrafish embryos at 24 hpf. Phenotypes are ordered from severe to normal. Class I: death. Other classes are as described in the main text. Lateral images: up, anterior.
(I) Distribution of phenotypes at 24 hpf dependent upon the sbMO dose injected. Y axis: percentage of embryos in this class. X axis: amount of TAB2 sbMO injected at the one-cell stage. At least 42 embryos were successfully injected for each dose. In total, over 230 embryos were successfully injected. Injection of 4 ng of cMO did not increase the frequency of abnormal phenotypes in comparison to noninjected or 0 ng-injected control embryos (not shown).
The molecular effect of sbMO injection on splicing of nascent tab2 RNA was investigated by PCR on cDNA from injected embryos (Figure 4B). Sequencing of RT-PCR products revealed that a transcript lacking wild-type exon 3 was formed upon this treatment, resulting in a frameshift and a premature stop codon starting at the seventh nucleotide of wild-type exon 4 (Figure 4A). This sbMO-generated transcript thus encodes a 95% truncated variant of the normal protein. As confirmed by the similar phenotype observed upon tbMO injection, this represents a loss-of-function situation. The relative amount of correctly spliced mRNA was assessed by rtqPCR on cDNA reverse transcribed from RNA that was extracted from sbMO-injected embryos at 20 hpf. This revealed that sbMO injection resulted in a dose-dependent reduction of normal splicing (Figure 4C). Phenotypes became apparent at a dose of 2 to 3 ng, corresponding to a 41% to 58% reduction of normal expression, thus showing that haploinsufficiency of tab2 causes developmental defects (Figures 4C–4I).
Sequence Analysis of TAB2 in Human Patients with Outflow Tract Defects
Because TAB2 has a relevant spatiotemporal expression pattern and a dosage-sensitive role in development, it was a very good candidate gene for explaining the CHDs evident upon deletion of 6q24-q25. To further confirm the role of TAB2 in CHDs, we analyzed the DNA sequence of TAB2 in 402 patients with outflow tract defects (Table S2). These patients were not analyzed by aCGH. Sequencing analysis revealed two heterozygous missense mutations in the TAB2 gene. One female (patient L) carried a c.622C>T mutation, causing a p.Pro208Ser mutation at the protein level (Figures 5A and 5C). She had a left-ventricular outflow tract obstruction, a subaortic stenosis (subAS) due to a fibromuscular shelf, residual aortic regurgitation, and atrial fibrillation and died at 61 years of age because of heart failure. Patient M carried a c.688C>A mutation, causing a p.Gln230Lys mutation at the protein level (Figure 5B). He had a bicuspid aortic valve and an aortic dilation. Both mutations alter highly conserved residues and are predicted to have detrimental effects on protein function (Figure 5C). Neither DNA samples nor phenotypic data were available for family members of patients L or M. These mutations were, however, absent from 658 ethnically matched control chromosomes.
Figure 5.
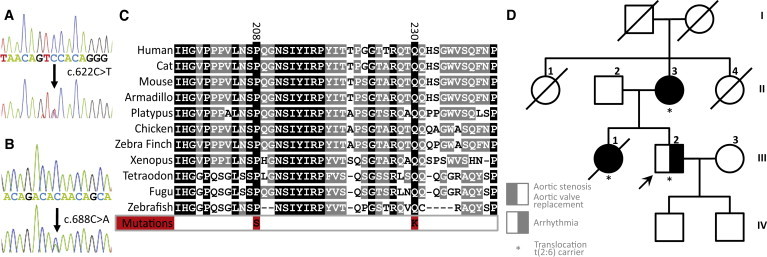
TAB2 Is Mutated and Disrupted in CHD Patients
(A and B) Partial TAB2 reference-sequence-read traces and corresponding traces of missense mutations as identified in patients L and M (phenotypes detailed in the main text).
(C) Conservation of mutated residues in several tetrapod and fish lineages. TAB2 is not found in lower lineages; the mutated residues are not conserved in the paralogous TAB3.
(D) Pedigree of family N. Translocation carriers are marked, and the breakpoint on chromosome 6 disrupts TAB2. Phenotypes are as annotated in the insert and are detailed in the main text.
