

Abstract
For more than a century, Jews and non-Jews alike have tried to define the relatedness of contemporary Jewish people. Previous genetic studies of blood group and serum markers suggested that Jewish groups had Middle Eastern origin with greater genetic similarity between paired Jewish populations. However, these and successor studies of monoallelic Y chromosomal and mitochondrial genetic markers did not resolve the issues of within and between-group Jewish genetic identity. Here, genome-wide analysis of seven Jewish groups (Iranian, Iraqi, Syrian, Italian, Turkish, Greek, and Ashkenazi) and comparison with non-Jewish groups demonstrated distinctive Jewish population clusters, each with shared Middle Eastern ancestry, proximity to contemporary Middle Eastern populations, and variable degrees of European and North African admixture. Two major groups were identified by principal component, phylogenetic, and identity by descent (IBD) analysis: Middle Eastern Jews and European/Syrian Jews. The IBD segment sharing and the proximity of European Jews to each other and to southern European populations suggested similar origins for European Jewry and refuted large-scale genetic contributions of Central and Eastern European and Slavic populations to the formation of Ashkenazi Jewry. Rapid decay of IBD in Ashkenazi Jewish genomes was consistent with a severe bottleneck followed by large expansion, such as occurred with the so-called demographic miracle of population expansion from 50,000 people at the beginning of the 15th century to 5,000,000 people at the beginning of the 19th century. Thus, this study demonstrates that European/Syrian and Middle Eastern Jews represent a series of geographical isolates or clusters woven together by shared IBD genetic threads.
Introduction
Jews originated as a national and religious group in the Middle East during the second millennium BCE1 and have maintained continuous genetic, cultural, and religious traditions since that time, despite a series of Diasporas.2 Middle Eastern (Iranian and Iraqi) Jews date from communities that were formed in the Babylon and Persian Empires in the fourth to sixth centuries BCE.3,4 Jewish communities in the Balkans, Italy, North Africa, and Syria were formed during classical antiquity and then admixed with Sephardic Jews who migrated after their expulsion from the Iberian Peninsula in the late 15th century.5 Ashkenazi Jews are thought to have settled in the Rhine Valley during the first millennium of the Common Era, then to have migrated into Eastern Europe between the 11th and 15th centuries, although alternative theories involving descent from Sorbs (Slavic speakers in Germany) and Khazars have also been proposed.6,7 Admixture with surrounding populations had an early role in shaping world Jewry, but, during the past 2000 years, may have been limited by religious law as Judaism evolved from a proselytizing to an inward-looking religion.8
Earlier genetic studies on blood groups and serum markers suggested that Jewish Diaspora populations had Middle Eastern origin, with greater genetic similarity between paired Jewish populations than with non-Jewish populations.9–11 These studies differed in their interpretation of the degree of admixture with local populations. Recent studies of Y chromosomal and mitochondrial DNA haplotypes have pointed to founder effects of both Middle Eastern and local origin, yet the issue of how to characterize Jewish people as mere coreligionists or as genetic isolates that may be closely or loosely related remains unresolved.12–16 To improve the understanding about the relatedness of contemporary Jewish groups, genome-wide analysis and comparison with neighboring populations was performed for representatives of three major groups of the Jewish Diaspora: Eastern European Ashkenazim; Italian, Greek, and Turkish Sephardim; and Iranian, Iraqi, and Syrian Mizrahim (Middle Easterners).
Material and Methods
Recruitment and Genotyping of Jewish Populations
Participants were recruited from the Iranian, Iraqi, Syrian, and Ashkenazi Jewish communities in the metropolitan New York region. Participants were recruited from the Turkish Sephardic Jewish community in Seattle, from the Greek Sephardic Jewish communities in Thessaloniki and Athens, and from the Italian Jewish community in Rome, the latter as previously described.17 All of the recruitments took place under a New York University School of Medicine Institutional Review Board-approved protocol (07-333 “Origins and Migrations of Jewish People”). Additional recruitment of Iraqi and Turkish Sephardic Jews occurred at Sheba Medical Centre in Tel Hashomer, Israel, under a local ethics committee- and an Israeli Ministry of Health Institutional Review Board-approved protocol. In every case, subjects provided informed consent. They were included only if all four grandparents came from the same Jewish community. Subjects were excluded if they were known first- or second-degree relatives of other participants or were found to have IBD coefficients ≥.30 by analysis of microarray data. Genotyping was performed with the Affymetrix Genome-Wide Human SNP Array 6.0 (Affy v 6) at the genomic facility at Albert Einstein College of Medicine.
DNA Preparation for SNP Array Analysis
DNA was prepared according to standard methods. Quality and quantity of genomic DNA was determined by agarose gel electrophoresis to assure that only high-molecule-weight DNA was present and by absorbance at 230, 260, and 280 nm to determine DNA concentration and assure that protein and organic contaminants were not present.
Genotyping
Genomic DNA samples were genotyped with the Affy v 6 in accordance with the manufacturer's protocols. A total of 305 Jewish samples were successfully genotyped at call rates >99% and with no gender mismatch. The resulting individuals were tested for relatedness via genome-wide IBD estimates. Samples were excluded if the IBD coefficients were ≥0.30, as this suggests hidden relatedness. To assure that members came from the stated community, the SMART PCA program from EIGENSOFT package used to remove genetic outliers (defined as having greater than six standard deviations from the mean PC position on at least one of the top ten eigenvectors). A total of 14 were observed and these samples were removed. Ultimately, 237 samples were used for comparative analyses. Results of samples of known European origin that were run on Affy v 6 arrays were included in the PCA analysis. These overlapped completely with the results from the current study, indicating the absence of a batch effect.
