

Abstract
Cranioectodermal dysplasia (CED) is a disorder characterized by craniofacial, skeletal, and ectodermal abnormalities. Most cases reported to date are sporadic, but a few familial cases support an autosomal-recessive inheritance pattern. Aiming at the elucidation of the genetic basis of CED, we collected 13 patients with CED symptoms from 12 independent families. In one family with consanguineous parents two siblings were affected, permitting linkage analysis and homozygosity mapping. This revealed a single region of homozygosity with a significant LOD score (3.57) on chromosome 3q21-3q24. By sequencing candidate genes from this interval we found a homozygous missense mutation in the IFT122 (WDR10) gene that cosegregated with the disease. Examination of IFT122 in our patient cohort revealed one additional homozygous missense change in the patient from a second consanguineous family. In addition, we found compound heterozygosity for a donor splice-site change and a missense change in one sporadic patient. All mutations were absent in 340 control chromosomes. Because IFT122 plays an important role in the assembly and maintenance of eukaryotic cilia, we investigated patient fibroblasts and found significantly reduced frequency and length of primary cilia as compared to controls. Furthermore, we transiently knocked down ift122 in zebrafish embryos and observed the typical phenotype found in other models of ciliopathies. Because not all of our patients harbored mutations in IFT122, CED seems to be genetically heterogeneous. Still, by identifying CED as a ciliary disorder, our study suggests that the causative mutations in the unresolved cases most likely affect primary cilia function too.
Main Text
The etiology of several developmental genetic human disorders such as Bardet-Biedl (MIM 209900), Senior-Loken (MIM 266900), Meckel (MIM 249000), Joubert (MIM 213300), Ellis van Creveld (MIM 225500), and Jeune syndrome (MIM 208500) has been shown to involve ciliary dysfunction.1,2 The clinical picture of these ciliopathies is very variable with a broad range of phenotypes comprising, among others, cystic kidney and/or liver, laterality defects, skeletal changes, and retinal degeneration.1,2
Patients with cranioectodermal dysplasia, also known as Sensenbrenner syndrome (CED [MIM 218330]), present with craniofacial, skeletal, and ectodermal abnormalities, which suggests that this disorder is also a ciliopathic condition.2,3 In particular, CED patients show similarity to Jeune syndrome patients, with whom they share short stature, limb shortening, short ribs, narrow chest, brachydactyly, renal failure, and hepatic fibrosis.4 Jeune syndrome is caused by mutations in the ciliogenesis genes IFT80 (MIM 611177) or DYNC2H1 (MIM 603297), but the underlying genetic defect in CED is unknown.5–7
Most of the 20 CED cases reported to date are sporadic, but a few familial cases support an autosomal-recessive inheritance pattern. For this study (which was performed in accordance with the ethical standards of the responsible committees on human experimentation) we recruited CED patients from 12 independent families (informed consent was obtained), including two consanguineous siblings with typical CED symptoms from a Polish family (CED-01).
Patient 1, an 8-year-old girl, was the first child of healthy parents who were distantly related. She was born at term after an uncomplicated pregnancy with a birth weight of 3.6 kg (50th centile), body length of 57 cm (97th centile), head circumference (OFC) of 35 cm (50th centile), and chest circumference of 34 cm (50th centile). Dysmorphic signs noticed at birth comprised short limbs, protuberant abdomen, brachydactyly, dolichcephaly, high forehead, prominent auricles, full cheeks, telecanthus, broad nasal bridge, and low-set prominent ears. Clinical examinations revealed additional ectodermal abnormalities such as small and abnormally shaped teeth, sparse hair, skin laxity, and abnormal nails, and she had bilateral inguinal hernias (Figure 1A and Table 1). In the first year of life she had frequent respiratory infections. Signs of renal insufficiency (diminished glomerular filtration rate) were detected for the first time at the age of 4 years. A renal ultrasound scan showed normal size of both kidneys with decreased corticomedullary differentiation. Stage 1 chronic kidney disease due to tubulo-interstitial nephropathy without liver abnormalities was diagnosed. At the age of 5 years, she had a weight of 14.5 kg (5th centile), a height of 104 cm (25th centile), and a head circumference of 49.5 cm (50th centile). Psychomotor development was normal and ophthalmological analysis revealed no evidence of retinal dystrophy. Chromosome analysis showed a normal female karyotype (46, XX).
Figure 1.
