

Abstract
Genomic rearrangements involving the peripheral myelin protein gene (PMP22) in human chromosome 17p12 are associated with neuropathy: duplications cause Charcot-Marie-Tooth disease type 1A (CMT1A), whereas deletions lead to hereditary neuropathy with liability to pressure palsies (HNPP). Our previous studies showed that >99% of these rearrangements are recurrent and mediated by nonallelic homologous recombination (NAHR). Rare copy number variations (CNVs) generated by nonrecurrent rearrangements also exist in 17p12, but their underlying mechanisms are not well understood. We investigated 21 subjects with rare CNVs associated with CMT1A or HNPP by oligonucleotide-based comparative genomic hybridization microarrays and breakpoint sequence analyses, and we identified 17 unique CNVs, including two genomic deletions, ten genomic duplications, two complex rearrangements, and three small exonic deletions. Each of these CNVs includes either the entire PMP22 gene, or exon(s) only, or ultraconserved potential regulatory sequences upstream of PMP22, further supporting the contention that PMP22 is the critical gene mediating the neuropathy phenotypes associated with 17p12 rearrangements. Breakpoint sequence analysis reveals that, different from the predominant NAHR mechanism in recurrent rearrangement, various molecular mechanisms, including nonhomologous end joining, Alu-Alu-mediated recombination, and replication-based mechanisms (e.g., FoSTeS and/or MMBIR), can generate nonrecurrent 17p12 rearrangements associated with neuropathy. We document a multitude of ways in which gene function can be altered by CNVs. Given the characteristics, including small size, structural complexity, and location outside of coding regions, of selected rare CNVs, their identification remains a challenge for genome analysis. Rare CNVs may potentially represent an important portion of “missing heritability” for human diseases.
Introduction
Genomic disorders are the pathologic conditions caused by rearrangements of the human genome.1–3 The 17p12 rearrangement-associated neuropathy is among the earliest identified genomic disorders: 17p12 duplications can lead to Charcot-Marie-Tooth disease type 1A (CMT1A [MIM 118220]) whereas deletions can lead to hereditary neuropathy with liability to pressure palsies (HNPP [MIM 162500]).4–7 The CMT1A neuropathy phenotype is caused by a gene dosage effect.8
Genomic rearrangements can be categorized into two major groups: recurrent and nonrecurrent rearrangements.9 Nonallelic homologous recombination (NAHR) between paralogous sequence repeats is the predominant mechanism underlying recurrent rearrangements with clustered breakpoints, whereas various mechanisms or models are implicated in nonrecurrent rearrangements with variable breakpoints.2,9 Notably, a previous study showed that the NAHR-mediated, recombination hotspot-associated, recurrent rearrangements are specific to meiosis, i.e., germline events.10
The recurrent 17p12 rearrangements associated with CMT1A or HNPP are generated by NAHR events between two low-copy repeats (LCRs), alternatively termed segmental duplications (SDs),11 specifically involving paralogous distal and proximal CMT1A-REP copies as homologous recombination substrates (Figure 1).12–14 The common region affected by recurrent 17p12 rearrangements is ∼1.4 Mb in length (Figure 1), and copy number variation (CNV) of the PMP22 (peripheral myelin protein 22 [MIM 601097]) gene that maps within this genomic interval is responsible for the CMT1A and HNPP neuropathy phenotypes. This contention is supported by the studies of both point mutations and altered gene dosage of PMP22.7,15–19
Figure 1.
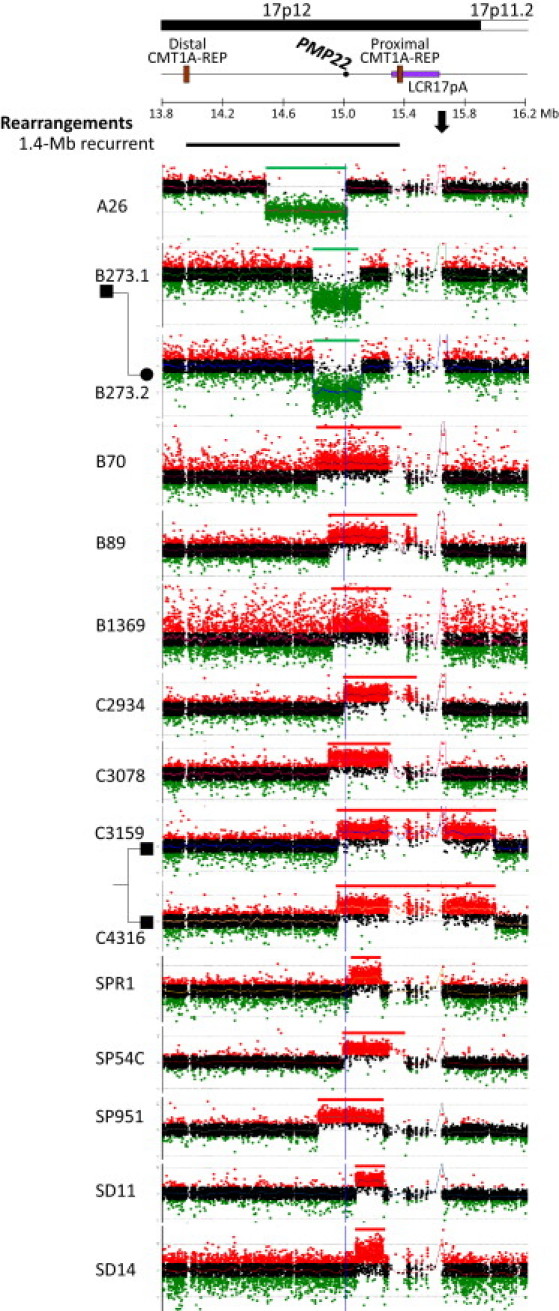
Oligonucleotide aCGH Analysis of Simple Nonrecurrent Genomic Rearrangements of 17p12 Associated with CMT1A or HNPP
Above, for reference, is a horizontal line showing the 1.4 Mb common CMT1A- or HNPP-associated rearrangement region and its flanking sequences on human chromosome 17 with cytogenetic bands depicted above and Megabase (Mb) genomic coordinates (NCBI build 36) below. Locations of the PMP22 gene, the distal and proximal CMT1A-REPs, and LCR17pA are shown. The black horizontal bar shows the location and size of common recurrent 17p12 rearrangements (both deletion and duplication) associated with CMT1A or HNPP. To the left are laboratory identification numbers of the subjects with nonrecurrent 17p12 rearrangements. The green (loss), black (no change), and red (gain) dots show the relative intensities (deviation from the horizontal line of log2Ratio = zero) and genomic locations of the oligonucleotide probes employed in our aCGH assay. The regions that lack unique probes correspond to the LCRs. The regions with copy number gains are indicated in red horizontal bars, and the losses are shown in green. Related subjects: deletions B273.1 and B273.2; duplications C3159 and C4316. The blue vertical lines indicate the location of the PMP22 gene. The arrow indicates the copy number change caused by known polymorphism in the control DNA (NA15510).
It has been shown that most (>99%) of the CMT1A- or HNPP-associated rearrangements in 17p12 are recurrent and mediated by NAHR.20 However, the role of nonrecurrent 17p12 rearrangements in neuropathy, and the rearrangement mechanism(s) for such nonrecurrent rearrangements, have not been extensively investigated and are therefore not well understood.