Reference Populations
HGDP Data Set
The Jewish data set was analyzed along with a selected HGDP data sets. The original HGDP data set had 1043 unrelated individuals from 52 world-wide populations.18 First, 28 extreme outliers identified by three independent preliminary PCA runs on a set of small randomly selected SNPs was removed. To reduce the size of the data set, members of related population groups were combined, including Pakistani (Balochi, Brahui, Burusho, Makrani, Pastun, Sindi, Uyghur), Southern American (Colombian, Karitiana, Maya, Pima, Surui), Central/Southern African (Bantu, Biaka, Mandenka, Mbuti Pygmy, Mozabite, San, Yoruba), and East Asian (Khmer, Dai, Daur, Northern Chinese Han, Southern Chinese Han, Hezhen, Japanese, Lahu, Miao, Mongolian, Naxi, Oroqen, She, Tu, Tujia, Xibo, Yi), and then 25 samples were selected randomly from each population. The final number of samples in this selected HGDP data set was 418. These came from 16 populations: North African (Mozabite), Central and South African, East Asian, Southern American, Pakistani_Hazara, Pakistani_Kalash, Pakistani_Other, Middle Eastern_Bedouin, Middle Eastern_Druze, Middle Eastern_Palestinian, Adygei, Russian, Basque, French, Northern Italian (Bergamo and Tuscan), and Sardinian. No significant differences were observed in the results when different data sets containing independent, randomly selected samples were used. To get a closer view of Jewish population structure, a localized data set was generated that combined the Jewish populations, three Middle Eastern non-Jewish populations, and six European populations. The HGDP samples were genotyped on the Illumina HumanHap650K Beadchips, as previously described. After filtering SNPs with call rate <95% and extracting overlapping SNP sets between two different platforms, 164,894 SNPs were used for further analysis.
PopRes Data Set
The Population Reference Sample (PopRes) project included more than 6000 individuals of African-American, East Asian, South Asian, Mexican, and European origin after quality control, of which 2407 individuals of unmixed ancestry were collected from a wide variety of European countries.19 These were genotyped on the Affymetrix 500K chip. To study the relationships between Jewish and European population, a localized data set was generated that combined the Jewish data set with selected PopRes data. First, 25 extreme outliers identified by three independent preliminary PCA runs on a small set of randomly selected SNPs were removed. Next, each of 2407 European subjects was assigned into one of 10 groups based on geographic region: South: Italy, Swiss-Italian; Southeast: Albania, Bosnia-Herzegovina, Bulgaria, Croatia, Greece, Kosovo, Macedonia, Romania, Serbia, Slovenia, Yugoslavia; Southwest: Portugal, Spain; East: Czech Republic, Hungary; East-Southeast: Cyprus, Turkey; Central: Austria, Germany, Netherlands, Swiss-German; West: Belgium, France, Swiss-French, Switzerland; North: Denmark, Norway, Sweden; Northeast: Finland, Latvia, Poland, Russia, Ukraine; Northwest: Ireland, Scotland, UK. To reduce the size of the data set, 50 samples were randomly selected from each geographic group whenever the sample size was greater than 50. The final number of samples in this selected PopRes data set was 383. No significant difference in the results was observed when different data sets containing independently selected samples were used. After filtering out SNPs with call rate < 95% and extracting overlapping SNP sets between two different platforms, 362,566 SNPs were used for further analysis. This sparser set of SNPs maintains ability to detect IBD, yet is larger than that used in a recent study of genetic structure of the Han Chinese (Figure S9 available online).21
Fst, Observed Heterozygosity, and Phylogenetic Analysis
Population divergence was measured with the pairwise FST statistic, calculated with the method of Weir and Cockerham.22 Confidence intervals of the FST were calculated by bootstrap resampling, with 500 replications. The genetic diversity across all loci within each population was assessed by using the observed heterozygosity (Ho), calculated from GenePop 4.0 (1 − Qinter). The neighbor-joining phylogenetic tree based on pairwise FST distance was constructed with MEGA4.23 A Sub-Saharan African population was used as an out-group to root the phylogenetic tree. Bootstrap analyses indicated that the phylogenetic tree is quite robust. Nonetheless, it is unlikely that these populations followed a strict tree-like model of evolution, given the abundance of admixture and gene flow between groups.
Principal Component and STRUCTURE Analysis
Principal component analysis was performed with the Smartpca program from the EIGENSOFT package (version 2.0).24 Except the initial run (to remove extreme genetic outliers), the analyses were performed without removal of outliers. To infer the population structure, a Bayesian model-based clustering method, implemented in the STRUCTURE version 2.2 software package was used for the global data set.29 To reduce the running time while still maintaining the information of population structure within the data set, a subset of 3904 SNPs with highest informativeness across all populations was used. SNPs informativeness was estimated by using average genetic distance difference (δ) among populations studied. For each population pair, δ was calculated as the sum of the absolute differences between allele frequencies. Markers were then ranked and top 5% of SNPs were selected for subsequent STRUCTURE analysis. The program was run 10 times for K values 2–6. All structure runs used 30,000 burn-in cycles followed by 30,000 MCMC iterations, assuming correlated allele frequencies and admixture model with separate alpha estimated for each population. The results from all replicates for each K were aligned with CLUMPP.25 Mean individual Q matrices were plotted with DISTRUCT.26
Differences between Subgroups Pairwise Fst, IBS, and ANOVA
Formal statistical t testing was performed of each pairwise Fst to demonstrate that they differed from zero (Tables S2 and S3). Permutation tests were performed for between-group identity-by-state (IBS) with 10,000 permutations for all pair-wise comparisons of 7 Jewish populations. The results showed that comparisons of individuals from the same Jewish populations were genetically much more similar than those from Jewish-non-Jewish and non-Jewish-non-Jewish populations (p values are generally smaller than 10−4—see Table S5 for details). In addition, analysis of variance (ANOVA) was applied to each subgroup's Eigenvalue PCA average to test whether paired populations were different.24
CNV Analysis
Informative CNVs were chosen based on the location of 164,894 SNPs that were used in our SNP analysis. One CNV upstream and one CNV downstream in close proximity to the candidate SNP were included in this analysis. In the cases of SNPs aggregation, CNVs from the regions flanking the SNPs were chosen. PCA analysis was performed on .cel files for 275,000 flanking CNVs from the 237 samples with JMP Genomics 4 (SAS Inc., Cary, NC). To account for biased copy numbers (more than 3 per locus), the values were reassigned new integral values: copy number 1 = 0, copy number 2 = 1, and ≥ copy number 3 = 2.
IBD Discovery
IBD segments were detected with the GERMLINE algorithm in Genotype Extension.27 GERMLINE identifies pairwise IBD shared segments in time proportional to the number of individuals processed. In brief, the algorithm rapidly seeks out short, exact pairwise matches between individuals, and then extends from these seeds to long, inexact matches that are indicative of IBD. The output of GERMLINE was used to detect unreported close relatives, who were omitted from the analysis. Two individuals were considered cryptic relatives if their total sharing is larger than 1500 cM and if the average segment length is more than 25 cM, suggesting an avuncular or closer relationship. The output was also used to produce sharing densities, sharing graphs, and sharing statistics.