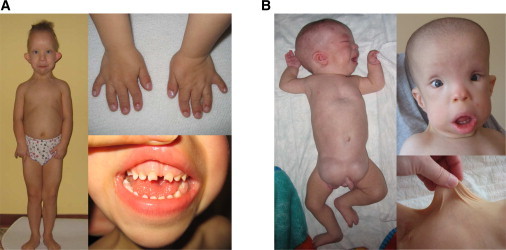
Consanguineous CED Patients
(A) CED-01, female patient 1 at the age of 4 years: note limb shortening, brachydactyly, short narrow thorax, protuberant abdomen, thin sparse hair, dolichocephaly, high prominent forehead, full cheeks, hypertelorism, small flat nose, full lower lip, prominent auricles, and small abnormally shaped teeth.
(B) CED-01, male patient 2 at the age of 4 months (left pane) and 1 year (right panes): note limb shortening, pectus excavatum, bilateral large inguinal hernias, characteristic face (similar to female), and skin laxity.
Table 1.
Clinical Features of CED Patients with and without IFT122 Mutations
| Family | CED-01 | CED-01 | CED-02 | CED-03 | CED-04 | CED-05 | CED-06 | CED-07 | CED-08 | CED-09 | CED-10 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
| Age at examination | 4–8 years | 0 months–4 years | 12 months | 4.5 years | 3 years | 15 months | 7 years | 5 years | 2 years | 4 years | 6 years |
| IFT122 mutation | V553G | V553G | S373F | W7C; c.502+5G>A | none | none | none | none | none | none | none |
| Sex | female | male | male | male | male | female | female | male | male | female | male |
| Psychomotor development | normal | normal | normal | normal | normal | normal | delayed | normal | normal | normal | normal |
| Dolichocephaly | + | + | + | + | + | + | − | + | + | + | + |
| Characteristic face | + | + | + | + | + | + | + | + | + | + | + |
| Dental anomalies | + | + | + | + | + | + | + | + | + | + | + |
| Sparse hair | + | + | + | − | + | + | + | + | + | + | + |
| Short/narrow thorax | + | + | + | + | + | + | + | + | + | + | + |
| Limb shortening | + | + | + | + | + | + | + | + | + | + | + |
| Short fingers | + | + | + | + | + | + | + | + | + | + | + |
| Bilateral inguinal hernias | + | + | + | + | − | − | − | − | − | − | − |
| Skin laxity | + | + | + | + | − | − | − | NA | NA | + | + |
| Renal failure | + | + | + | + | + | + | + | + | + | + | + |
| Liver disease | − | hepatomegaly | NA | hepatic fibrosis | − | − | − | − | − | − | − |
| Retinal dystrophy | − | − | − | − | + | − | − | NA | − | − | − |
“+” indicates feature present; “−” indicates feature not reported; NA indicates data not available.
Her brother (patient 2) was born after an uncomplicated pregnancy with a birth weight of 3.5 kg (50th centile) and body length of 54 cm (90th centile). Dysmorphic features were nearly identical to those of his sister (Figure 1B and Table 1). Blood tests at the age of 29 days revealed elevated creatinine values. Renal function deteriorated rapidly over the following months, and by the age of 3.5 years he had developed end-stage renal failure, and peritoneal dialysis was started. Ultrasound scans showed decreased size of both kidneys with increased echogenicity. The liver was enlarged and showed structural heterogeneity with hypoechogenic streaked foci along the strongly hyperechogenic portal vessels. Mental and motor development were normal. Ophthalmological examination showed no evidence of retinal dystrophy. Chromosome analysis revealed a normal male karyotype (46, XY).
In order to localize the disease-causing genetic defect in this family, we first performed linkage analysis and homozygosity mapping by using the Affymetrix GeneChip human Mapping 10K Array8 for genotyping both affected children and their parents. Genome-wide multipoint linkage analysis was performed with the Merlin9 software under the assumption of a fourth-cousin marriage, a fully penetrant autosomal-recessive trait with a disease frequency of 0.001, and no phenocopies as described previously.10 This revealed a single region of homozygosity with a significant LOD score (3.57) on chromosome 3q21-3q24 (Figure 2A; see Figure S1 available online for haplotype information), spanning 16.7 Mbp (between rs977683 and rs1992093) and containing 124 RefSeq protein-coding genes.
Figure 2.