To study the underlying mechanism(s) for 17p12 rearrangements and the critical gene(s) for the neuropathy phenotypes, we investigated 21 subjects with either CMT1A or HNPP neuropathy that were shown by previous assays to have apparent rare CNVs of atypical size in 17p12. We examined these genomic rearrangements by high-density oligonucleotide-based array comparative genomic hybridization (aCGH) and breakpoint sequence analyses. Our observations suggest that various mechanisms, including nonhomologous end joining (NHEJ), Alu-Alu-mediated recombination, and the newly proposed replication-based mechanisms, are involved in the CMT1A- or HNPP-associated nonrecurrent rearrangements. Furthermore, the studies confirm that PMP22, by either altered dosage or dysregulation, is the major gene responsible for the neuropathy phenotypes of CMT1A and HNPP. These studies document the multitude of structural changes that can alter gene function. Our findings implicate rare CNV in both Mendelian traits and sporadic diseases as well as being potentially responsible for some fraction of the missing heritability of apparent complex traits.
Subjects and Methods
Subjects
Twenty-one nonrecurrent rearrangements of 17p12 associated with CMT1A or HNPP were studied and summarized together with another nine nonrecurrent 17p12 rearrangements that we recently published.20 The subjects were initially screened by the following conventional assays:20–24 multiplex ligation-dependent probe amplification (MLPA): A23, A26, A29, B1369; restriction fragment length polymorphism (RFLP) genotyping with probes showing the recombination hotspots: B70, B89, B273.1, B273.2; microsatellite genotyping and MLPA: C1292, C2405, C2934, C3011, C3078, C3159, C4316; and Southern blot and MLPA: SPR1, SPR2, SP54C, SP951, SD11, SD14. Samples from CMT1A or HNPP subjects were obtained with informed consent approved by the Institutional Review Board for Human Subject Research at Baylor College of Medicine and/or collaborative institutions. Anonymous genomic DNAs (A23, A26, and A29) were provided by Athena Diagnostics (Worcester, MA). The female control DNA (NA15510) was obtained from Coriell Cell Repositories.
Oligonucleotide-Based aCGH Analysis
We designed high-density oligonucleotide-based microarrays for a comparative genomic hybridization assay to finely examine the location, size, genomic content, and breakpoint interval of the 17p12 rearrangements associated with CMT1A or HNPP. This array is based on the Agilent 8 × 15K format. Approximately 15,000 oligonucleotide probes were selected from the Agilent eArray system to interrogate the 1.4 Mb common CMT1A or HNPP rearrangement region and its 1 Mb flanking regions with a genome resolution of ∼300 bp. In subject C3011 whose rearrangement extends outside the array coverage of the 8 × 15K array, another 4 × 44K custom-designed CGH array covering the short arm of the human chromosome 17 was employed.20 Probes having sequences complementary to more than one genomic locus have been purged and only unique sequence probes were employed. After digestion with AluI and RsaI, the test DNAs were labeled with Cy5-dCTP and control DNA was labeled with Cy3-dCTP by means of the BioPrime Array CGH genomic labeling kit (Invitrogen Corporation, Carlsbad, CA). Purification of labeling products, array hybridization, washing, scanning, and data analysis were conducted by following the Agilent oligonucleotide aCGH protocol (version 5.0).
Long-Range PCR Amplification
The oligonucleotide aCGH data were used to initially pinpoint approximate breakpoint positions in the genome. We next designed outward-facing primers for presumed tandem duplications and used inward-facing primers for deletions to amplify rearrangement breakpoint junction.25 Different orientations and combinations of primers were also tested for breakpoint analyses considering the potential for complex rearrangements. Long-range PCR was conducted with TaKaRa LA Taq polymerase. A 50 μl PCR reaction was performed with 2.5 U TaKaRa LA Taq polymerase with 1 × PCR buffer, 0.4 mM dNTP, 10 pmol of each primer, 1 μl DMSO, and 200 ng DNA template. The PCR conditions were as follows: 98°C for 30 s, 32 cycles at 94°C for 1 min, 65°C for 20 s, and 68°C for 20 min, followed by 68°C for 10 min.
Breakpoint Sequence Analysis
PCR products that potentially contained breakpoint junctions were submitted to SeqWright DNA Technology Services (Houston, TX) for sequencing by the Sanger dideoxy method. DNA sequences were analyzed by comparing to the human genome reference assembly (NCBI Build 36) with the BLAT tool from the UCSC Genome Browser.
Results
Twenty-one subjects with CMT1A or HNPP were initially found to have rare CNVs of atypical size in 17p12 by microsatellite genotyping, MLPA, RFLP, and/or Southern blot. These conventional assays are both locus-specific and “low-resolution” genome analysis tools for assessing CNVs. We employed high-density oligonucleotide-based aCGH for copy number determination and long-range PCR amplification for breakpoint determination and subsequent sequence analysis in this study to comprehensively examine the “genomotype” (i.e., location, size, genomic content, and simple or complex type of genomic rearrangement)26 and breakpoint interval of the nonrecurrent 17p12 rearrangements associated with CMT1A or HNPP.
Genomic Deletion Rearrangements Involving PMP22
Three simple genomic deletions, two of which are from related subjects, were identified in this study (Figure 1). The deletion rearrangement in subject A26 is 536 kb in length, and the proximal breakpoint maps within the PMP22 gene. Therefore, only the 3′ end portion of PMP22, including coding exons 4 and 5, was deleted. Some genome rearrangements can have microhomologies at breakpoints, i.e., one or more nucleotides shared between distal and proximal reference sequences at the rearrangement ends.9 However, no microhomology was detected at the breakpoint sequence of A26 (Figure S1 available online). As expected, the remaining two deletions in related subjects B273.1 and B273.2 (father and daughter) are the same. The 17p12 deletion in the B273 family is ∼320 kb, which includes the entire PMP22 gene. We surmise that B273.1 transmitted the deletion to B273.2. Breakpoint interval amplification was not achieved and no breakpoint sequence is available for the deletion in the B273 family. Because the rearrangement breakpoints in subjects B273.1 and B273.2 are apparently located outside LCR regions, as evidenced by a transition from normal copy to a loss relative to the control (i.e., a deletion CNV; Figure 1), a LCR-associated failure of specific amplification can probably be excluded. However, aCGH provides neither orientation nor genome positional information of DNA segments in the investigated genomic rearrangements. Considering that our long-range PCR assay can amplify DNA segments up to 15–20 kb in size, we hypothesize that a large insertion at the deletion breakpoint, or other complex rearrangement that is not resolvable in the aCGH assay, were potential causes precluding our ability to capture the breakpoint by PCR amplification of the perceived breakpoint interval in B273.1 and B273.2.
Genomic Duplication Rearrangements Involving PMP22
Eight unique genomic duplications, varying from ∼400 to 1048 kb in length, were found to have increased copy number of the entire PMP22 gene (Figure 1). Related subjects C3159 and C4316 are brothers. The aCGH analysis showed that the duplications in C3159 and C4316 were the same. Therefore, we hypothesized that this duplication was inherited from one of their parents. However, no parental DNA sample is available for further study of parental origin. Breakpoint sequence analysis showed that both distal and proximal breakpoints of this duplication map within AluY elements (Figure S1).
Interestingly, six out of the remaining seven 17p12 duplications (B70, B89, B1369, C2934, C3078, and SP54C) have proximal breakpoints in LCRs, i.e., LCR17pA or proximal CMT1A-REP (Figure 1),27 whereas their distal breakpoints are in unique genomic regions. Therefore, it can be challenging to amplify the specific breakpoint intervals in these subjects because of the inability to uniquely identify a specific genomic location for a PCR assay. Long-range PCR amplification was achieved only in subject SP54C and the breakpoint sequencing data revealed a recombination event between two AluSg elements (Figure S1).