Inference of Population History
To estimate population parameters, data were simulated with Genome, with default parameters of recombination rate and block size.28 Population size and timing of founder/split events were attempted as described in Figure S10 to best fit observed data. Theoretical analysis suggests that the number of IBD segments of a particular length L resulting from a shared ancestor k generations ago, decreases, for a fixed k, as an exponential function of L. A history of rapid expansion after a recent bottleneck implies that a large fraction of IBD segments are due to the bottleneck generation, consistent with the exponential decay of shared segments as a function of L, that is observed in Ashkenazi samples. In contrast, a fixed-size population will have segments resulting from ancestors at different generations, producing a different decay pattern, as a sum of exponentials.
Statistical Analyses of Interpopulation Differences and Neighbor Joining Trees
To identify whether there are significant genetic differences between Jewish populations, PLINK was used to run permutation test (10,000 permutations) for between-group IBS differences. The neighbor joining tree was generated by using pairwise FST. To assess the reliability of the NJ tree, SNP loci were randomly sampled 500 times and distance matrix were generated from each sampling data set. The function “Neighbor” from PHYLIP was used to construct all bootstrap trees, and then “Consense” was used to get bootstrap consensus tree and bootstrap support values for each node.42
GERMLINE Analysis
Genotype Extension
In its latest version, GERMLINE can be used in Haplotype Extension or Genotype Extension.27 The Haplotype Extension is intended to process well-phased data, where it performs with near-perfect accuracy; however, performance can suffer when the data is phased poorly, as can be the case when trio or family data are unavailable. This analysis was performed with the Genotype Extension, where heterozygous markers are treated as wildcards and IBD segments are detected with long segments of mutually homozygous markers. The first stage of GERMLINE searches for seed matches of completely identical haplotypes in computationally phased data. Seeds of k = 128 common SNPs were used for Affy v 6 SNP 6.0 data, and k = 32 common SNPs for the sparser set of SNPs in the intersection this SNP array with the HGDP SNP set. Genotype extension then attempts to extend each seed match between a pair of samples by assuming a Hidden Markov Model (HMM) would well describe the genotypes of the IBD pair along the IBD segment extending the match. This HMM have been previously used in standard tools, such as PLINK (–segment option), or other work for the entire process of IBD detection.41 A speedup of the HMM analysis was implemented that advances 64 SNPs at a time and requires assumptions on genotyping accuracy. In this analysis, one inconsistency was allowed for Affy v 6 SNP 6.0 data and zero inconsistencies for the sparser data.
Filtering Regions for Informative SNPs
GERMLINE output was filtered to ensure consistency across genotyping platforms and to remove noise by filtering out regions of low information content. SNP density in sliding, nonoverlapping blocks across the genome was used to filter shared segments that spanned SNP-sparse regions, particularly the edges of the centromere and telomere. Specifically, regions that presented less than 100 SNPs per megabase or 100 SNPs per centimorgan were identified and excised and, subsequently, shared segments that were shorter than 3 cM were removed.
Sharing Densities
Histograms of post-processed sharing densities were represented by Manhattan-style plots, where the y axis represents the chance of a random pair of individuals having a shared segment at a SNP: all pairs of individuals sharing a segment across that position were counted and normalized by the total number of potential pairs. Within populations, the normalization factor was equal to, where n is the population size. Between populations, it was the product of the respective sizes.
Sharing Graphs
The amount of sharing for the analyzed data set was visualized with the ShareViz software. Individuals were represented as nodes, grouped into populations of origin. The thickness of the edges between nodes represent the total amount of sharing (in centimorgans) between each pair of individuals. For presenting populations geographically, planar quasi-isometric embedding (ISOMAP) was used, where distances between populations were defined as inverse of the populations' pairwise average.
Sharing Statistics
To compute the average total sharing between populations I and J, the following expression was used:
where Wij is the total sharing between individuals i and j from populations I and J, respectively, and n and m are the number of individuals in populations I and J. The average lengths of the shared segments across populations were computed through the arithmetic mean of the shared segments for each pair of populations. To compute the distribution of longest segments (Table 1, Figure 3), the longest shared segments for all possible pairs was considered. The observed probability of a pair sharing a longest segment of a specified length was computed normalizing the observed counts by the number of possible pairs within or between the considered populations. The counts for all the histograms were obtained through floor rounding of the values.
Table 1.
Genetic Diversity of Jewish, European, and Middle Eastern Non-Jewish Populations
| Populations | n | Hoa | IRN | IRQ | SYR | ASH | ITJ | GRK | TUR | N. Italian | Sardinian | French | Basque | Adygei | Russian | Palestinian | Druze | Bedouin |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IRN | 28 | 0.291 | 0.015 | 0.015 | 0.017 | 0.018 | 0.015 | 0.014 | 0.018 | 0.027 | 0.022 | 0.030 | 0.018 | 0.028 | 0.017 | 0.017 | 0.021 | |
| IRQ | 37 | 0.293 | 4.906 | 0.008 | 0.013 | 0.012 | 0.009 | 0.008 | 0.012 | 0.019 | 0.016 | 0.024 | 0.013 | 0.023 | 0.010 | 0.012 | 0.015 | |
| SYR | 25 | 0.296 | 0.999 | 3.145 | 0.008 | 0.008 | 0.004 | 0.003 | 0.007 | 0.014 | 0.010 | 0.018 | 0.010 | 0.018 | 0.007 | 0.009 | 0.012 | |
| ASH | 34 | 0.294 | 0.746 | 0.827 | 1.926 | 0.009 | 0.006 | 0.005 | 0.008 | 0.014 | 0.009 | 0.017 | 0.012 | 0.016 | 0.011 | 0.012 | 0.016 | |
| ITJ | 37 | 0.294 | 0.609 | 0.857 | 1.566 | 3.093 | 0.005 | 0.005 | 0.008 | 0.014 | 0.011 | 0.018 | 0.012 | 0.018 | 0.010 | 0.011 | 0.015 | |