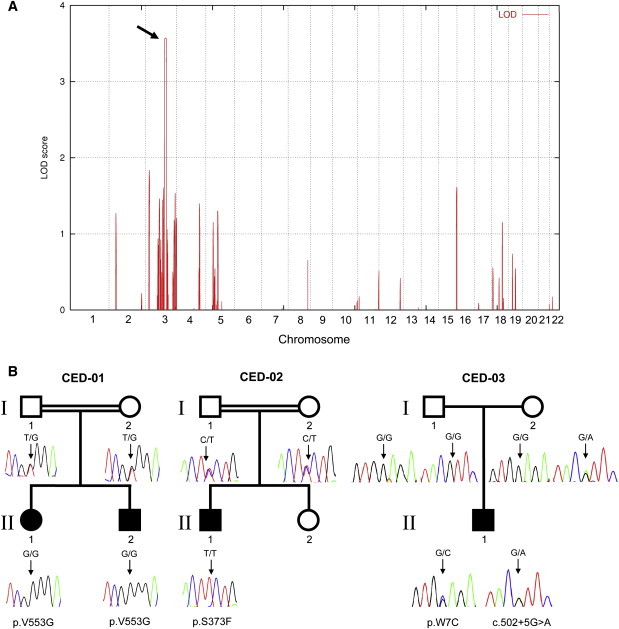
Linkage Interval, Pedigrees, and IFT122 Mutations
(A) Linkage interval on chromosome 3q21-3q24 (arrow) with a significant LOD score (3.57).
(B) Simplified pedigrees and IFT122 mutations. CED-01: Consanguineous Polish family with two affected siblings (patients 1 and 2) homozygous for p.V553G. CED-02: Consanguineous Norwegian family (patient 3 homozygous for p.S373F). CED-03: Sporadic patient 4, who is a compound heterozygote for a splice-site mutation (c.502+5G>A) and p.W7C. Filled symbols indicate CED patient status.
Sequencing of the coding regions and exon-intron boundaries of 79 candidate genes from this interval (Table S1) revealed a homozygous missense mutation (p.V553G) in the IFT122 (WDR10 [MIM 606045]) gene that cosegregated with the disease (Figure 2B, CED-01).
The main transcript of IFT122 encodes a 1292 amino acid cytoplasmic protein that has a molecular weight of approximately 140 kDa and contains seven WD40 domains, which are involved in the formation of β-propeller structures acting as platform for the association of binding partners.11 As a component of the intraflagellar transport complex A, the IFT122 protein is involved in retrograde ciliary transport, which returns proteins from the tip of the cilia to the cell body and plays an important role in the assembly and maintenance of eukaryotic cilia and flagella.12,13
We next examined IFT122 in the patients from the remaining 11 families of our cohort and identified one additional homozygous missense change (p.S373F) in a patient from a second consanguineous family14 (Figure 2B, CED-02, patient 3). In addition, we found putative compound heterozygosity for two different IFT122 mutations in patient 4, a previously described sporadic case15 (Figure 2B, CED-03, patient 4). This patient carried a donor splice-site change in intron 6 (c.502+5G>A) that was inherited from the mother in combination with a missense change in exon 1 (p.W7C) that was not found in either parent. Because paternity could be confirmed by an independent genotyping experiment with DNA from the father, this mutation can be considered to have arisen de novo. To test our hypothesis that this change affects the paternal IFT122 allele, leading to compound heterozygosity with the maternally inherited splice-donor change (c.502+5G>A), we performed RT-PCR with a series of primer combinations on RNA extracted from patient blood (Table S3).
Human IFT122 is composed of 31 exons with at least four alternatively spliced variants that are expressed in multiple tissues (Genome Browser, Human Protein Reference Database). Our experiment showed that in blood at least three transcripts are expressed, including the known isoforms 3 (comprising exons 1, 2, 3, 4, 6, 7, 8, 10, and 11) and 4 (comprising exons 1, 2, 3, 7, 8, 10, and 11) as well as a new transcript also without exon 6 (comprising exons 1, 2, 3, 4, 7, 8, 10, and 11) (Figures S2 and S3).
Subsequent sequencing revealed that in patient 4 the amplicon specific for isoform 3 was homozygous for p.W7C (Figure 3A), suggesting that this isoform is expressed from only one (paternal) allele. Amplicons corresponding to the new transcript without exon 6 or to isoform 4 were both heterozygous for the corresponding missense change, showing that these variants are expressed from both alleles (Figures 3B and 3C).
Figure 3.