Investigation of a Family with a t(2;6) Translocation and CHD
A family with CHD segregating with a t(2;6)(q21;q25) translocation was previously identified.14 In this family (family N), all three translocation carriers have a history of CHD (Figure 5D). Patient N-II.2 was diagnosed with aortic stenosis and atrial fibrillation at age 67 and required an aortic valve replacement. Patient N-III.1 was diagnosed with aortic stenosis at the age of 2 yrs. Her aortic valve was surgically replaced at age 25. At age 34 she was hospitalized for paroxysmal supraventricular tachycardia. She died at age 49 from a myocardial infarction. Patient N-III.2 was diagnosed— like his mother and sister—with cardiac rhythm problems (sick sinus syndrome, tachycardia). However, he refused to participate in further clinical examinations and was not investigated by echocardiography.
The translocation segregating in this family was balanced, as demonstrated by aCGH on 1Mb arrays and on chromosome 6 and 2 tiling microarrays (data not shown). FISH studies positioned the translocation breakpoint on 2q21 to a 14.2 kb region at chromosome position 131,691,033-131,705,253, disrupting the POTEE gene (encoding prostate, ovary, testis-expressed protein E). The position of the translocation breakpoint in 6q25 lies within a 17 kb region at chromosome position 149,678,240-149,695,219, within the first intron, first exon, or promoter region of TAB2 (Figure S4).
Discussion
We identified a locus for CHDs on 6q24-q25, which is deleted in seven individuals with a CHD (Figure 1A). In search of genes causing the CHDs in this region, we prioritized all 105 genes from chromosome 6q24 and 6q25 for their involvement in heart development, using a tool based on an established genomic data fusion algorithm.16 This strategy ensures gene selection that is influenced to only a minor extent by a researcher's prior knowledge and preferences. TAB2 ranked first (p = 4.17 × 10−7) among all candidate genes (Figure 1C). Importantly, none of the other ten genes in the commonly deleted region generated a significant signal for their involvement in heart development with the use of this method (p > 0.03 for all).
Support for the involvement of TAB2 in human heart development was further provided by the expression of this gene in developing human and zebrafish embryos. TAB2 is expressed in the endothelial lining of the developing human heart: we detected expression in endothelial cells lining the trabeculae and the developing aortic valves (Figure 2). Endocardial cushions are involved in endothelial-mesenchymal transformation, an important event during development of the cardiac valves and outflow tract.25 The conservation of Tab2 expression in the developing cardiovascular system of the mouse26 and the zebrafish (Figure 3) further supports the functional relevance of TAB2 in cardiovascular development.
TAB2 (TAK1 binding protein 2) encodes a protein that is studied mainly for its role in the inflammatory response. TAB2 causes autophosphorylation and activation of mitogen-activated protein kinase kinase kinase 7 (MAP3K7, also known as TAK1 [MIM 602614]),27 thus (through mediators such as TNF-receptor-associated factors [TRAFs] and receptor-interacting proteins [RIPs]) relaying signals from receptors for chemokines (TNF, LPS, IL1) and other extracellular signaling molecules (TGF-β and Wnt) to downstream proteins such as IKK, p38, JNK, RCAN, and NLK. TAK1 consequently modulates the activity of molecules such as NF-κB, β-catenin, NFAT, and HDAC3 and their downstream target genes. Tak1−/− mice die around embryonic day 10.5 (E10.5) to E12.5 and have neural tube and cardiovascular defects.28,29 Xie and colleagues (2006)30 generated mice with a cardiac-specific expression of a dominant-negative Tak1, showing that inhibiting Tak1 function in the heart leads to altered electrical conduction (shortened PR interval), impaired ventricular filling, and cardiac hyperthropy. Interestingly, the authors also discussed unpublished results that showed that cardiac-specific deletion of Tak1 causes midgestation death.30 Zhang and colleagues (2000)31 reported on mice with cardiac-specific expression of activated Tak1, showing that they develop cardiac hypertrophy. Combined, these results demonstrate that altered Tak1 signaling in the heart leads to cardiac disorders. Of interest, in our aCGH screening study that led to the identification of the presented TAB2 deletion, we identified a 650 kb TAK1 duplication in a girl with a pulmonary stenosis (data not shown). This duplication was inherited from her mother, who has no cardiac defect. Her brother had died early in childhood from a severe pulmonary valve stenosis as well, but no DNA was available for study.