| GRK | 42 | 0.296 | 0.564 | 0.773 | 1.570 | 2.153 | 2.476 | 0.001 | 0.004 | 0.010 | 0.007 | 0.014 | 0.009 | 0.015 | 0.006 | 0.008 | 0.011 | |
| TUR | 34 | 0.297 | 0.747 | 1.043 | 2.049 | 2.954 | 2.411 | 2.556 | 0.004 | 0.010 | 0.007 | 0.014 | 0.008 | 0.014 | 0.005 | 0.007 | 0.010 | |
| N_Italian | 21 | 0.295 | 0.675 | 0.740 | 0.865 | 1.015 | 0.978 | 0.906 | 0.899 | 0.007 | 0.002 | 0.008 | 0.008 | 0.009 | 0.010 | 0.011 | 0.016 | |
| Sardinian | 28 | 0.289 | 0.675 | 0.683 | 0.970 | 1.098 | 0.852 | 0.955 | 0.946 | 1.386 | 0.009 | 0.013 | 0.019 | 0.020 | 0.017 | 0.017 | 0.022 | |
| French | 28 | 0.296 | 0.498 | 0.623 | 0.999 | 1.012 | 0.948 | 0.889 | 0.937 | 1.361 | 1.353 | 0.007 | 0.009 | 0.005 | 0.014 | 0.014 | 0.020 | |
| Basque | 24 | 0.291 | 0.584 | 0.662 | 0.854 | 1.153 | 0.862 | 0.935 | 0.905 | 1.427 | 1.472 | 2.078 | 0.018 | 0.015 | 0.021 | 0.021 | 0.027 | |
| Adygei | 17 | 0.298 | 0.604 | 0.504 | 0.655 | 0.738 | 0.748 | 0.805 | 0.699 | 0.840 | 0.647 | 0.831 | 1.073 | 0.012 | 0.012 | 0.012 | 0.019 | |
| Russian | 25 | 0.295 | 0.470 | 0.524 | 0.623 | 0.913 | 0.822 | 0.642 | 0.811 | 1.236 | 0.933 | 1.460 | 1.205 | 0.905 | 0.021 | 0.021 | 0.028 | |
| Palestinian | 39 | 0.303 | 0.530 | 0.642 | 0.597 | 0.580 | 0.659 | 0.609 | 0.708 | 0.514 | 0.670 | 0.549 | 0.590 | 0.562 | 0.480 | 0.009 | 0.009 | |
| Druze | 36 | 0.296 | 0.656 | 0.638 | 0.754 | 0.778 | 0.671 | 0.738 | 0.752 | 0.658 | 0.713 | 0.595 | 0.675 | 0.797 | 0.568 | 0.623 | 0.013 | |
| Bedouin | 40 | 0.301 | 0.572 | 0.606 | 0.564 | 0.576 | 0.567 | 0.541 | 0.590 | 0.606 | 0.545 | 0.529 | 0.542 | 0.386 | 0.371 | 1.013 | 0.649 | |
| Total Sharing | 41.947 | 33.360 | 17.261 | 11.620 | 28.446 | 6.005 | 4.458 | 2.366 | 10.839 | 1.629 | 15.966 | 6.285 | 5.799 | 25.504 | 49.590 | 25.361 |
Pairwise Fstb is shown in the upper triangle. Pairwise sharing distance between populations, defined as the total centiMorgan length of IBD segments >3cM each averaged across all pairs of samples from the respective populations, is shown in the lower triangle.
Ho is observed heterozygosity.
Confidence intervals are listed in Table S2.
Figure 3.
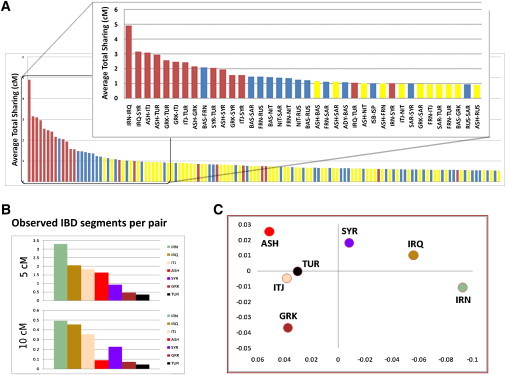
Analysis of Shared Informative by Descent Segments across Jewish and Non-Jewish Populations
(A) Average total sharing across populations. The genome-wide average IBD sharing (y axis) for any two individuals sampled from different Mediterranean and European population pairs (x axis: top 50% sharing pairs, detail on top 15% pairs) was computed. The population pairs have been grouped into Jewish-Jewish (red bars), Jewish-non-Jewish (yellow bars), and non-Jewish-non-Jewish (blue bars).
(B) Distribution of segment lengths within each Jewish population. The expected number of IBD segments shared within each Jewish population (y axis) for segments of length 5 cM and 10 cM were computed.
(C) Planar embedding of Jewish populations, with their inverse distances corresponding to average IBD between them (see Material and Methods).
Sharing between Remote Relatives
Siblings share, on average, the length of one haploid genome IBD. At each locus, sharing persists for an additional meiotic transmission with probability ½. Cousins therefore share a total of ¼ of the genome length on average, and k-th cousins share (1/4)k of the genome. The length of a segment shared by k-th cousins is the length between adjacent crossover sites along any of the transmissions from the shared ancestor of the segment. This length is distributed exponentially, with mean inversely proportional to the number of transmissions involved. For k-th cousins, this mean is 50 cM/(k + 1).
Selecting Loci with Significantly Excessive Sharing
We defined a locus as excessively sharing if the frequency of shared segments there exceeded 4 standard deviations beyond the mean genome-wide sharing.
Results
Jewish Populations Form Distinctive Clusters with Genetic Proximity to European and Middle Eastern Groups
Affy v 6 data were generated for 237 unrelated individuals (51.1% female) from the seven Jewish populations (Table S1). To examine the population genetic structure of Jewish populations in the global and regional contexts, the SNP data were merged with selected data sets from the Human Genome Diversity Panel (HGDP). The first two principal components of worldwide populations showed that the Jewish populations clustered with the European groups (Figure 1A). When compared only to the European and Middle Eastern, non-Jewish populations (Bedouins, Druze, Palestinians), each of the Jewish populations formed its own distinctive cluster, indicating the shared ancestry and relative genetic isolation of the members of each of those groups (Figures 1B and 1C). Pairwise FST analysis indicated that each of these clusters was distinct and statistically different from all of the others (Table 1, top; Tables S2 and S3). ANOVA on the PCA Eigenvalues confirmed that the populations differed from one another (p < 0.0001) as did the permutation testing of between-group IBD for all pairwise comparisons of the seven Jewish populations (Tables S4 and S5). PC1 distinguished Northern and Southern European and Jewish and Middle Eastern populations. Along this axis, Europeans were closest to Ashkenazi Jews, followed by Sephardic, Italian, Syrian, and Middle Eastern Jews. Of the European populations, the Northern Italians showed the greatest proximity to the Jews, followed by Sardinians and French (Figure 1B), an observation that was confirmed by FST (Table 1). Also along this axis, the Adygei, a Caucasian population, showed proximity to the Ashkenazi Jews. The Druze, Bedouins, and Palestinians, respectively, were closest to the Middle Eastern (Iranian and Iraqi) and Syrian Jews (Figure 1C). PC2 distinguished the Middle Eastern Jewish and non-Jewish populations (Figure 1C). Along PC2, the clusters of the Iranian, Iraqi, and Syrian Jews and Druze, Bedouins, and Palestinians followed a north to south distribution that was reminiscent of their geographic separation in the Middle East (Figures 1B and 1C). Virtually identical results were observed when the Jewish groups were compared with the European national groups of the Population Reference Sample (PopRes) (Figures S1A and S1B). The observations with SNPs tended to be confirmed by CNVs. The principal component analysis of CNVs demonstrated distinctive clusters for all of the Jewish populations, except Iraqi Jews (Figure 1D). The stability of these clusters was determined by using different numbers of CNVs, representing the tails of the genetic distance distributions (Figure S2).