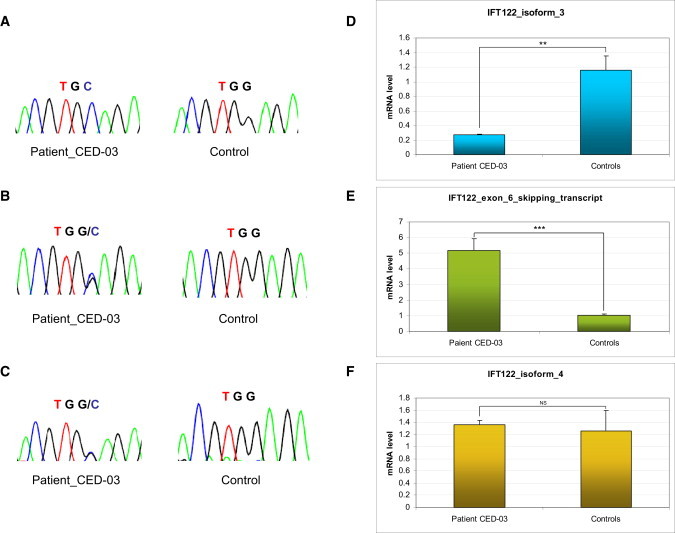
Compound Heterozygosity for c.502+5G>A and p.W7C in Patient 4, CED-03
Chromatograms show sequencing results corresponding to the affected codon of p.W7C obtained with exon 6 specific primers (A) as well as primers specific for the new transcript with exon 6 skipping (B) and for isoform 4, which lacks exon 6 by default (C). Results of qPCR experiments for the corresponding amplicons show a significant (Student's t test, p < 0.01) loss of (exon 6-containing) isoform 3 (D) and a highly significant (p < 0.001) increase in expression of the transcript with exon 6 skipping (E) in RNA extracted from patient blood. Expression of isoform 4 was unaltered (F). Error bars represent the standard deviation (n = 3). Expression of the GAPDH gene was used for normalization.
To quantify the RT-PCR results we performed real-time PCR (see Supplemental Data, available online, for details) with primer combinations that were specific for isoform 3, isoform 4, and the new transcript of IFT122 (Table S4).
The experiments were carried out on an Applied Biosystems 7900 instrument, in 96-well optical reaction plates (Applied Biosystems, Foster City, CA). Additional dissociation curves of the products were created. Each amplicon was analyzed in triplicate. Expression of the GAPDH gene was used for normalization.
This analysis showed that in patient cells IFT122 isoform 3 expression was significantly reduced whereas the transcript resulting from the skipping of exon 6 showed highly significant (p < 0.001) enrichment as compared to controls (Figures 3D and 3E). Expression of isoform 4 that does not contain exon 6 by default and is not affected by the splice-site mutation was unaltered (Figure 3F).
These observations are in line with the assumption that c.502+5G>A leads to skipping of exon 6 and thus to an out-of-frame transcript with premature termination of translation of the affected allele. Because all sequences that could be obtained from exon 6-containing transcripts exclusively contained p.W7C, they must be derived from the other allele, thus proving the assumed compound heterozygosity in patient 4. The p.W7C change, however, like the two homozygous mutations p.V553G and p.S373F, affects a highly conserved amino acid in all four alternatively spliced variants. All three missense mutations are located within or close to a WD40 domain (Figure S2) and were predicted to have a deleterious effect on IFT122 function by the PolyPhen software. None of the changes were found in 340 chromosomes from ethnically matched controls. Clinical features reported for patients with and without mutations in IFT122 are summarized in Table 1. The four patients with IFT122 mutations did not show grave phenotypic differences from the nine patients without mutations, and the preponderant overlap of clinical features between all patients suggests that CED, like other ciliary syndromes, is genetically heterogeneous.
To understand the impact of the mutant IFT122 transcripts and corresponding protein, we investigated the abundance and morphology of primary cilia in fibroblasts from patient 2 (the male patient from family CED-01) under different culture conditions.
Control fibroblasts from three independent, healthy German individuals were provided by the Department of Human Molecular Genetics of the Max Planck Institute for Molecular Genetics. Control and patient fibroblasts (patient 2) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, glutamine, and penicillin and streptomycin. Cells were plated onto coverslips coated with gelatin (Chemicon) or fibronectin (R&D Systems) at 105/cm2. Medium was replaced with OptiMem (GIBCO) and cultured for 72 hr. Cells were then fixed, blocked in 10% serum, and stained with mouse anti-acetylated α-tubulin (Sigma, 1:2500) at 4°C overnight. Cells were then washed and incubated overnight with Cy2-labeled secondary antibodies (Jackson ImmunoResearch) and TO-PRO-3 (Invitrogen) at 4°C, washed again, and mounted. Three coverslips for each of six cell lines (three control and three CED lines) were imaged with a PerkinElmer RS3 Spinning Disk Confocal Microscope with a Hamamatsu C9100-13 EMCCD camera. For frequency analysis, α-tubulin-stained cilia and TO-PRO-3-labeled nuclei were counted from 15 projected Z-stack images (five per coverslip) for each cell line. The average frequency across the three cell lines per genotype was determined and statistical significance was assessed by z-score analysis. Cilia lengths were measured with the National Institutes of Health (NIH) ImageJ software, and values were subjected to t tests.