Tab2−/− mice die around E12.5 and display severe liver degeneration with increased hepatocyte apoptosis. No immunological or hematological problems were described in patients with TAB2 disruption. Interestingly, however, 70% of Tab2+/− mice die within 1 wk after birth. The reason for this increased mortality was not established.32 Although this obviously precludes a direct phenotypic comparison with human hemizygous patients, it does demonstrate that Tab2 is dosage sensitive in the mouse and is an excellent candidate for explaining the CHDs observed in the patients.
To further investigate the dosage sensitivity of tab2, we titrated a knockdown of tab2 gene expression through MO injection. We observed phenotypic changes when the fraction of normal tab2 mRNA approximately halved, demonstrating that the tab2 gene is dosage sensitive also in the zebrafish. The morphants displayed early embryological defects (delayed epibolic movements during gastrulation; Figure 4F) and further developed into larvae that were shorter and had a dysfunctional heart, a curved tail, abnormal somites, and a small head in comparison to control larvae. However, the early defects observed in these embryos preclude a conclusive analysis on the involvement of tab2 in heart development: although the hearts of morphant embryos were abnormally structured and heart failure was evident by the enlarged pericardial sac (Figure 4E), we cannot exclude that these effect are secondary to the earlier defects in gastrulation. Other reports have similarly shown that knockout of genes involved in isolated CHDs in humans can cause a much more pleiotropic phenotype in zebrafish.33 This is probably attributable to species-specific differences; for example, in the epibolic movements that have no direct correlate in mammalian development. Nevertheless, the clear dosage sensitivity of tab2 in zebrafish again demonstrates that a proper tab2 dosage is critical for a normal development in vertebrates.
Because of the above findings and for confirmation of the role of TAB2 in CHD, we investigated the presence of germline mutations in CHD patients. This revealed two missense mutations affecting evolutionarily highly conserved residues (Figures 5A–5C). These conserved residues are not in any recognized protein domain, nor do they affect any of the hitherto described protein binding sites; future studies will need to establish the function of this conserved domain. The observed paucity in mutations was not unexpected given the known etiological and genetic heterogeneity of CHDs: mutations in other genes with an established role in CHD development are similarly found in only a small (< 1%) fraction of CHD patients.9 Furthermore, because not all CHD phenotypes were investigated, we cannot exclude that TAB2 mutations are associated with additional CHD phenotypes. Moreover, we identified a family in which CHD and cardiomyopathy cosegregate with a balanced translocation between 2q21 and 6q25. Family members that do not carry this translocation do not have a CHD. The breakpoint in 2q21 disrupts the POTEE gene (Figure S4), one of seven poorly conserved paralogues of a recently expanded gene family.34 Given that these genes are not expressed in the mammalian heart and that POTEE is variable in copy number in the normal human population, it is unlikely that disruption of POTEE causes the observed CHDs in this family. The breakpoint on 6q25 is located within TAB2, which leads to its disruption, further supporting a role for TAB2 in the pathogenesis of CHD. Taken together, these results demonstrate that perturbations and mutations of TAB2 are indeed causing CHDs.
Although most of the described CHDs are outflow tract defects, we do not exclude that the phenotypic spectrum extends further. Indeed, patient C presented with an atrial septum defect (ASD) and patient E with a ventricular septal defect (VSD) (Table S1). This is not surprising, given the large variation in CHD phenotypes that has been described in most genetic syndromes. A classic example is 22q11 deletion syndrome, in which CHD phenotypes include Tetralogy of Fallot (ToF), pulmonary stenosis (PS), interrupted aortic arch, VSD, ASD, and other cardiac malformations.35 Other examples include Wolf-Hirchhorn syndrome, in which patients may have ASD, PS, VSD, or patent ductus arteriosus,36 and Smith-Magenis syndrome, in which valvular abnormalities, ASD, VSD, and ToF have been reported.37 Variable CHD phenotypes are also observed in patients with gene mutations. Examples are plenty and include patients with JAG1 mutations, in which PS, aortic stenosis, VSD, and ASD have been reported,38 and patients with CITED2 mutations, who can present with ASD, VSD, ToF, or transposition of the great arteries.39 Patients with NOTCH1 mutations can present with AS, VSD, ToF, or a double outlet right ventricle, but they are also at risk for late-onset complications such as aortic valve calcification and AS, requiring valve replacement,40 as was observed in patient N-II.3.