Figure 1.
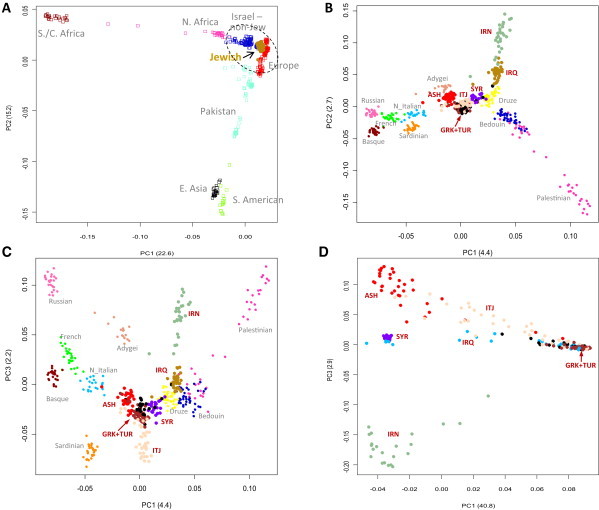
Principal Component Analysis of Jewish and Non-Jewish Populations
Principal components analysis of Jewish populations in a global (A) and regional context (B, PC1 versus PC2; C, PC1 versus PC3) CNVs (D, PC1 versus PC3). ASH, Ashkenazi Jews; IRN, Iranian Jews; IRQ, Iraqi Jews; SYR, Syrian Jews; ITJ, Italian Jews; GRK, Greek Jews; TUR, Turkey Jews. N. Italian is a combined set comprising Bergamo and Tuscan Italians. In (A), Middle Eastern non-Jewish populations are in blue, Jewish populations are in brown, and European populations are in red.
These findings demonstrated that the most distant and differentiated of the Jewish populations were Iranian Jews followed by Iraqi Jews (average FST to all other Jewish populations 0.016 and 0.011, respectively). The closest genetic distance was between Greek and Turkish Sephardic Jews (FST = 0.001) who, in turn, were close to Italian, Syrian, and Ashkenazi Jews. Thus, two major groups were identifiable that could be characterized as Middle Eastern Jews and European/Syrian Jews, an observation that was supported by pairwise FST and by phylogenetic tree analysis (Figure 2C). Notably, the Iranian and Iraqi Jews were grouped together with strong statistical support. The European and Syrian Jews shared a common branch that included non-Jewish European populations. The Druze, Palestinian, and Bedouins were on branches distinctive from the other populations. The robustness of this phylogenetic tree was demonstrated by the fact that a majority of major branching was supported by greater than 75% of bootstrap replications.
Figure 2.
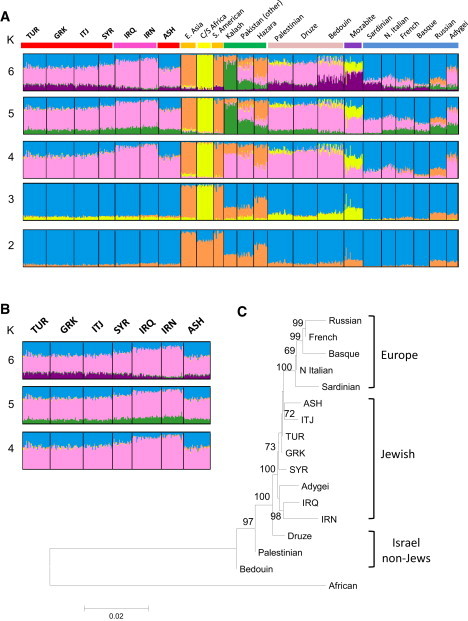
STRUCTURE and Phylogenetic Analysis of Jewish Populations
(A) STRUCTURE results for K = 2 to 6 for Jewish populations combined with selected HGDP worldwide populations. Each individual is represented by a vertical line, partitioned into colored segments that correspond to membership coefficients in the subgroups. The analysis is based on 3904 SNPs with potentially high informativeness in revealing population structure (see Material and Methods).
(B) Expanded view of STRUCTURE results for Jewish populations for K = 4 to 6.
(C) Neighbor-joining tree of Jewish, European, and Israel non-Jews populations with Central/Southern African population as outgroup. Pairwise Fst distances were used for constructing the tree. Major population groups are indicated by right bracket. 500 bootstrap replications were performed to obtain confidence value for each interior node. Only bootstrap values above 50% are shown.
The structure analysis was compatible with the Iranian and Iraqi Jews having predominant Middle Eastern/Central Asian ancestry and the European and Syrian Jews having both Middle Eastern/Central Asian and European ancestry with the proportion of European ancestry ranging between 20% and 40% when K ranged from 4 to 6. The Sephardic, Italian, and Syrian Jews all showed a low level component (8%–11%) that was shared with the North African Mozabite population when K equaled 6 (Figures 2A and 2B). This component was less apparent among the Ashkenazi and Middle Eastern Jews (Figure 2B; Figure S3).
Jewish Communities Show High Levels of IBD
IBD between Jewish individuals exhibited high frequencies of shared segments (Figure 3A; Figure S4). The median pair of individuals within a community shared a total of 50 cM IBD (quartiles: 23.0 cM and 92.6 cM)—such levels are expected to be shared by 4th or 5th cousins in a completely outbred population. However, the typical shared segments in these communities were shorter than expected between 5th cousins (8.33 cM length), suggesting multiple lineages of more remote relatedness between most pairs of Jewish individuals (Figure S5).