In subject SP951, sequence complexity was identified in breakpoint sequence analysis (Figure S1). A 23 bp fragment (TAAAATTATCTTTTAGTCATTAA) was inserted at the join point between the distal and proximal breakpoints. This insertion can be copied from the DNA template that is only a few nucleotides adjacent to the distal breakpoint. These findings suggest the potential involvement of the serial replication slippage (SRS) mechanism in generating the complex duplication in SP951.28,29 Alternatively, the sequence complexity is also consistent with multiple NHEJ events. The enzymatic features of both replication slippage and NHEJ mechanisms underlying genomic rearrangements have been summarized by Lieber (2010).30
Genomic Duplication Involving Only the Sequences Upstream of PMP22
In addition to the genomic rearrangements inclusive of PMP22, it was recently reported that a genomic duplication affecting only the upstream region of PMP22 can also lead to CMT1A potentially by altering the PMP22 gene expression.24 Two previously reported subjects (SD11 and SD14) with this 186 kb duplication24 were also studied by oligonucleotide-based aCGH in this study and the rare CNV was confirmed. This duplication CNV affects only the upstream sequence with one breakpoint mapping ∼34 kb proximal to the PMP22 gene (Figure 2).24 A 1 bp microhomology was identified at the breakpoint (Figure S1).
Figure 2.
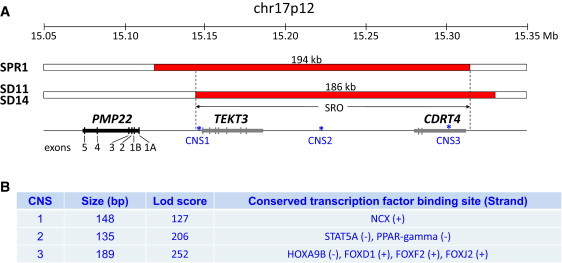
CMT1A-Associated 17p12 Duplications Exclusive of the PMP22 Coding Region but Involving the Potential Upstream Regulatory Sequence
(A) Two duplications (red bars) of the upstream region of PMP22 were identified in subject SPR1 and related subjects SD11 and SD14. The SRO involves the TEKT3 and CDRT4 genes, whereas PMP22 is intact and has normal copy number. Both the noncoding exons (1A and 1B) and coding exons (2 to 5) are shown. Three ultra CNSs associated with conserved human transcription factor binding sites were present in SRO.
(B) The information of CNS size, lod score, and transcription factor binding site was provided. Details are provided in Figures S2 and S3.
The other CMT1A-associated duplication devoid of the PMP22 coding region was newly identified in subject SPR1 to be 194 kb in length with one breakpoint only ∼9 kb proximal to the PMP22 gene (Figure 2). The breakpoint sequence analysis showed a 5 bp microhomology (TCTCT) at the junctions (Figure S1).
It has been hypothesized that the CMT1A-associated duplications exclusive of the PMP22 coding region may affect a conserved region upstream of PMP22 and potentially cause dysregulation of PMP22 gene expression.24 The above two different duplications share a 168 kb smallest region of overlap (SRO; chr17:15,143,663-15,311,619, NCBI build 36) with one end that is located only 34 kb proximal to the PMP22 gene. Several highly conserved noncoding sequences (CNSs) are located within this duplication SRO interval (Figure 2; Figure S2). These observations support the potential involvement of altered dosage or CNV of the regulatory regions for PMP22 in the CMT1A-associated duplications exclusive of PMP22 coding sequences.
Clinical Findings in PMP22 Upstream Duplication CNVs
No differentiating clinical features were observed specific to the CMT subjects with PMP22 upstream duplications. However, their clinical phenotypes, although variable between individuals, appear milder than those usually mediated by PMP22 gene duplications.
As for the 186 kb PMP22 upstream duplication previously identified in multiple unrelated families (including subjects SD11 and SD14 in this study), the phenotype is variable between and within affected families.24 However, milder phenotypes than those in classic CMT1A caused by PMP22 duplications were observed in most cases, including relatively late age of onset, normal to brisk reflexes, and mildly reduced nerve conduction velocities (NCVs).24
A relatively mild phenotype was also observed in subject SPR1 that was newly identified in this study to have a 194 kb PMP22 upstream duplication CNV. Motor NCVs of upper and lower extremities were mildly reduced (median nerve right 44.5 m/s, ulnar nerve right 44 m/s, peroneal nerve right 30 m/s).
Complex Rearrangements Involving PMP22
In addition to the simple types (either duplication or deletion) of nonrecurrent 17p12 rearrangements, we also identified three subjects with CMT1A-associated complex genomic rearrangements involving the entire PMP22 gene (Figure 3). Subjects C1292 and C2405 are related; C2405 is the sister of C1292's maternal grandfather. High-density aCGH analysis revealed a 1294 kb complex rearrangement in 17p12 with aCGH data showing a pattern consistent with deletion-normal-duplication wherein the entire PMP22 gene was duplicated (Figure 3). Breakpoint sequence analysis showed an AluY-AluY-mediated recombination between the distal end of the deletion and the proximal end of the duplication, whereas a 22 bp insertion of unknown origin was identified at the breakpoint interval between the distal end of duplication and the proximal end of deletion (Figure S1).
Figure 3.
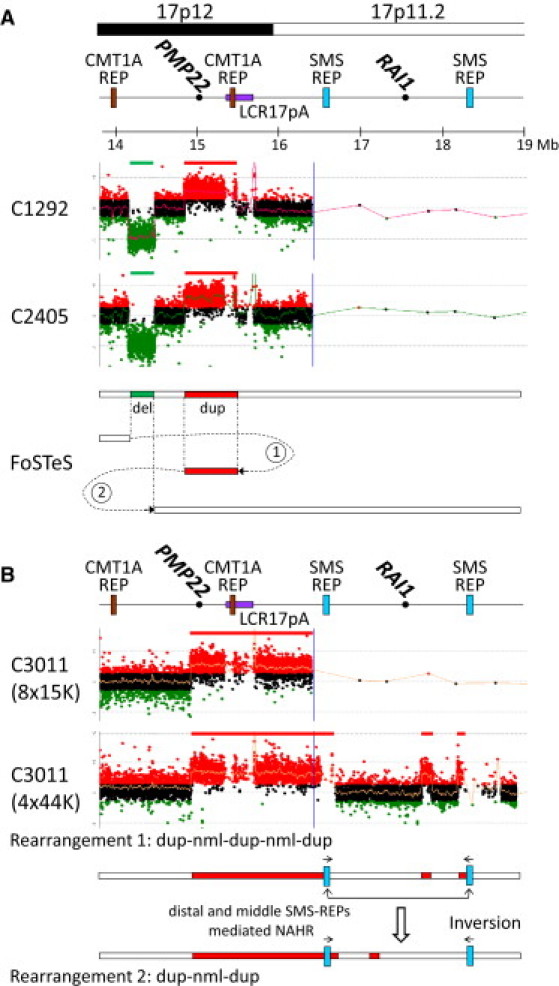
Oligonucleotide aCGH Analysis Revealed Complex Genomic Rearrangements in 17p12
(A) Subjects C1292 and C2405 are related (C2405 is the sister of C1292's maternal grandfather). Two proposed FoSTeS events (FoSTeS × 2) consistent with this complex rearrangement were shown.
(B) In C3011, the 17p CGH array (4 × 44k format) revealed additional duplications in 17p11.2 that is not covered by the CMT1A array (8 × 15k format). Rearrangement abbreviations: nml, normal; dup, duplication. This complex rearrangement can be alternatively interpreted as duplication-normal-duplication-normal-duplication or duplication-normal-duplication that is accompanied by an inversion polymorphism mediated by NAHR between two inverted LCRs (distal and proximal SMS-REPs).