In conclusion, we demonstrated the ability of a positional cloning strategy for the identification of genes involved in human heart defects. The strategy presented here enables a rapid, generic, and powerful positional cloning and led to the identification of a gene involved in CHD, TAB2. This gene is expressed in the developing heart, is dosage sensitive in zebrafish development, and is mutated, deleted, or disrupted by a translocation in CHD patients.
Acknowledgments
B.T. is supported by a Ph.D. fellowship from the Agentschap voor Innovatie door Wetenschap en Technologie (IWT). P.V.L. is a postdoctoral researcher, K.D. a senior clinical investigator, and J.B. an aspirant investigator of the Research Foundation-Flanders (FWO). L.A.L. is supported by The Danish Heart Foundation and The Novo Nordisk Foundation. This work was supported by OT/O2/40, GOA/2006/12, Centre of Excellence SymBioSys (EF/05/007) from the University of Leuven and from the Belgian program of Interuniversity Poles of Attraction (IUAP), ProMeta, GOA Ambiorics, GOA MaNet, START 1, FWO (G.0318.05, G.0254.05, G.0553.06, G.0302.07, ICCoS, ANMMM, MLDM, G.0733.09, G.082409), IWT (Silicos, SBO-BioFrame, SBO-MoKa, TBM-IOTA3), IUAP P6/25 BioMaGNet, ERNSI (FP7-HEALTH CHeartED). The Wilhelm Johannsen Centre for Functional Genome Research is established by the Danish National Research Foundation. We thank D. Stainier for sharing the transgenic flk-GFP zebrafish, S. Forschhammer and A. Ilgun for technical assistance, and the Leuven Aquatic Facility for excellent fish care.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BioMart, www.ensembl.org/biomart
CHDWiki, homes.esat.kuleuven.be/∼bioiuser/chdwiki
Endeavour, www.esat.kuleuven.be/endeavour
Gene expression omnibus (GEO), www.ncbi.nlm.nih.gov/geo
Homologene, www.ncbi.nlm.nih.gov/homologene
Inparanoid, inparanoid.sbc.su.se
Online Mendelian Inheritance in Man (OMIM), www.ncbi.nlm.nih.gov/Omim
Accession Numbers
The GenBank accession numbers for human TAB2 cDNA and protein sequences reported in this paper are NM_015093.3 and NP_055908.1. Human and zebrafish genome coordinates are numbered according to the March 2006 human reference sequence (NCBI build 36.1) and the July 2007 zebrafish (Danio rerio) Zv7 assembly.