Within the different Jewish communities, three distinct patterns were observed (Figure 3B; Table 1; Figures S4 and S5). The Greek and Turkish Jews had relatively modest levels of IBD, similar to that observed in the French HGDP samples. The Italian, Syrian, Iranian, and Iraqi Jews demonstrated the high levels of IBD that would be expected for extremely inbred populations. Unlike the other populations, the Ashkenazi Jews exhibited increased sharing of segments at the shorter end of the range (i.e., 5 cM length), but decreased sharing at the longer end (i.e., 10 cM) (Figure S5)
Frequent IBD between Different Jewish Populations Reflects Their Genetic Proximity
As expected, the vast majority of long shared segments (89% of 15 cM segments, 78% of 10 cM segments) were shared within communities. However, the genetic connections between the Jewish populations became evident from the frequent IBD across these Jewish groups (63% of all shared segments). The web of relatedness between the 27,966 pairs of individuals in this study was intricate, even if restricted only to the 2,166 pairs sharing a total 50 cM or more, a level of sharing among third cousins (Figure S6). When population averages were examined, this network of IBD was consistent with the geographic distances between populations, with planar embedding (Figure 3C) representing 93% of the initial information content. The notable exception was that of Turkish and Italian Jews who were nearest neighbors in terms of IBD, but more distant on the geographical map, potentially reflecting their shared Sephardic ancestry. Jewish populations shared more and longer segments with one another than with non-Jewish populations, highlighting the commonality of Jewish origin. Among pairs of populations ordered by total sharing, 12 out of the top 20 were pairs of Jewish populations, and none of the top 30 paired a Jewish population with a non-Jewish one (Figure 3A).
Specific Regions of the Genome Are Frequently Shared between Jewish Populations
Shared regions spanned the entire genome, but none (longer than 5 cM) was shared among all the Jewish populations. Between Jewish populations, spikes of frequently shared segments were observed relative to the lower background sharing (Figures S7 and S8). Loci that demonstrated significantly excessive (≥4 standard deviations) sharing between Jewish populations are listed in Table S6. These loci spanning >20 million bases in total were not spanned by single LD blocks, nor did they include single haplotypes of high frequency (Figure S8). Gene content along these regions was slightly higher (p < 0.013) than the genome-wide average (Table S6).
Timing of the Middle Eastern-European Jewish Divergence
As a first step, population simulation was performed to estimate the ancestral population size for the Jewish and Middle-Eastern non-Jewish cohorts in this study. The ancient (before the introduction of agriculture, 5000 years before present) ancestral population size was set to a smaller and realistic 1000 individuals per simulated population size, although this result does not change significantly, as shown by the fact that the fraction of IBS pairs is affected almost exclusively by recent generations. Ashkenazi Jewish samples were excluded from this analysis, because the sharing in this population was inconsistent with a near-constant recent population size. The ancestral population size was then used in two simulations to estimate the time splits between Middle-Eastern and European (Italian) Jews. Under these assumptions, the split was consistent with 100–150 generations, or during the first millennium BCE, assuming a generation time of 20 years. The split between Middle-Eastern Jews and non-Jews was inconsistent with these simulation assumptions, suggesting a more complex history than a simple split of a single ancestral population.
Discussion
This study touches upon an issue that was raised over a century ago by Maurice Fishberg, Joseph Jacobs, and others about whether the Jews constitute a race, a religious group, or something else.29,30 In this study, Jewish populations from the major Jewish Diaspora groups—Ashkenazi, Sephardic, and Mizrahi—formed a distinctive population cluster by PCA analysis, albeit one that is closely related to European and Middle Eastern, non-Jewish populations. Within the study, each of the Jewish populations formed its own cluster as part of the larger Jewish cluster. Each group demonstrated Middle Eastern ancestry and variable admixture with European populations. This was observed in the structure plots and in the Fst analysis by the proximity of all Jewish populations one to another, to non-Jewish Middle Eastern populations, and to non-Jewish Southern European (French, Northern Italian, and Sardinian) populations. The patterns of relatedness were similar, albeit with higher resolution to what was reported in a recent study of fewer Jewish populations via microsatellite markers.31 Earlier investigators who studied fewer autosomal markers with less resolution and more recent investigators who studied Y chromosomal markers had similar observations. All noted that a major difference in Jewish groups was in the extent of admixture with local populations.7–11,13,14,17
Two major differences among the populations in this study were the high degree of European admixture (30%–60%) among the Ashkenazi, Sephardic, Italian, and Syrian Jews and the genetic proximity of these populations to each other compared to their proximity to Iranian and Iraqi Jews. This time of a split between Middle Eastern Iraqi and Iranian Jews and European/Syrian Jews, calculated by simulation and comparison of length distributions of IBD segments, is 100–150 generations, compatible with a historical divide that is reported to have occurred more than 2500 years ago.2,5 The Middle Eastern populations were formed by Jews in the Babylonian and Persian empires who are thought to have remained geographically continuous in those locales. In contrast, the other Jewish populations were formed more recently from Jews who migrated or were expelled from Palestine and from individuals who were converted to Judaism during Hellenic-Hasmonean times, when proselytism was a common Jewish practice. During Greco-Roman times, recorded mass conversions led to 6 million people practicing Judaism in Roman times or up to 10% of the population of the Roman Empire. Thus, the genetic proximity of these European/Syrian Jewish populations, including Ashkenazi Jews, to each other and to French, Northern Italian, and Sardinian populations favors the idea of non-Semitic Mediterranean ancestry in the formation of the European/Syrian Jewish groups and is incompatible with theories that Ashkenazi Jews are for the most part the direct lineal descendants of converted Khazars or Slavs.32 The genetic proximity of Ashkenazi Jews to southern European populations has been observed in several other recent studies.33–36
Admixture with local populations, including Khazars and Slavs, may have occurred subsequently during the 1000 year (2nd millennium) history of the European Jews. Based on analysis of Y chromosomal polymorphisms, Hammer estimated that the rate might have been as high as 0.5% per generation or 12.5% cumulatively (a figure derived from Motulsky), although this calculation might have underestimated the influx of European Y chromosomes during the initial formation of European Jewry.15 Notably, up to 50% of Ashkenazi Jewish Y chromosomal haplogroups (E3b, G, J1, and Q) are of Middle Eastern origin,15 whereas the other prevalent haplogroups (J2, R1a1, R1b) may be representative of the early European admixture.20 The 7.5% prevalence of the R1a1 haplogroup among Ashkenazi Jews has been interpreted as a possible marker for Slavic or Khazar admixture because this haplogroup is very common among Ukrainians (where it was thought to have originated), Russians, and Sorbs, as well as among Central Asian populations, although the admixture may have occurred with Ukrainians, Poles, or Russians, rather than Khazars.12,35 In support of the ancestry observations reported in the current study, the major distinguishing feature between Ashkenazi and Middle Eastern Jewish Y chromosomes was the absence of European haplogroups in Middle Eastern Jewish populations.37 Four founder mitochondrial haplogroups of Middle Eastern origins comprise approximately 40% of the Ashkenazi Jewish genetic pool, whereas the remainder is comprised of other haplogroups, many of European origin and supporting the degree of admixture observed in the current study.13 Evidence for founder females of Middle Eastern origin has been observed in other Jewish populations based on nonoverlapping mitochondrial haplotypes with coalescence times >2000 years.14 The number of founders and their relative proportions from one population to another is variable. These Y chromosomal and mitochondrial haplogroup studies along with the population-specific genetic clusters and prevalent within and between-population IBD segments of the current study, and Mendelian genetic disease mutation studies all point to local founder effects with subsequent genetic drift that caused genetic differentiation.38 The differential pattern of IBD observed only among Ashkenazi Jews in which older IBD segments became shorter and few new ones were created is consistent with a population bottleneck followed by rapid expansion (see Material and Methods). This corresponds to the so-called demographic miracle of Ashkenazi Jewish history discussed earlier.6
The Iranian and Iraqi Jews are the most differentiated with the greatest genetic distances from the other populations and the least distances from each other, as well as the least sharing of the “European” component in Structure. Similar differentiation was observed for mitochondrial haplotypes.14 The high rate of IBD within these groups (and in Italian and Syrian Jews) demonstrates a high coefficient of inbreeding. Yet, the sharing of Iranian and Iraqi Jews of a branch on the phylogenetic tree with the Adygei suggests that a certain degree of admixture may have occurred with local populations not included in this study.