The genomic rearrangement in subject C3011 is the largest one among the 17 unique CMT1A or HNPP rearrangements. The proximal end of the genomic rearrangement in C3011 extended outside the coverage of our 8 × 15K CGH microarray interrogating the 1.4 Mb common 17p12 rearrangement region and its flanking sequences (1 Mb on each side; Figure 3). Therefore, a 4 × 44K CGH array covering the entire short arm of the human chromosome 17 was used to re-examine the genomic rearrangement in C3011. Interestingly, two additional segments with copy number gains were identified. These observations indicated a complex rearrangement of ∼3.4 Mb in C3011 (Figure 3). Amplification of potential breakpoint intervals was achieved only between the distal and proximal ends of the small duplication from 17.73 to 17.86 Mb on the human chromosome 17 (NCBI build 36) and a 23 bp insertion of unknown origin was identified at the breakpoints (Figure S1).
Exonic Rearrangements Deleting One or Several Exons of PMP22
Three exonic deletions were identified by our high-density aCGH analysis. The 5 kb deletion in subject A23 and the 13 kb deletion in subject A29 affect the coding exons 2 and 3 of PMP22, whereas the 17 kb deletion in subject SPR2 involves only the PMP22 exon 4 (Figure 4). Breakpoint sequence analysis showed microhomologies at the breakpoints of both subjects A23 (A) and A29 (TC). No microhomology was identified at the breakpoint of subject SPR2, but instead a 3 bp short sequence (CAT) that did not match the reference human genome sequence was found at the breakpoint (Figure S1).
Figure 4.
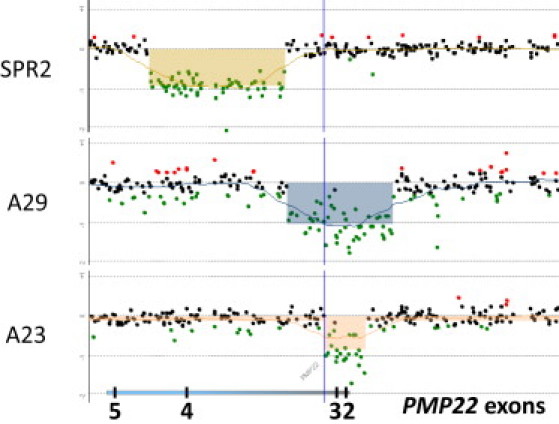
Oligonucleotide aCGH Analysis of Exonic Rearrangements of PMP22
The deleted regions are shadowed. Below, the locations of the PMP22 gene and its coding exons (2 to 5) are shown. Exon 4 is deleted in subject SPR2. Both subjects A23 and A29 have deletions of exons 1–3.
Discussion
Genomic rearrangements of 17p12 previously identified by conventional molecular assays in 21 subjects with CMT1A or HNPP were newly investigated by high-resolution genome analysis with an oligonucleotide-based aCGH assay and breakpoint sequence analysis. Together with another nine (seven unrelated) subjects with nonrecurrent 17p12 rearrangements that were recently reported,20 we now summarize the observations of 24 unique rare CNVs of 17p12 in 30 subjects with neuropathy (Table 1). All 30 subjects were ascertained by virtue of a neuropathy phenotype. By these analyses we narrow the critical region for CMT1A and HNPP, study the breakpoint characteristics, and infer the rearrangement mechanisms. We also show a multitude of ways that CNVs at this locus can cause neuropathy, including genomotypes that do not even include any PMP22 coding exons, i.e., upstream duplication CNVs.
Table 1.
Summary of Nonrecurrent 17p12 Rearrangements with Breakpoint Characteristics and Underlying Mechanisms in 30 Subjects with Neuropathy
| Subject | Rearrangement | Size (kb) |
Breakpoint Characteristics |
Mechanism | ||
|---|---|---|---|---|---|---|
| Distal | Proximal | Microhomology | ||||
| Complex Rearrangement | ||||||
| C3011 | dup-nml-dup-nml-dup | ∼3400a | AluJo | N.A. | RBM | |
| Alu/L2 | N.A. | |||||
| SMS-REP | SMS-REP | N.A. | ||||
| C1292, C2405 | del-nml-dup | 1294a | AluY | AluY | 27 bp | RBM |
| L1 | N.A. | |||||
| A2,b A9b | dup-tri-dup-nml-dup | 520a | Alu/L1 | N.A. | RBM | |
| Alu/L1/L2 | N.A. | |||||
| LCR17pA | LCR17pA | N.A. | ||||
| A15,b A15.2b |
del-nml-del-nml-dup |
9a |
L1 | AACA | RBM (FoSTeS × 3) |
|
| L1 | AACCT | |||||
| AAG | ||||||
| Duplication | ||||||
| C3159, C4316 | dup | 1048 | AluY | AluY | 24 bp | Alu-Alu/RBM |
| B89 | dup | ∼590 | LCR17pA | N.A. | N.A. | |
| B70b | dup | ∼500 | LCR17pA | N.A. | N.A. | |
| C2934 | dup | ∼500 | Alu/L1 | LCR17pA | N.A. | N.A. |
| SP951 | dup | 437 | L1 | L2 | none | NHEJ/RBM |
| L1 | L2 | TTTA | ||||
| SP54C | dup | 412 | AluSg | AluSg/LCR17pA | GTTTCACCAT | Alu-Alu/RBM |
| B1369 | dup | ∼400 | LCR17pA | N.A. | N.A. | |
| C3078 | dup | ∼400 | Alu/L1 | LCR17pA | N.A. | N.A. |
| SPR1 | dup | 194 | TCTCT | NHEJ/RBM | ||
| SD11, SD14 | dup | 186 | AluSq/x | A | NHEJ/RBM | |
| Deletion | ||||||
| A26 | del | 536 | none | NHEJ | ||
| B273.1, B273.2 | del | ∼320 | Alu/L1 | N.A. | N.A. | |
| A10b | del | 17 | AluJo | GATT | NHEJ/RBM | |
| SPR2 | del | 17 | AluJo | none | NHEJ | |
| A29 | del | 13 | TC | NHEJ/RBM | ||
| A21b | del | 12 | C | NHEJ/RBM | ||
| A11b | del | 7 | AC | NHEJ/RBM | ||
| A23 | del | 5 | A | NHEJ/RBM | ||
| A12b | del | <1 | GACG | NHEJ/RBM | ||
| A14b | del | <1 | GC | NHEJ/RBM | ||
Note: Related subjects are listed in the same line.
Abbreviations: del, deletion; nml, normal; dup, duplication, tri, triplication; NHEJ, nonhomologous end joining; RBM, replication-based mechanism; N.A., not available.
The sizes of the entire genomic region involved in complex rearrangements are shown.
These subjects have been previously published.20,21 Subject A15 and her affected sister A15.2 have the same complex exonic PMP22 deletion, and their healthy mother is mosaic (both germ line and somatic) for the identical rearrangement.20 B70 is the patient of the LF26 family published by Palau et al.21
PMP22 Is Critical for the 17p12 Rearrangement-Associated Neuropathy Phenotypes
In the 1.4 Mb common recurrent rearrangements associated with CMT1A or HNPP, the PMP22 gene is the dosage-sensitive gene involved in conveying the neuropathy phenotype. All 24 unique nonrecurrent types of 17p12 rearrangements mediate neuropathy by affecting (1) the entire PMP22 gene (i.e., gene dosage), (2) individual PMP22 exon(s), or (3) ultraconserved noncoding sequences upstream of PMP22 (Figures 1–4). The SRO of the PMP22 upstream duplications potentially reflects the genomic regions important to the regulation of PMP22 gene expression (Figure 2).