References
- 1.Hoffman J.I., Kaplan S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002;39:1890–1900. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- 2.Thom T., Haase N., Rosamond W., Howard V.J., Rumsfeld J., Manolio T., Zheng Z.J., Flegal K., O'Donnell C., Kittner S., American Heart Association Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics—2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2006;113:e85–e151. doi: 10.1161/CIRCULATIONAHA.105.171600. [DOI] [PubMed] [Google Scholar]
- 3.Jenkins K.J., Correa A., Feinstein J.A., Botto L., Britt A.E., Daniels S.R., Elixson M., Warnes C.A., Webb C.L., American Heart Association Council on Cardiovascular Disease in the Young Noninherited risk factors and congenital cardiovascular defects: current knowledge: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115:2995–3014. doi: 10.1161/CIRCULATIONAHA.106.183216. [DOI] [PubMed] [Google Scholar]
- 4.Calcagni G., Digilio M.C., Sarkozy A., Dallapiccola B., Marino B. Familial recurrence of congenital heart disease: an overview and review of the literature. Eur. J. Pediatr. 2007;166:111–116. doi: 10.1007/s00431-006-0295-9. [DOI] [PubMed] [Google Scholar]
- 5.Manning N., Archer N. A study to determine the incidence of structural congenital heart disease in monochorionic twins. Prenat. Diagn. 2006;26:1062–1064. doi: 10.1002/pd.1556. [DOI] [PubMed] [Google Scholar]
- 6.Pierpont M.E., Basson C.T., Benson D.W., Jr., Gelb B.D., Giglia T.M., Goldmuntz E., McGee G., Sable C.A., Srivastava D., Webb C.L., American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115:3015–3038. doi: 10.1161/CIRCULATIONAHA.106.183056. [DOI] [PubMed] [Google Scholar]
- 7.Burn J., Brennan P., Little J., Holloway S., Coffey R., Somerville J., Dennis N.R., Allan L., Arnold R., Deanfield J.E. Recurrence risks in offspring of adults with major heart defects: results from first cohort of British collaborative study. Lancet. 1998;351:311–316. doi: 10.1016/s0140-6736(97)06486-6. [DOI] [PubMed] [Google Scholar]
- 8.Whittemore R., Wells J.A., Castellsague X. A second-generation study of 427 probands with congenital heart defects and their 837 children. J. Am. Coll. Cardiol. 1994;23:1459–1467. doi: 10.1016/0735-1097(94)90392-1. [DOI] [PubMed] [Google Scholar]
- 9.Posch M.G., Perrot A., Schmitt K., Mittelhaus S., Esenwein E.M., Stiller B., Geier C., Dietz R., Gessner R., Ozcelik C., Berger F. Mutations in GATA4, NKX2.5, CRELD1, and BMP4 are infrequently found in patients with congenital cardiac septal defects. Am. J. Med. Genet. A. 2008;146A:251–253. doi: 10.1002/ajmg.a.32042. [DOI] [PubMed] [Google Scholar]
- 10.Erdogan F., Larsen L.A., Zhang L., Tümer Z., Tommerup N., Chen W., Jacobsen J.R., Schubert M., Jurkatis J., Tzschach A. High frequency of submicroscopic genomic aberrations detected by tiling path array comparative genome hybridisation in patients with isolated congenital heart disease. J. Med. Genet. 2008;45:704–709. doi: 10.1136/jmg.2008.058776. [DOI] [PubMed] [Google Scholar]
- 11.Thienpont B., Mertens L., de Ravel T., Eyskens B., Boshoff D., Maas N., Fryns J.P., Gewillig M., Vermeesch J.R., Devriendt K. Submicroscopic chromosomal imbalances detected by array-CGH are a frequent cause of congenital heart defects in selected patients. Eur. Heart J. 2007;28:2778–2784. doi: 10.1093/eurheartj/ehl560. [DOI] [PubMed] [Google Scholar]
- 12.Nowaczyk M.J., Carter M.T., Xu J., Huggins M., Raca G., Das S., Martin C.L., Schwartz S., Rosenfield R., Waggoner D.J. Paternal deletion 6q24.3: a new congenital anomaly syndrome associated with intrauterine growth failure, early developmental delay and characteristic facial appearance. Am. J. Med. Genet. A. 2008;146:354–360. doi: 10.1002/ajmg.a.32144. [DOI] [PubMed] [Google Scholar]
- 13.van der Velde E.T., Vander V.E., Vriend J.W., Mannens M.M., Uiterwaal C.S., Brand R., Mulder B.J. CONCOR, an initiative towards a national registry and DNA-bank of patients with congenital heart disease in the Netherlands: rationale, design, and first results. Eur. J. Epidemiol. 2005;20:549–557. doi: 10.1007/s10654-005-4264-9. [DOI] [PubMed] [Google Scholar]