These experiments showed significantly reduced cilia frequency and length in patient fibroblasts as compared to three independent controls (Figures 4A–4D).
Figure 4.
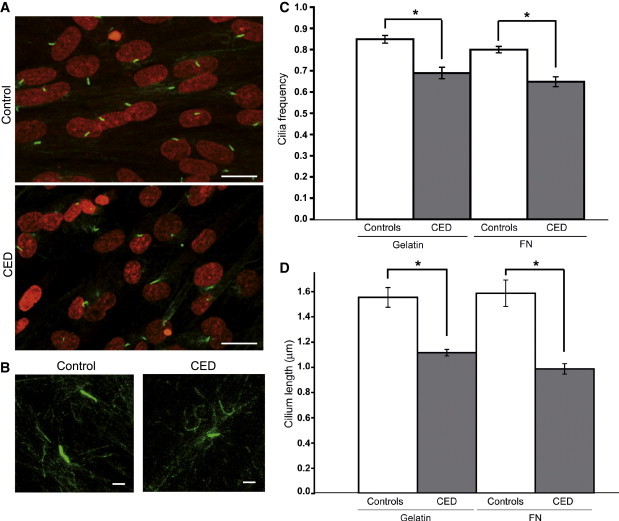
Primary Cilia in Control and CED Patient Fibroblasts
(A) Primary cilia from serum-starved primary fibroblasts stained with anti-acetylated-tubulin (green). Nuclei are counterstained with TO-PRO-3 (red). The scale bar represents 10 μm.
(B) High magnification of cilia showing difference in axonemal lengths. The scale bar represents 1 μm.
(C) Significant diffrences (p < 0.001) in average cilia frequency between control and CED patient fibroblast lines plated on gelatin and fibronectin (FN). Error bars represent the standard error of the mean (SEM).
(D) Significant diffrences (p < 0.05) in average axomenal lengths between patient and control. Error bars represent the SEM.
To corroborate our hypothesis that the IFT122 mutations we identified are functionally relevant and therefore responsible for the patient phenotype, we next transiently knocked down ift122 in zebrafish embryos, by using an antisense morpholino against the 5′UTR (5′-AAATCACGGCGTTTCCGAAAGCCGC-3′) immediately upstream of the start ATG as well as a second one targeting the exon 1-intron 1 splice boundary (5′-TATTGGTCTCTTTACCAGAACTCTG-3′) of Danio rerio ift122 (Figure S4).
Wild-type fish (AB × Tup Longfin) were staged and maintained according to standard protocols.16 Embryos were injected with 4–8 ng of morpholino at the 1–2 cell stage and incubated at 28.5°C until the desired age. Fluorescent antibody staining for α- and γ-Tubulin (Sigma, T6793 and T6667, 1:500) was used to mark the cilia and centrosomes, respectively, and was performed on 4% PFA fixed embryos with the appropriate Alexa secondary antibodies (1:1000). In situ hybridization for shha7 and ptc18 was performed via standard protocols.17 Fluorescent images were captured on a Zeiss LSM710 confocal microscope.
In 80 hr postfertilization (hpf) embryos injected with either morpholino, we observed several defects including shortened body axis and curvature (consistent with a convergent extension defect), cardiac edema, and small eyes (Figure 5A). In addition, at 5 dpf, morphants displayed pronephric cysts and a distended cranium consistent with hydrocephalus and otolith defects (Figure 5A). Thus, ift122 morphant zebrafish embryos display the typical phenotype observed in many other ciliopathies models (e.g., Bardet-Biedl syndrome, Jeune syndrome, and nephronophthisis [MIM 256100]).18–20
Figure 5.