Half (12 CNVs, 50%) of the 24 rare CNVs in 17p12 did not perturb the coding sequence integrity, but instead altered the gene copy number by deletion (two related subjects, B273.1 and B273.2), duplication (10 unique duplication CNVs), or even triplication (two related subjects, A2 and A9) of the entire coding region of PMP22 (Figures 1 and 3; Table 1).
Ten (42%) out of 24 rare CNVs in 17p12 are partial deletions of PMP22, including subjects A23, A26, A29, and SPR2 in this study and subjects A10, A11, A12, A14, A15, and A21 in our previous study.20 These deletions affect only portions of the PMP22 gene, i.e., only one or several exons. These observations suggest that not only deletions of the entire PMP22 gene and point mutations in PMP22, but partial PMP22 deletions involving only one or a few exons can also potentially result in loss-of-function mutations and haploinsufficiency of the PMP22 protein and cause neuropathy.
No partial duplication of PMP22 has been identified in this study. The bias of prevalent deletion versus rare duplication for exonic PMP22 rearrangements in the subjects with neuropathies can be potentially explained by the argument that a portion of exonic PMP22 duplications could be benign as indicated by the fact that these duplications do not alter PMP22 gene dosage by creating an additional complete copy of PMP22. Interestingly, a 25 kb duplication involving PMP22 exons 4 and 5 has been reported in the African (Yoruba) population of HapMap.31 However, some PMP22 exonic duplications may be pathogenic mutations through exon shuffling, insertional translocation, or other molecular mechanisms.32,33
Interestingly, Weterman et al.24 recently reported identical 186 kb duplications in 11 subjects from 6 seemingly unrelated Dutch families. This duplication proximal to but exclusive of the coding region of PMP22 can also lead to the CMT1A phenotype that is usually associated with copy number gain of PMP22. It was suggested by the haplotype study of Weterman et al.24 that the neuropathy subjects with the 186 kb duplication share an ancestral mutation. The breakpoint sequence analysis of this current study showed that LCRs as NAHR substrates were absent at the breakpoints of the 186 kb duplication; therefore, the possibility of recurrent duplication events mediated by NAHR can be excluded. The 186 kb duplications from unrelated families probably have a common ancestral origin rather than multiple independent occurrences.
In addition to this 186 kb duplication, another duplication involving the upstream region of PMP22 was identified during this study in subject SPR1. This 194 kb duplication of SPR1 is located closer to PMP22 than that found in subjects SD11 and SD14 (9 kb versus 34 kb). There are two protein coding genes located in the SRO of the above two duplications: TEKT3 (MIM 612683) and CDRT4 (Figure 2). TEKT3 encodes a putative testicular microtubule-associated protein that is primarily expressed in male germ cells,34 whereas no molecular function has been reported for CDRT4. Instead, both increased dosage4,8 and point mutations of PMP2215,19 have been previously reported to cause CMT1A. Therefore, the PMP22 gene is the most likely gene responsible for the neuropathy phenotype in these two upstream duplications. The hypothesized dysregulation of PMP22 is supported by the observations of three ultraconserved noncoding sequences comprising conserved transcription factor binding sites in the SRO (Figure 2; Figures S2 and S3).
These findings in this study and the previous report suggest that not only PMP22 gene duplications but duplication of the adjacent genomic region upstream of the PMP22 gene can also lead to CMT1A potentially through altering PMP22 gene expression. Notably, in both of the cases reported by Weterman et al.24 and the different sized one reported herein, the duplications of upstream regulatory sequences conveyed a less severe phenotype than did PMP22 gene duplications. Our observations in the neuropathy-associated 17p12 rearrangements involving the coding region or upstream regulatory region of the PMP22 gene further confirm that PMP22 is the critical gene for CMT1A and HNPP. Interestingly, in another dosage-sensitive gene (PLP1 [MIM 300401]), the gene duplication of which causes the central nervous system dysmyelinating disorder Pelizaeus-Merzbacher disease (PMD [MIM 312080]), the duplication of downstream genomic region adjacent to PLP1 is also associated with phenotypic consequences.35 Thus, CNV either upstream or downstream from a dosage-sensitive gene may perturb gene regulation, perhaps through altering chromatin structure, remodeling, or other position effects underlying long-range control of gene expression.36
Complex Rearrangements in 17p12 and DNA Replication-Based Mechanisms
In addition to the simple types (e.g., deletion or duplication) of nonrecurrent rearrangements, complex rearrangements also exist and play an important role in genomic disorders;25,37 for example, complex rearrangements in 17p11.2 account for 57% of the nonrecurrent rearrangements associated with Potocki-Lupski syndrome (PTLS [MIM 610883]).20 In our current study, we also identified complexities in four subjects: C3011; two related cases, C1292 and C2405, based on the aCGH assay (Figure 3); and SP951, based on the breakpoint sequence analysis (Figure S1). Taking the previously identified complex 17p12 rearrangements (related subjects A2 and A9 and related individuals A15 and A15.2 from family HOU1109)20 into account, a sum of at least 21% (5/24) was identified to be complex in the rare 17p12 rearrangements associated with neuropathy. For six unique aCGH-based simple rearrangements (6/24, 25%), we could not obtain breakpoints; therefore, potential complexities may not have been identified at the level of resolution afforded by aCGH.
Notably, these complex CNVs cannot readily be explained by a simple rearrangement event mediated by the long-established DNA recombination mechanisms, for example, NAHR or NHEJ.2,9,38 To explain the observations of both complexity and microhomology at the breakpoints, we proposed a replication fork stalling and template switching (FoSTeS) mechanism involving DNA replication errors in human subjects.25 Studies from both human subjects and other model organisms including bacteria and yeast further delineate the molecular details and proposed microhomology-mediated break-induced replication (MMBIR).39 The MMBIR mechanism proposes: (1) fork stalling by a collapsed replication fork; as the replication fork proceeds through a DNA single-stranded nick and generates a one-ended, double-stranded DNA, that must be processed distinctly from a two-ended, double-stranded break that is the usual substrate for double-stranded break repair, and (2) template switching, as part of a break-induced replication, resulting in microhomology at the “join point” reflecting the priming of DNA replication on the new “template switched” fork. DNA replication-based mechanisms (RBMs) include serial replication slippage (SRS),28,29 FoSTeS, and/or MMBIR,20,25,39 microhomology/microsatellite-induced replication,40 and other similar models. The details of these mechanisms have been reviewed recently.41,42
Microhomology, as a hallmark of RBM, can be traced at the breakpoints of complex rearrangements. In the related subjects C1292 and C2405, a microhomology of 27 bp shared by two AluY elements was detected at one of their breakpoints (Table 1; Figure S1), which was alternatively consistent with Alu-Alu-mediated recombination43,44 and/or RBMs,26,41 though the feature of rearrangement complexity in C1292 and C2405 is more parsimonious with the latter. In subject SP951, sequence-based complexity was identified at breakpoints, i.e., a 23 bp fragment that can be copied from an adjacent DNA template and inserted at the breakpoint by SRS28,29 or other RBMs (Figure S1). However, only one microhomology of TTTA was identified at the breakpoint of SP951 (Table 1; Figure S1), whereas two microhomologies were expected according to the SRS model. The potential involvement of more complex rearrangement events cannot be excluded; for example, multiple NHEJ processes might generate such an event.30,41,45 Two NHEJ events having no microhomology at the first breakpoint and a microhomology of TTTA at the second breakpoint can potentially cause the sequence complexity in SP951 (Figure S1).
Various Mechanisms Involved in Nonrecurrent Rearrangements of 17p12
Distinct from one predominant NAHR mechanism in recurrent rearrangements, various mechanisms have been implicated in nonrecurrent rearrangements: NHEJ, Alu-Alu-mediated recombination, and RBMs have been shown to be involved in the nonrecurrent rearrangements in 17p12.