- 14.Bache I., Hjorth M., Bugge M., Holstebroe S., Hilden J., Schmidt L., Brondum-Nielsen K., Bruun-Petersen G., Jensen P.K., Lundsteen C. Systematic re-examination of carriers of balanced reciprocal translocations: a strategy to search for candidate regions for common and complex diseases. Eur. J. Hum. Genet. 2006;14:410–417. doi: 10.1038/sj.ejhg.5201592. [DOI] [PubMed] [Google Scholar]
- 15.Menten B., Maas N., Thienpont B., Buysse K., Vandesompele J., Melotte C., de Ravel T., Van Vooren S., Balikova I., Backx L. Emerging patterns of cryptic chromosomal imbalance in patients with idiopathic mental retardation and multiple congenital anomalies: a new series of 140 patients and review of published reports. J. Med. Genet. 2006;43:625–633. doi: 10.1136/jmg.2005.039453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aerts S., Lambrechts D., Maity S., Van Loo P., Coessens B., De Smet F., Tranchevent L.C., De Moor B., Marynen P., Hassan B. Gene prioritization through genomic data fusion. Nat. Biotechnol. 2006;24:537–544. doi: 10.1038/nbt1203. [DOI] [PubMed] [Google Scholar]
- 17.Motoike T., Loughna S., Perens E., Roman B.L., Liao W., Chau T.C., Richardson C.D., Kawate T., Kuno J., Weinstein B.M. Universal GFP reporter for the study of vascular development. Genesis. 2000;28:75–81. doi: 10.1002/1526-968x(200010)28:2<75::aid-gene50>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 18.Thisse C., Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- 19.Caselli R., Mencarelli M.A., Papa F.T., Uliana V., Schiavone S., Strambi M., Pescucci C., Ariani F., Rossi V., Longo I. A 2.6 Mb deletion of 6q24.3-25.1 in a patient with growth failure, cardiac septal defect, thin upperlip and asymmetric dysmorphic ears. Eur. J. Med. Genet. 2007;50:315–321. doi: 10.1016/j.ejmg.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Bisgaard A.M., Kirchhoff M., Tümer Z., Jepsen B., Brøndum-Nielsen K., Cohen M., Hamborg-Petersen B., Bryndorf T., Tommerup N., Skovby F. Additional chromosomal abnormalities in patients with a previously detected abnormal karyotype, mental retardation, and dysmorphic features. Am. J. Med. Genet. A. 2006;140:2180–2187. doi: 10.1002/ajmg.a.31425. [DOI] [PubMed] [Google Scholar]
- 21.Osoegawa K., Vessere G.M., Utami K.H., Mansilla M.A., Johnson M.K., Riley B.M., L'Heureux J., Pfundt R., Staaf J., van der Vliet W.A. Identification of novel candidate genes associated with cleft lip and palate using array comparative genomic hybridisation. J. Med. Genet. 2008;45:81–86. doi: 10.1136/jmg.2007.052191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagamani S.C., Erez A., Eng C., Ou Z., Chinault C., Workman L., Coldwell J., Stankiewicz P., Patel A., Lupski J.R., Cheung S.W. Interstitial deletion of 6q25.2-q25.3: a novel microdeletion syndrome associated with microcephaly, developmental delay, dysmorphic features and hearing loss. Eur. J. Hum. Genet. 2009;17:573–581. doi: 10.1038/ejhg.2008.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pirola B., Bortotto L., Giglio S., Piovan E., Janes A., Guerrini R., Zuffardi O. Agenesis of the corpus callosum with Probst bundles owing to haploinsufficiency for a gene in an 8 cM region of 6q25. J. Med. Genet. 1998;35:1031–1033. doi: 10.1136/jmg.35.12.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barriot R., Breckpot J., Thienpont B., Brohée S., Van Vooren S., Coessens B., Tranchevent L.C., Van Loo P., Gewillig M., Devriendt K., Moreau Y. Collaboratively charting the gene-to-phenotype network of human congenital heart defects. Genome Med. 2010;2:16. doi: 10.1186/gm137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirby M.L. Oxford University Press; Oxford, New York: 2007. Cardiac Development. [Google Scholar]
- 26.Orelio C., Dzierzak E. Identification of 2 novel genes developmentally regulated in the mouse aorta-gonad-mesonephros region. Blood. 2003;101:2246–2249. doi: 10.1182/blood-2002-07-2260. [DOI] [PubMed] [Google Scholar]