Besides Southern European groups, the closest genetic neighbors to most Jewish populations are the Palestinians, Bedouins, and Druze. The observed differentiation of these groups reflects their histories of within-group endogamy.39 Yet, their genetic proximity to one another and to European and Syrian Jews suggests a shared genetic history of related Middle Eastern and non-Semitic Mediterranean ancestors who chose different religious and tribal affiliations. These observations are supported by the significant overlap of Y chromosomal haplogroups between Israeli and Palestinian Arabs with Ashkenazi and non-Ashkenazi Jewish populations that has been described previously.37 Likewise, a study comparing 20 microsatellite markers in Israeli Jewish, Palestinian, and Druze populations demonstrated the proximity of these two non-Jewish populations to Ashkenazi and Iraqi Jews.40
This study demonstrates that the studied Jewish populations represent a series of geographical isolates or clusters with genetic threads that weave them together. These threads are observed as IBD segments that are shared within and between Jewish groups. Over the past 3000 years, both the flow of genes and the flow of religious and cultural ideas have contributed to Jewishness.
Acknowledgments
This study was supported, in part, by the Lewis and Rachel Rudin Foundation, the Iranian-American Jewish Federation of New York, the U.S.-Israel Binational Science Foundation, the US National Institutes of Health (5 U54 CA121852), and Ruth and Sidney Lapidus. The authors thank Alon Keinan, Alkes Price, Yongzhao Shao, Carlos Bustamante, and David Goldstein for useful technical discussions; Lanchi U, Malka Sasson, Bridget Riley, Christopher Campbell, Jidong Shan, David Reynolds, Michael Bamshad, Nora Iny, Robert Ohebshalom, Lana Bahhash, Rabbi Salomon Cohen-Scali, Ino Angel, and Avram Fortis for technical contributions; and Lawrence Schiffman, Robert Chazen, Aron Rodrigue, and Harvey Goldberg for informative historical discussions.
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
Jewish Hapmap Project, http://pediatrics.med.nyu.edu/genetics/research/jewish-hapmap-project
References
- 1.Biran A., Naveh J. An Aramaic stele fragment from Tel Dan. Isr. Explor. J. 1993;43:81–98. [Google Scholar]
- 2.Ben-Sasson H.H. Harvard University Press; Cambridge: 1976. A History of the Jewish People. [Google Scholar]
- 3.Levy H. Mazda Publishers; Costa Mesa, CA: 1999. Comprehensive History of the Jews of Iran. [Google Scholar]
- 4.Rejwan N. Westview Press; Boulder: 1985. The Jews of Iraq: 3000 Years of History and Culture. [Google Scholar]
- 5.Baron S.W. Columbia University Press; New York: 1937. Social and Religious History of the Jews. [Google Scholar]
- 6.Weinryb B. Jewish Publication Society of America; Philadelphia: 1973. A History of the Jews in Poland. [Google Scholar]
- 7.Wexler P. Mouton de Gruyter; Berlin, New York: 2002. Two-Tiered Relexification in Yiddish: Jews, Sorbs, Khazars, and the Kiev-Polessian Dialect. [Google Scholar]
- 8.Cohen S.J.D. University of California Press; Berkeley: 1999. The Beginnings of Jewishness. [Google Scholar]
- 9.Livshits G., Sokal R.R., Kobyliansky E. Genetic affinities of Jewish populations. Am. J. Hum. Genet. 1991;49:131–146. [PMC free article] [PubMed] [Google Scholar]
- 10.Karlin S., Kenett R., Bonne-Tamir B. Analysis of biochemical genetic data on Jewish populations: II. Results and interpretations of heterogeneity indices and distance measures with respect to standards. Am. J. Hum. Genet. 1979;31:341–365. [PMC free article] [PubMed] [Google Scholar]
- 11.Carmelli D., Cavalli-Sforza L.L. The genetic origin of the Jews: A multivariate approach. Hum. Biol. 1979;51:41–61. [PubMed] [Google Scholar]
- 12.Behar D.M., Garrigan D., Kaplan M.E., Mobasher Z., Rosengarten D., Karafet T.M., Quintana-Murci L., Ostrer H., Skorecki K., Hammer M.F. Contrasting patterns of Y chromosome variation in Ashkenazi Jewish and host non-Jewish European populations. Hum. Genet. 2004;114:354–365. doi: 10.1007/s00439-003-1073-7. [DOI] [PubMed] [Google Scholar]
- 13.Behar D.M., Metspalu E., Kivisild T., Achilli A., Hadid Y., Tzur S., Pereira L., Amorim A., Quintana-Murci L., Majamaa K. The matrilineal ancestry of Ashkenazi Jewry: Portrait of a recent founder event. Am. J. Hum. Genet. 2006;78:487–497. doi: 10.1086/500307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Behar D.M., Metspalu E., Kivisild T., Rosset S., Tzur S., Hadid Y., Yudkovsky G., Rosengarten D., Pereira L., Amorim A. Counting the founders: The matrilineal genetic ancestry of the Jewish Diaspora. PLoS ONE. 2008;3:e2062. doi: 10.1371/journal.pone.0002062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hammer M.F., Redd A.J., Wood E.T., Bonner M.R., Jarjanazi H., Karafet T., Santachiara-Benerecetti S., Oppenheim A., Jobling M.A., Jenkins T. Jewish and Middle Eastern non-Jewish populations share a common pool of Y-chromosome biallelic haplotypes. Proc. Natl. Acad. Sci. USA. 2000;97:6769–6774. doi: 10.1073/pnas.100115997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nebel A., Filon D., Faerman M., Soodyall H., Oppenheim A. Y chromosome evidence for a founder effect in Ashkenazi Jews. Eur. J. Hum. Genet. 2005;13:388–391. doi: 10.1038/sj.ejhg.5201319. [DOI] [PubMed] [Google Scholar]