In the NHEJ events, an “information scar” of cleavage or addition of several nucleotides from or to the ends of double-strand break can be left at breakpoints,9,38 which are characteristic and can help distinguish the NHEJ-mediated rearrangements from the rearrangement products of other mechanisms. In this study, no microhomology is identified at the breakpoints of subjects A26 and SPR2. Instead, a 3 bp mismatched sequence (CAT) was identified at the breakpoint of SPR2. These observations, consistent with the nucleotide addition or cleavage, suggest that the NHEJ mechanism generated the nonrecurrent rearrangements in these two subjects.
Alu-Alu-mediated recombination is another mechanism for nonrecurrent rearrangement.43,44 Different from previously reported uncommon NHEJ events that join two Alu elements together and generate a longer fused breakpoint sequence,46 Alu-Alu-mediated recombination can cause a recombinant Alu with microhomology shared by two repetitive Alu elements. In related subjects C3159 and C4316, the breakpoint interval was mediated between two AluY elements, wherein a 24 bp microhomology was shared (Figure S1). In subject SP54C, the breakpoints are mediated between two AluSg elements, which shared a 10 bp microhomology (Figure S1). These above two nonrecurrent rearrangements are apparently consistent with an Alu-Alu-mediated recombination mechanism. However, considering the microhomologies that were identified at breakpoints, the involvement of RBMs (e.g., DNA template switching for only one time in the FoSTeS event resulting in a deletion or a duplication)20 cannot be excluded.
In five subjects (A23, A29, SPR1, SD11, and SD14) with nonrecurrent rearrangements, microhomologies of 1 to 5 bp were identified at breakpoints, which can alternatively be explained by either NHEJ or RBMs.
Prevalence of Repeat and Repetitive Sequences at Breakpoints of Nonrecurrent 17p12 Rearrangements
Distinct from the location of recurrent rearrangement breakpoints clustering in LCRs,9 no such restriction has been reported for the breakpoints of nonrecurrent rearrangements. However, the presence of both repeat and repetitive sequences at breakpoint junctions and the proximity of complex LCRs to breakpoint grouping have been reported in nonrecurrent duplications of MECP2, RAI1, and many other loci.27,37,47 Similarly, the prevalence of repeats (i.e., LCRs or SDs) and repetitive sequences (including long interspersed elements [LINEs] and short interspersed elements [SINEs]) was observed in the neuropathy-associated genomic rearrangements of this study, especially in the genomic duplications and complex rearrangements (Tables 1 and 2).
Table 2.
Prevalence of Repeat and Repetitive Sequences at Breakpoints of 24 Unique Nonrecurrent Rearrangements in 17p12
| Rearrangement Type | Number |
No. (%) of Breakpoints in Repeat or Repetitive Sequences |
|
|---|---|---|---|
| Distal | Proximal | ||
| (1) Nonrecurrent Rearrangements of 17p12 | |||
| Deletion | 2 | 0 (0%) | 1 (50%) |
| Duplication | 10 | 5 (50%) | 9 (90%) |
| Complex | 3 | 3 (100%) | 3 (100%) |
| All | 15 | 8 (53%) | 13 (87%) |
| (2) Exonic Rearrangement of PMP22 | |||
| Deletion | 8 | 2 (25%) | 0 (0%) |
| Complex | 1 | 0 (0%) | 1 (100%) |
| All | 9 | 2 (22%) | 1 (11%) |
The percentages >50% are shown in bold.
We identified 24 unique CNVs in the 30 neuropathy-associated nonrecurrent rearrangements summarized in this study, including 15 large genomic rearrangements of 17p12 and 9 exonic rearrangements of the PMP22 gene. The statistics of the breakpoints located in repetitive or repeat sequences are shown in Table 2. Interestingly, the prevalence of SINEs (e.g., Alu families), LINEs (e.g., L1 and L2), and LCRs (also rich in SINEs and LINEs) was observed in the breakpoints of large genomic rearrangements of 17p12, especially those of genomic duplications (distal, 50%; proximal 90%) and at least one breakpoint of each complex rearrangement (both distal and proximal, 100%) (Table 2), which is much higher than the composition of ∼34% for LINEs and SINEs in the human genome.48 Notably, 7 (54%) out of these 10 genomic duplications and 3 complex rearrangements of 17p12 have proximal breakpoints in LCR17pA. This finding is consistent with evolutionary studies of the proximal 17p region in primates, suggesting that LCRs or SDs acted as the seeds of serial segmental duplication events and led to more complex genomic architecture in 17p.49,50 This finding is also consistent with the observed breakpoint grouping of nonrecurrent rearrangements seen at other genomic disorder loci, such as PLP1 and MECP2.25,37
These observations reflect the special genomic architecture prone to genome instability. Because the repetitive sequences have been found to be associated with double-strand breaks or stalled replication, subsequent DNA repair via NHEJ or restarting DNA replication by template switching can be involved and lead to genomic rearrangements.51–53
The prevalence of repeats or repetitive sequences is not obvious at the breakpoints of small exonic PMP22 rearrangements (Table 2). This phenomenon is possibly due to the below-average content of SINEs and LINEs in the PMP22 gene region (24% in PMP22 versus genome-wide 34%).
Missing Heritability
Although tremendous efforts have been expended to dissect the genetic factors underlying human diseases, the genetic code accounts for only <20% of the known disease-associated variations in the human genome.54 The genetic variation observed is only as good as the method used to detect it. Many of the variants described to be small, complex, and/or exclusive of gene coding regions in the present study are challenging and may not be detected by currently implemented clinical assays for the CMT1A duplication and HNPP deletion. Rare CNVs caused by genomic rearrangements can be one source of variation potentially responsible for the “missing heritability” of human diseases.
In our previous studies,26 we introduced a new concept of genomotype (different from the traditional genotype) to describe CNVs of genomic segments and the study of such variations or changes in the context of genomic disorder phenotypes to determine genomotype-phenotype correlations as a way to unravel the genomic code. In this study, we also showed how genomic alterations, such as deletion, duplications, and complex rearrangements involving exons, noncoding upstream sequences, the entire PMP22 gene, or together with the flanking genomic regions, can lead to the neuropathy phenotypes of CMT1A and HNPP. Our study of rare CNVs with different sizes and genomic content in 17p12 further dissected the heritability associated with the neuropathy phenotype that is usually manifested by the 1.4 Mb common CNV in 17p12. Genomotype-phenotype correlations will be particularly relevant to the elucidation of the genomic code, especially in the instances where the CNV does not involve coding sequences, because coding sequences account for <2% of the human genome.48
In summary, our study documents that various mechanisms, including NHEJ, Alu-Alu-mediated recombination, and RBMs (e.g., SRS, FoSTeS, and/or MMBIR), are implicated in the nonrecurrent 17p12 rearrangements associated with neuropathy phenotypes. We also further document that rare CNVs, even those exclusive of coding sequences, can cause human diseases and suggest that CNVs that do not either involve genes or include coding sequences can nevertheless effect gene regulation. We speculate that CNVs involving both coding and noncoding sequences may be a type of variation responsible for some fraction of missing heritability.