- 27.Xia Z.P., Sun L., Chen X., Pineda G., Jiang X., Adhikari A., Zeng W., Chen Z.J. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature. 2009;461:114–119. doi: 10.1038/nature08247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shim J.H., Xiao C., Paschal A.E., Bailey S.T., Rao P., Hayden M.S., Lee K.Y., Bussey C., Steckel M., Tanaka N. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 2005;19:2668–2681. doi: 10.1101/gad.1360605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jadrich J.L., O'Connor M.B., Coucouvanis E. The TGF beta activated kinase TAK1 regulates vascular development in vivo. Development. 2006;133:1529–1541. doi: 10.1242/dev.02333. [DOI] [PubMed] [Google Scholar]
- 30.Xie M., Zhang D., Dyck J.R., Li Y., Zhang H., Morishima M., Mann D.L., Taffet G.E., Baldini A., Khoury D.S., Schneider M.D. A pivotal role for endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc. Natl. Acad. Sci. USA. 2006;103:17378–17383. doi: 10.1073/pnas.0604708103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang D., Gaussin V., Taffet G.E., Belaguli N.S., Yamada M., Schwartz R.J., Michael L.H., Overbeek P.A., Schneider M.D. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat. Med. 2000;6:556–563. doi: 10.1038/75037. [DOI] [PubMed] [Google Scholar]
- 32.Sanjo H., Takeda K., Tsujimura T., Ninomiya-Tsuji J., Matsumoto K., Akira S. TAB2 is essential for prevention of apoptosis in fetal liver but not for interleukin-1 signaling. Mol. Cell. Biol. 2003;23:1231–1238. doi: 10.1128/MCB.23.4.1231-1238.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roessler E., Ouspenskaia M.V., Karkera J.D., Vélez J.I., Kantipong A., Lacbawan F., Bowers P., Belmont J.W., Towbin J.A., Goldmuntz E. Reduced NODAL signaling strength via mutation of several pathway members including FOXH1 is linked to human heart defects and holoprosencephaly. Am. J. Hum. Genet. 2008;83:18–29. doi: 10.1016/j.ajhg.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bera T.K., Zimonjic D.B., Popescu N.C., Sathyanarayana B.K., Kumar V., Lee B., Pastan I. POTE, a highly homologous gene family located on numerous chromosomes and expressed in prostate, ovary, testis, placenta, and prostate cancer. Proc. Natl. Acad. Sci. USA. 2002;99:16975–16980. doi: 10.1073/pnas.262655399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryan A.K., Goodship J.A., Wilson D.I., Philip N., Levy A., Seidel H., Schuffenhauer S., Oechsler H., Belohradsky B., Prieur M. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J. Med. Genet. 1997;34:798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Battaglia A., Filippi T., Carey J.C. Update on the clinical features and natural history of Wolf-Hirschhorn (4p-) syndrome: experience with 87 patients and recommendations for routine health supervision. Am. J. Med. Genet. C. Semin. Med. Genet. 2008;148C:246–251. doi: 10.1002/ajmg.c.30187. [DOI] [PubMed] [Google Scholar]
- 37.Potocki L., Shaw C.J., Stankiewicz P., Lupski J.R. Variability in clinical phenotype despite common chromosomal deletion in Smith-Magenis syndrome [del(17)(p11.2p11.2)] Genet. Med. 2003;5:430–434. doi: 10.1097/01.gim.0000095625.14160.ab. [DOI] [PubMed] [Google Scholar]
- 38.McElhinney D.B., Krantz I.D., Bason L., Piccoli D.A., Emerick K.M., Spinner N.B., Goldmuntz E. Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome. Circulation. 2002;106:2567–2574. doi: 10.1161/01.cir.0000037221.45902.69. [DOI] [PubMed] [Google Scholar]
- 39.Sperling S., Grimm C.H., Dunkel I., Mebus S., Sperling H.P., Ebner A., Galli R., Lehrach H., Fusch C., Berger F., Hammer S. Identification and functional analysis of CITED2 mutations in patients with congenital heart defects. Hum. Mutat. 2005;26:575–582. doi: 10.1002/humu.20262. [DOI] [PubMed] [Google Scholar]
- 40.Garg V., Muth A.N., Ransom J.F., Schluterman M.K., Barnes R., King I.N., Grossfeld P.D., Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.