- 17.Oddoux C., Guillen-Navarro E., Ditivoli C., Dicave E., Cilio M.R., Clayton C.M., Nelson H., Sarafoglou K., McCain N., Peretz H. Mendelian diseases among Roman Jews: Implications for the origins of disease alleles. J. Clin. Endocrinol. Metab. 1999;84:4405–4409. doi: 10.1210/jcem.84.12.6268. [DOI] [PubMed] [Google Scholar]
- 18.Rosenberg N.A., Pritchard J.K., Weber J.L., Cann H.M., Kidd K.K., Zhivotovsky L.A., Feldman M.W. Genetic structure of human populations. Science. 2002;298:2381–2385. doi: 10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
- 19.Nelson M.R., Bryc K., King K.S., Indap A., Boyko A.R., Novembre J., Briley L.P., Maruyama Y., Waterworth D.M., Waeber G. The Population Reference Sample, POPRES: A resource for population, disease, and pharmacological genetics research. Am. J. Hum. Genet. 2008;83:347–358. doi: 10.1016/j.ajhg.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 21.Xu S., Yin X., Li S., Jin W., Lou H., Yang L., Gong X., Wang H., Shen Y., Pan X. Genomic dissection of population substructure of Han Chinese and its implication in association studies. Am. J. Hum. Genet. 2009;85:762–774. doi: 10.1016/j.ajhg.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weir B.S., Cockerham C.C. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 23.Tamura K., Dudley J., Nei M., Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 24.Patterson N., Price A.L., Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jakobsson M., Rosenberg N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–1806. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- 26.Rosenberg N.A. Distruct: A program for the graphical display of population structure. Mol. Ecol. Notes. 2004;4:137–138. [Google Scholar]
- 27.Gusev A., Lowe J.K., Stoffel M., Daly M.J., Altshuler D., Breslow J.L., Friedman J.M., Pe'er I. Whole population, genome-wide mapping of hidden relatedness. Genome Res. 2009;19:318–326. doi: 10.1101/gr.081398.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang L., Zollner S., Abecasis G.R. GENOME: A rapid coalescent-based whole genome simulator. Bioinformatics. 2007;23:1565–1567. doi: 10.1093/bioinformatics/btm138. [DOI] [PubMed] [Google Scholar]
- 29.Fishberg M. Charles Scribner's Sons; New York: 1911. The Jews: A Study of Race and Environment. [Google Scholar]
- 30.Jacobs J. D. Nutt; London: 1891. Jewish Statistics: Social, Vital, Anthropometric. [Google Scholar]
- 31.Kopelman N.M., Stone L., Wang C., Gefel D., Feldman M.W., Hillel J., Rosenberg N.A. Genomic microsatellites identify shared Jewish ancestry intermediate between Middle Eastern and European populations. BMC Genet. 2009;10:80. doi: 10.1186/1471-2156-10-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koestler A. Random House; New York: 1976. The Thirteenth Tribe. [Google Scholar]
- 33.Tian C., Plenge R.M., Ransom M., Lee A., Villoslada P., Selmi C., Klareskog L., Pulver A.E., Qi L., Gregersen P.K. Analysis and application of European genetic substructure using 300 K SNP information. PLoS Genet. 2008;4:e4. doi: 10.1371/journal.pgen.0040004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Price A.L., Butler J., Patterson N., Capelli C., Pascali V.L., Scarnicci F., Ruiz-Linares A., Groop L., Saetta A.A., Korkolopoulou P. Discerning the ancestry of European Americans in genetic association studies. PLoS Genet. 2008;4:e236. doi: 10.1371/journal.pgen.0030236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Need A.C., Kasperaviciute D., Cirulli E.T., Goldstein D.B. A genome-wide genetic signature of Jewish ancestry perfectly separates individuals with and without full Jewish ancestry in a large random sample of European Americans. Genome Biol. 2009;10:R7. doi: 10.1186/gb-2009-10-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olshen A.B., Gold B., Lohmueller K.E., Struewing J.P., Satagopan J., Stefanov S.A., Eskin E., Kirchhoff T., Lautenberger J.A., Klein R.J. Analysis of genetic variation in Ashkenazi Jews by high density SNP genotyping. BMC Genet. 2008;9:14. doi: 10.1186/1471-2156-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nebel A., Filon D., Brinkmann B., Majumder P.P., Faerman M., Oppenheim A. The Y chromosome pool of Jews as part of the genetic landscape of the Middle East. Am. J. Hum. Genet. 2001;69:1095–1112. doi: 10.1086/324070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ostrer H. A genetic profile of contemporary Jewish populations. Nat. Rev. Genet. 2001;2:891–898. doi: 10.1038/35098506. [DOI] [PubMed] [Google Scholar]
- 39.Curtis M., American Academic Association for Peace in the Middle East . Transaction Books; New Brunswick, N.J.: 1975. The Palestinians: People, History, Politics. [Google Scholar]
- 40.Rosenberg N.A., Woolf E., Pritchard J.K., Schaap T., Gefel D., Shpirer I., Lavi U., Bonne-Tamir B., Hillel J., Feldman M.W. Distinctive genetic signatures in the Libyan Jews. Proc. Natl. Acad. Sci. USA. 2001;98:858–863. doi: 10.1073/pnas.98.3.858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Felsenstein J. PHYLIP—Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.