Acknowledgments
We thank all participating subjects and families for their kind cooperation in the study. We also thank Drs. E. Ehler, V. Farhanová, R. Mazanec, and J. Zvolská for sending patients to the CMT1A DNA diagnostics. This work was supported by the National Institute of Neurological Disorders and Stroke (NINDS, NIH) grant R01NS058529 to J.R.L.; Texas Children's Hospital General Clinical Research Center (GCRC) grant M01RR00188; and Intellectual and Developmental Disabilities Research Centers (IDDRC) grant P30HD024064. F.Z. is supported by Shanghai Pujiang Program and the Ministry of Education of China grant NCET-09-0322. P.S. is supported by Ministry of Health of Czech Republic grant IGA NS 10554-3. P.D.J. and V.T. are supported by a Methusalem grant of the University of Antwerp, the Fund for Scientific Research (FWO-Flanders), and the Interuniversity Attraction Poles program (P6/43) of the Belgian Federal Science Policy Office (BELSPO). B.R. is supported by the BMBF grant 01ES0815. J.R.L. is a consultant for Athena Diagnostics, 23andMe, and Ion Torrent Systems Inc., and holds multiple US and European patents for DNA diagnostics. Furthermore, the Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular diagnostic testing (Medical Genetics Laboratories).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Agilent Technologies eArray, http://earray.chem.agilent.com/earray/
Coriell Cell Repositories, http://ccr.coriell.org/
Database of Genomic Variants (DGV), http://projects.tcag.ca/variation/
Medical Genetics Laboratories at Baylor College of Medicine, http://www.bcm.edu/geneticlabs/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
References
- 1.Lupski J.R. Genomic disorders: Structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 2.Stankiewicz P., Lupski J.R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 3.Lupski J.R. Genomic disorders ten years on. Genome Med. 2009;1:42. doi: 10.1186/gm42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lupski J.R., de Oca-Luna R.M., Slaugenhaupt S., Pentao L., Guzzetta V., Trask B.J., Saucedo-Cardenas O., Barker D.F., Killian J.M., Garcia C.A. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- 5.Chance P.F., Alderson M.K., Leppig K.A., Lensch M.W., Matsunami N., Smith B., Swanson P.D., Odelberg S.J., Disteche C.M., Bird T.D. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143–151. doi: 10.1016/0092-8674(93)90058-x. [DOI] [PubMed] [Google Scholar]
- 6.Raeymaekers P., Timmerman V., Nelis E., De Jonghe P., Hoogendijk J.E., Baas F., Barker D.F., Martin J.J., De Visser M., Bolhuis P.A. Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a) Neuromuscul. Disord. 1991;1:93–97. doi: 10.1016/0960-8966(91)90055-w. [DOI] [PubMed] [Google Scholar]
- 7.Lupski J.R., Chance P.F. Hereditary motor and sensory neuropathies involving altered dosage or mutation of PMP22: The CMT1A duplication and HNPP deletion. In: Dyck P.J., Thomas P.K., editors. Peripheral Neuropathy. Elsevier Science; Philadelphia: 2005. pp. 1659–1680. [Google Scholar]
- 8.Lupski J.R., Wise C.A., Kuwano A., Pentao L., Parke J.T., Glaze D.G., Ledbetter D.H., Greenberg F., Patel P.I. Gene dosage is a mechanism for Charcot-Marie-Tooth disease type 1A. Nat. Genet. 1992;1:29–33. doi: 10.1038/ng0492-29. [DOI] [PubMed] [Google Scholar]
- 9.Gu W., Zhang F., Lupski J.R. Mechanisms for human genomic rearrangements. PathoGenetics. 2008;1:4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner D.J., Miretti M., Rajan D., Fiegler H., Carter N.P., Blayney M.L., Beck S., Hurles M.E. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat. Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bailey J.A., Gu Z., Clark R.A., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 12.Pentao L., Wise C.A., Chinault A.C., Patel P.I., Lupski J.R. Charcot-Marie-Tooth type 1A duplication appears to arise from recombination at repeat sequences flanking the 1.5 Mb monomer unit. Nat. Genet. 1992;2:292–300. doi: 10.1038/ng1292-292. [DOI] [PubMed] [Google Scholar]
- 13.Reiter L.T., Murakami T., Koeuth T., Gibbs R.A., Lupski J.R. The human COX10 gene is disrupted during homologous recombination between the 24 kb proximal and distal CMT1A-REPs. Hum. Mol. Genet. 1997;6:1595–1603. doi: 10.1093/hmg/6.9.1595. [DOI] [PubMed] [Google Scholar]
- 14.Reiter L.T., Hastings P.J., Nelis E., De Jonghe P., Van Broeckhoven C., Lupski J.R. Human meiotic recombination products revealed by sequencing a hotspot for homologous strand exchange in multiple HNPP deletion patients. Am. J. Hum. Genet. 1998;62:1023–1033. doi: 10.1086/301827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valentijn L.J., Baas F., Wolterman R.A., Hoogendijk J.E., van den Bosch N.H., Zorn I., Gabreels-Festen A.W., de Visser M., Bolhuis P.A. Identical point mutations of PMP-22 in Trembler-J mouse and Charcot-Marie-Tooth disease type 1A. Nat. Genet. 1992;2:288–291. doi: 10.1038/ng1292-288. [DOI] [PubMed] [Google Scholar]
- 16.Patel P.I., Roa B.B., Welcher A.A., Schoener-Scott R., Trask B.J., Pentao L., Snipes G.J., Garcia C.A., Francke U., Shooter E.M. The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot-Marie-Tooth disease type 1A. Nat. Genet. 1992;1:159–165. doi: 10.1038/ng0692-159. [DOI] [PubMed] [Google Scholar]
- 17.Timmerman V., Nelis E., Van Hul W., Nieuwenhuijsen B.W., Chen K.L., Wang S., Ben Othman K., Cullen B., Leach R.J., Hanemann C.O. The peripheral myelin protein gene PMP-22 is contained within the Charcot-Marie-Tooth disease type 1A duplication. Nat. Genet. 1992;1:171–175. doi: 10.1038/ng0692-171. [DOI] [PubMed] [Google Scholar]
- 18.Valentijn L.J., Bolhuis P.A., Zorn I., Hoogendijk J.E., van den Bosch N., Hensels G.W., Stanton V.P., Jr., Housman D.E., Fischbeck K.H., Ross D.A. The peripheral myelin gene PMP-22/GAS-3 is duplicated in Charcot-Marie-Tooth disease type 1A. Nat. Genet. 1992;1:166–170. doi: 10.1038/ng0692-166. [DOI] [PubMed] [Google Scholar]
- 19.Roa B.B., Garcia C.A., Suter U., Kulpa D.A., Wise C.A., Mueller J., Welcher A.A., Snipes G.J., Shooter E.M., Patel P.I. Charcot-Marie-Tooth disease type 1A. Association with a spontaneous point mutation in the PMP22 gene. N. Engl. J. Med. 1993;329:96–101. doi: 10.1056/NEJM199307083290205. [DOI] [PubMed] [Google Scholar]
- 20.Zhang F., Khajavi M., Connolly A.M., Towne C.F., Batish S.D., Lupski J.R. The DNA replication FoSTeS/MMBIR mechanism can generate human genomic, genic, and exonic complex rearrangements. Nat. Genet. 2009;41:849–853. doi: 10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palau F., Lofgren A., De Jonghe P., Bort S., Nelis E., Sevilla T., Martin J.J., Vilchez J., Prieto F., Van Broeckhoven C. Origin of the de novo duplication in Charcot-Marie-Tooth disease type 1A: Unequal nonsister chromatid exchange during spermatogenesis. Hum. Mol. Genet. 1993;2:2031–2035. doi: 10.1093/hmg/2.12.2031. [DOI] [PubMed] [Google Scholar]
- 22.Timmerman V., Rautenstrauss B., Reiter L.T., Koeuth T., Lofgren A., Liehr T., Nelis E., Bathke K.D., De Jonghe P., Grehl H. Detection of the CMT1A/HNPP recombination hotspot in unrelated patients of European descent. J. Med. Genet. 1997;34:43–49. doi: 10.1136/jmg.34.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seeman P., Mazanec R., Zidar J., Hrusakova S., Ctvrteckova M., Rautenstrauss B. Charcot-Marie-Tooth disease type 1A (CMT1A) and hereditary neuropathy with liability to pressure palsies (HNPP): reliable detection of the CMT1A duplication and HNPP deletion using 8 microsatellite markers in 2 multiplex PCRs. Int. J. Mol. Med. 2000;6:421–426. doi: 10.3892/ijmm.6.4.421. [DOI] [PubMed] [Google Scholar]