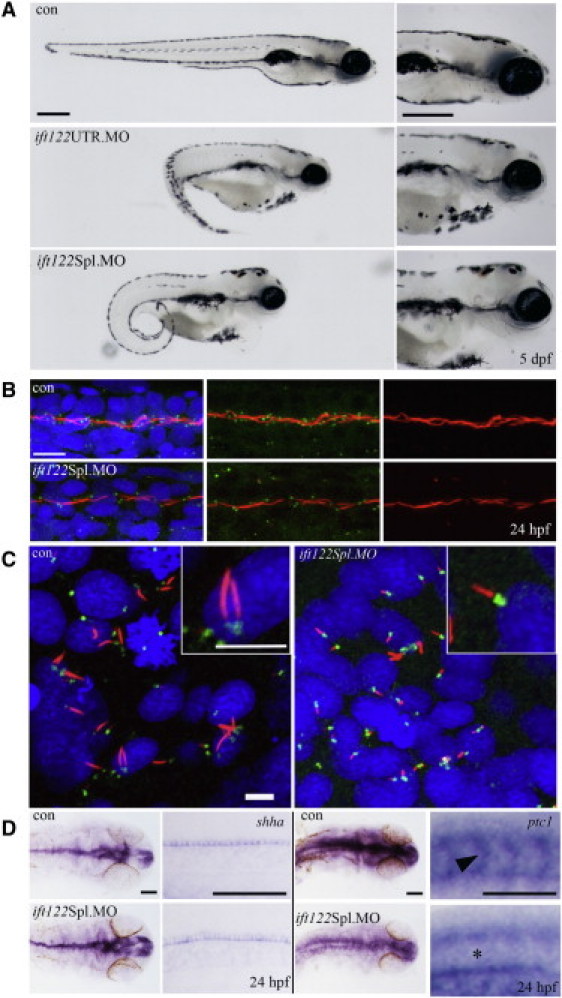
Loss of ift122 in Zebrafish Results in a Ciliopathy-Related Phenotype
(A) Gene-specific knockdown by morpholino modified antisense oligonucleotides to either the 5′UTR or to the exon 1-intron 1 splice boundary of ift122 caused reduced ocular development, melanocyte mislocalization, curly tails, renal cysts, and heart edemas by 5 dpf. All common features related to loss of ciliary genes in zebrafish. The scale bar represents 400 μm.
(B) Ift122 morphants display reduced and shortened motile cilia. Immunofluorescence staining for cilia (acetylated α-tubulin, red), centrosomes (γ-tubulin, green), and nuclei (DAPI, blue) in the pronephric duct (PND) shows reduced centrosome numbers and interrupted and/or shortened cilia in morphant compared to control embryos. The scale bar represents 10 μm.
(C) Cilia in the KV of ift122 Spl. Morphants are visibly shorter than uninjected controls. The scale bar represents 5 μm.
(D) A mild reduction in Hh signaling was observed in morphant embryos. Embryos were analyzed for either shha or ptc1 by in situ hybridization at 24 hpf; images of the head and somites (at approximately the somite 15 position) are displayed for comparison. Shha accumulates in the floor plate of the neural tube and in the notochord (at low levels) at the 24 hpf stage; this expression remains unperturbed in morphant embryos. Ptc1 appears to be globally reduced in ift122 morphants, most notably in the somites (see asterisk) where ptc1 is normally abundant in uninjected control embryos (see arrowhead). The scale bar represents 100 μm.
Because CED patients suffer from renal failure and cilia dysfunction and/or immotility is thought to underlie the pronephric duct dilation and cyst formation, we next examined the cilia in the pronephric duct (PND) of morphant embryos. Whereas cilia and their basal bodies were abundant in control embryos (24 hpf), their numbers in morphants were dramatically reduced (Figure 5B). Consistent with the patient fibroblast analysis, primary cilia shortening was observed in the zebrafish Kupffer's vesicle (Figure 5C).
Another consistent feature in CED is brachydactyly, which can be caused by impairment of the sonic hedgehog (shh) signaling pathway.21 Because shh signal transduction involves intraflagellar transport,13,22 we investigated shh signaling in our zebrafish morphants and found the expression of the directly regulated target gene ptc1 diminished and expanded (Figure 5D). This confirms the requirement of Ift122 for shh signal transduction and provides an explanation for the skeletal features observed in CED patients. We recognize that the use of antisense morpholinos is not without limitations and ideally a zebrafish mutant would be preferable; however, such a line is not yet available. One concern of morpholinos is the potential for off-target effects, especially upregulation of p53. We have excluded this possibility by using p53 morpholinos, in whose morphants we do not see any difference from fish injected with target morpholino alone (data not shown). As for determining the specificity of our ift122 ATG-morpholino, in the absence of a suitable zebrafish antibody we designed two additional splice-site morpholinos. Both of these gave rise to a phenotype identical to the ATG-morpholino, suggesting that we are successfully targeting ift122 and that the remaining possibility for some aspects of the morphant phenotypes to be nonspecific in origin is rather small.
Defects in cilia formation and function lead to broad pleiotropic phenotypes, which is probably due to their ubiquitous presence in many different cell types. In a previously described mouse model, Ift122 null mutant embryos show multiple developmental defects that result in a lethal phenotype.23 In embryonic fibroblasts from these mice, cilia are completely absent. However, a second mouse Ift122 null allele on a different strain background causes a ciliary phenotype more similar to the one we describe in patient fibroblasts: murine embryonic fibroblasts from these animals generate cilia, although at a reduced frequency. Such cilia are somewhat shortened and exhibit dramatic swelling at their tips with an accumulation of anterograde intraflagellar-transport proteins (e.g., Ift88), which is consistent with disturbed retrograde intraflagellar transport (J. Qin and J.E., unpublished data).