- 24.Weterman M.A., van Ruissen F., de Wissel M., Bordewijk L., Samijn J.P., van der Pol W.L., Meggouh F., Baas F. Copy number variation upstream of PMP22 in Charcot-Marie-Tooth disease. Eur. J. Hum. Genet. 2010;18:421–428. doi: 10.1038/ejhg.2009.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee J.A., Carvalho C.M., Lupski J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 26.Bi W., Sapir T., Shchelochkov O.A., Zhang F., Withers M.A., Hunter J.V., Levy T., Shinder V., Peiffer D.A., Gunderson K.L. Increased LIS1 expression affects human and mouse brain development. Nat. Genet. 2009;41:168–177. doi: 10.1038/ng.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stankiewicz P., Shaw C.J., Dapper J.D., Wakui K., Shaffer L.G., Withers M., Elizondo L., Park S.S., Lupski J.R. Genome architecture catalyzes nonrecurrent chromosomal rearrangements. Am. J. Hum. Genet. 2003;72:1101–1116. doi: 10.1086/374385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J.M., Chuzhanova N., Stenson P.D., Ferec C., Cooper D.N. Meta-analysis of gross insertions causing human genetic disease: Novel mutational mechanisms and the role of replication slippage. Hum. Mutat. 2005;25:207–221. doi: 10.1002/humu.20133. [DOI] [PubMed] [Google Scholar]
- 29.Chen J.M., Chuzhanova N., Stenson P.D., Ferec C., Cooper D.N. Complex gene rearrangements caused by serial replication slippage. Hum. Mutat. 2005;26:125–134. doi: 10.1002/humu.20202. [DOI] [PubMed] [Google Scholar]
- 30.Lieber M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010 doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kidd J.M., Cooper G.M., Donahue W.F., Hayden H.S., Sampas N., Graves T., Hansen N., Teague B., Alkan C., Antonacci F. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang S.H., Shaw C., Ou Z., Eng P.A., Cooper M.L., Pursley A.N., Sahoo T., Bacino C.A., Chinault A.C., Stankiewicz P. Insertional translocation detected using FISH confirmation of array-comparative genomic hybridization (aCGH) results. Am. J. Med. Genet. A. 2010;152A:1111–1126. doi: 10.1002/ajmg.a.33278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stankiewicz P., Pursley A.N., Cheung S.W. Challenges in clinical interpretation of microduplications detected by array CGH analysis. Am. J. Med. Genet. A. 2010;152A:1089–1100. doi: 10.1002/ajmg.a.33216. [DOI] [PubMed] [Google Scholar]
- 34.Roy A., Yan W., Burns K.H., Matzuk M.M. Tektin3 encodes an evolutionarily conserved putative testicular microtubules-related protein expressed preferentially in male germ cells. Mol. Reprod. Dev. 2004;67:295–302. doi: 10.1002/mrd.20025. [DOI] [PubMed] [Google Scholar]
- 35.Lee J.A., Madrid R.E., Sperle K., Ritterson C.M., Hobson G.M., Garbern J., Lupski J.R., Inoue K. Spastic paraplegia type 2 associated with axonal neuropathy and apparent PLP1 position effect. Ann. Neurol. 2006;59:398–403. doi: 10.1002/ana.20732. [DOI] [PubMed] [Google Scholar]
- 36.Kleinjan D.A., van Heyningen V. Long-range control of gene expression: emerging mechanisms and disruption in disease. Am. J. Hum. Genet. 2005;76:8–32. doi: 10.1086/426833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carvalho C.M., Zhang F., Liu P., Patel P., Sahoo T., Bacino C.A., Shaw C., Peacock S., Pursley A., Tavyev Y.J. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum. Mol. Genet. 2009;18:2188–2203. doi: 10.1093/hmg/ddp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lieber M.R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- 39.Hastings P.J., Ira G., Lupski J.R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Payen C., Koszul R., Dujon B., Fischer G. Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet. 2008;4:e1000175. doi: 10.1371/journal.pgen.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang F., Carvalho C.M., Lupski J.R. Complex human chromosomal and genomic rearrangements. Trends Genet. 2009;25:298–307. doi: 10.1016/j.tig.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hastings P.J., Lupski J.R., Rosenberg S.M., Ira G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 2009;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bailey J.A., Liu G., Eichler E.E. An Alu transposition model for the origin and expansion of human segmental duplications. Am. J. Hum. Genet. 2003;73:823–834. doi: 10.1086/378594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sen S.K., Han K., Wang J., Lee J., Wang H., Callinan P.A., Dyer M., Cordaux R., Liang P., Batzer M.A. Human genomic deletions mediated by recombination between Alu elements. Am. J. Hum. Genet. 2006;79:41–53. doi: 10.1086/504600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gajecka M., Gentles A.J., Tsai A., Chitayat D., Mackay K.L., Glotzbach C.D., Lieber M.R., Shaffer L.G. Unexpected complexity at breakpoint junctions in phenotypically normal individuals and mechanisms involved in generating balanced translocations t(1;22)(p36;q13) Genome Res. 2008;18:1733–1742. doi: 10.1101/gr.077453.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elliott B., Richardson C., Jasin M. Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol. Cell. 2005;17:885–894. doi: 10.1016/j.molcel.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 47.Vissers L.E., Bhatt S.S., Janssen I.M., Xia Z., Lalani S.R., Pfundt R., Derwinska K., de Vries B.B., Gilissen C., Hoischen A. Rare pathogenic microdeletions and tandem duplications are microhomology-mediated and stimulated by local genomic architecture. Hum. Mol. Genet. 2009;18:3579–3593. doi: 10.1093/hmg/ddp306. [DOI] [PubMed] [Google Scholar]
- 48.Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 49.Inoue K., Dewar K., Katsanis N., Reiter L.T., Lander E.S., Devon K.L., Wyman D.W., Lupski J.R., Birren B. The 1.4-Mb CMT1A duplication/HNPP deletion genomic region reveals unique genome architectural features and provides insights into the recent evolution of new genes. Genome Res. 2001;11:1018–1033. doi: 10.1101/gr.180401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stankiewicz P., Shaw C.J., Withers M., Inoue K., Lupski J.R. Serial segmental duplications during primate evolution result in complex human genome architecture. Genome Res. 2004;14:2209–2220. doi: 10.1101/gr.2746604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wells R.D. Non-B DNA conformations, mutagenesis and disease. Trends Biochem. Sci. 2007;32:271–278. doi: 10.1016/j.tibs.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 52.Argueso J.L., Westmoreland J., Mieczkowski P.A., Gawel M., Petes T.D., Resnick M.A. Double-strand breaks associated with repetitive DNA can reshape the genome. Proc. Natl. Acad. Sci. USA. 2008;105:11845–11850. doi: 10.1073/pnas.0804529105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Voineagu I., Narayanan V., Lobachev K.S., Mirkin S.M. Replication stalling at unstable inverted repeats: Interplay between DNA hairpins and fork stabilizing proteins. Proc. Natl. Acad. Sci. USA. 2008;105:9936–9941. doi: 10.1073/pnas.0804510105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., McCarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.