That the IFT122 mutations found in our patients are still compatible with life is probably due to the fact that they do not completely abolish the IFT122 gene product but rather lead to hypomorphic forms of the protein, which impair cilia formation and function.
Taken together, our results show that hypomorphic mutations in IFT122 cause a ciliary phenotype in homozygous carriers, which is reflected in zebrafish morphants. As in other ciliary disorders, the CED clinical phenotype is pleiotropic because of the involvement of cilia in a multitude of tissues and during many developmental stages, as well as because of the role of cilia in morphogenetic signal transduction.13,22,24
Because only 4 out of the 13 CED patients in our cohort harbored mutations in IFT122, it must be assumed that this disorder is genetically heterogeneous. This is only the second IFT gene in which human mutations have been discovered, and it highlights the diverse nature of the resulting phenotype compared with IFT80. One of the key differences distinguishing CED from Jeune syndrome is the presence of skin and dental dysplasia, and so it remains to be determined whether primary cilia and IFT in particular are required for ectoderm development.
Acknowledgments
We are grateful to all affected individuals and their families for their participation in the study. We thank Daniela Pilz, Ian D. Young, Krystyna Chrzanowska, Satoshi Sasaki, Han Brunner, Helen Fryssira, and Ewa Obersztyn for providing DNA samples. We thank Lia Abbasi Moheb and Lars Riff Jensen for continuous support, Marianne Schlicht and Susanne Freier for technical assistance, and Joseph Goodhouse for help with confocal microscopy. This work was supported by grants from the EU Framework 7 EUCILIA (HEALTH-F2-2007-201804) and the Max-Planck-Innovation funds. Philip Beales is a Wellcome Trust Senior Research Fellow.
Contributor Information
Joanna Walczak-Sztulpa, Email: jsztulpa@umed.poznan.pl.
Andreas W. Kuss, Email: kuss_a@molgen.mpg.de.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Genome Browser, http://genome.ucsc.edu/
Human Protein Reference Database, http://www.hprd.org/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PolyPhen, http://genetics.bwh.harvard.edu/pph/
References
- 1.Badano J.L., Mitsuma N., Beales P.L., Katsanis N. The ciliopathies: An emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 2.Lehman J.M., Michaud E.J., Schoeb T.R., Aydin-Son Y., Miller M., Yoder B.K. The Oak Ridge Polycystic Kidney mouse: Modeling ciliopathies of mice and men. Dev. Dyn. 2008;237:1960–1971. doi: 10.1002/dvdy.21515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konstantinidou A.E., Fryssira H., Sifakis S., Karadimas C., Kaminopetros P., Agrogiannis G., Velonis S., Nikkels P.G., Patsouris E. Cranioectodermal dysplasia: A probable ciliopathy. Am. J. Med. Genet. A. 2009;149A:2206–2211. doi: 10.1002/ajmg.a.33013. [DOI] [PubMed] [Google Scholar]
- 4.Eke T., Woodruff G., Young I.D. A new oculorenal syndrome: Retinal dystrophy and tubulointerstitial nephropathy in cranioectodermal dysplasia. Br. J. Ophthalmol. 1996;80:490–491. doi: 10.1136/bjo.80.5.490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beales P.L., Bland E., Tobin J.L., Bacchelli C., Tuysuz B., Hill J., Rix S., Pearson C.G., Kai M., Hartley J. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 2007;39:727–729. doi: 10.1038/ng2038. [DOI] [PubMed] [Google Scholar]
- 6.Dagoneau N., Goulet M., Geneviève D., Sznajer Y., Martinovic J., Smithson S., Huber C., Baujat G., Flori E., Tecco L. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am. J. Hum. Genet. 2009;84:706–711. doi: 10.1016/j.ajhg.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merrill A.E., Merriman B., Farrington-Rock C., Camacho N., Sebald E.T., Funari V.A., Schibler M.J., Firestein M.H., Cohn Z.A., Priore M.A. Ciliary abnormalities due to defects in the retrograde transport protein DYNC2H1 in short-rib polydactyly syndrome. Am. J. Hum. Genet. 2009;84:542–549. doi: 10.1016/j.ajhg.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsuzaki H., Loi H., Dong S., Tsai Y.Y., Fang J., Law J., Di X., Liu W.M., Yang G., Liu G. Parallel genotyping of over 10,000 SNPs using a one-primer assay on a high-density oligonucleotide array. Genome Res. 2004;14:414–425. doi: 10.1101/gr.2014904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 10.Garshasbi M., Motazacker M.M., Kahrizi K., Behjati F., Abedini S.S., Nieh S.E., Firouzabadi S.G., Becker C., Rüschendorf F., Nürnberg P. SNP array-based homozygosity mapping reveals MCPH1 deletion in family with autosomal recessive mental retardation and mild microcephaly. Hum. Genet. 2006;118:708–715. doi: 10.1007/s00439-005-0104-y. [DOI] [PubMed] [Google Scholar]
- 11.Orlicky S., Tang X., Willems A., Tyers M., Sicheri F. Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell. 2003;112:243–256. doi: 10.1016/s0092-8674(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 12.Tsao C.C., Gorovsky M.A. Tetrahymena IFT122A is not essential for cilia assembly but plays a role in returning IFT proteins from the ciliary tip to the cell body. J. Cell Sci. 2008;121:428–436. doi: 10.1242/jcs.015826. [DOI] [PubMed] [Google Scholar]
- 13.Pedersen L.B., Rosenbaum J.L. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr. Top. Dev. Biol. 2008;85:23–61. doi: 10.1016/S0070-2153(08)00802-8. [DOI] [PubMed] [Google Scholar]
- 14.Fry A.E., Klingenberg C., Matthes J., Heimdal K., Hennekam R.C.M., Pilz D.T. Connective tissue involvement in two patients with features of cranioectodermal dysplasia. Am. J. Med. Genet. 2009;149A:2212–2215. doi: 10.1002/ajmg.a.33027. [DOI] [PubMed] [Google Scholar]
- 15.Zaffanello M., Diomedi-Camassei F., Melzi M.L., Torre G., Callea F., Emma F. Sensenbrenner syndrome: A new member of the hepatorenal fibrocystic family. Am. J. Med. Genet. 2006;140A:2336–2340. doi: 10.1002/ajmg.a.31464. [DOI] [PubMed] [Google Scholar]
- 16.Westerfield M. University of Oregon Press; Eugene, Oregon: 1995. The Zebrafish Book - A Guide for the Laboratory Use of Zebrafish (Danio rerio) [Google Scholar]
- 17.Coutelle O., Blagden C.S., Hampson R., Halai C., Rigby P.W., Hughes S.M. Hedgehog signalling is required for maintenance of myf5 and myoD expression and timely terminal differentiation in zebrafish adaxial myogenesis. Dev. Biol. 2001;236:136–150. doi: 10.1006/dbio.2001.0193. [DOI] [PubMed] [Google Scholar]
- 18.Tobin J.L., Beales P.L. Restoration of renal function in zebrafish models of ciliopathies. Pediatr. Nephrol. 2008;23:2095–2099. doi: 10.1007/s00467-008-0898-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerdes J.M., Liu Y., Zaghloul N.A., Leitch C.C., Lawson S.S., Kato M., Beachy P.A., Beales P.L., DeMartino G.N., Fisher S. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 2007;39:1350–1360. doi: 10.1038/ng.2007.12. [DOI] [PubMed] [Google Scholar]
- 20.Oishi I., Kawakami Y., Raya A., Callol-Massot C., Izpisúa Belmonte J.C. Regulation of primary cilia formation and left-right patterning in zebrafish by a noncanonical Wnt signaling mediator, duboraya. Nat. Genet. 2006;38:1316–1322. doi: 10.1038/ng1892. [DOI] [PubMed] [Google Scholar]
- 21.Niedermaier M., Schwabe G.C., Fees S., Helmrich A., Brieske N., Seemann P., Hecht J., Seitz V., Stricker S., Leschik G. An inversion involving the mouse Shh locus results in brachydactyly through dysregulation of Shh expression. J. Clin. Invest. 2005;115:900–909. doi: 10.1172/JCI200523675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eggenschwiler J.T., Anderson K.V. Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol. 2007;23:345–373. doi: 10.1146/annurev.cellbio.23.090506.123249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cortellino S., Wang C., Wang B., Bassi M.R., Caretti E., Champeval D., Calmont A., Jarnik M., Burch J., Zaret K.S. Defective ciliogenesis, embryonic lethality and severe impairment of the Sonic Hedgehog pathway caused by inactivation of the mouse complex A intraflagellar transport gene Ift122/Wdr10, partially overlapping with the DNA repair gene Med1/Mbd4. Dev. Biol. 2009;325:225–237. doi: 10.1016/j.ydbio.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tran P.V., Haycraft C.J., Besschetnova T.Y., Turbe-Doan A., Stottmann R.W., Herron B.J., Chesebro A.L., Qiu H., Scherz P.J., Shah J.V. THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat. Genet. 2008;40:403–410. doi: 10.1038/ng.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.