

Abstract
Deep sequencing will soon generate comprehensive sequence information in large disease samples. Although the power to detect association with an individual rare variant is limited, pooling variants by gene or pathway into a composite test provides an alternative strategy for identifying susceptibility genes. We describe a statistical method for detecting association of multiple rare variants in protein-coding genes with a quantitative or dichotomous trait. The approach is based on the regression of phenotypic values on individuals' genotype scores subject to a variable allele-frequency threshold, incorporating computational predictions of the functional effects of missense variants. Statistical significance is assessed by permutation testing with variable thresholds. We used a rigorous population-genetics simulation framework to evaluate the power of the method, and we applied the method to empirical sequencing data from three disease studies.
Introduction
GWAS have successfully identified hundreds of loci harboring common variants that are reproducibly associated with complex traits. However, common variants identified to date typically explain only a small fraction of overall heritability, motivating interest in low-frequency or rare variants that may contribute to genetic risk.1,2 Technological advances in high-throughput sequencing platforms will soon make it possible to extend association studies to low-frequency and rare variants, particularly in targeted resequencing of exons.3,4 Rare variants are predicted to be enriched for functional alleles and to exhibit stronger effect sizes than common variants, consistent with the view that functional allelic variants are subject to purifying selection pressure.5–7 Deep-resequencing studies of candidate genes have already demonstrated the effect of rare alleles on several complex traits of biomedical relevance.8–14
The statistical power to detect phenotypic association with an individual rare variant is limited, due to the small number of observations for any given variant and a more stringent multiple-test correction as compared to common variants. This motivates analytical approaches that test the combined effect of multiple rare variants, but this requires prior specification of which variants to combine into the test. To date, most candidate-gene resequencing studies have compared the number of individuals carrying alleles exclusive to either of the phenotypic extremes. This strategy effectively eliminates common alleles from the test because they would be present in individuals at both extremes unless they have enormous effect. For large sample sizes, however, limiting the association analysis to exclusive alleles may unnecessarily reduce the statistical power of the test.
A recently proposed approach is to pick a fixed allele-frequency threshold and perform an association test on the set of variants below that threshold, giving them each equal weight (more generally, variants can be collapsed into multiple frequency bins).15 Another approach is to weight counts of each variant on the basis of the estimated variance under the null hypothesis of no association.16 This scheme applies much higher weights to very rare variants, and it implicitly assumes that the log odds ratio is approximately inversely proportional to the square root of the allele frequency, as we show below.
Using population-genetics simulations informed by empirical sequencing data, we analyzed the relationship between the phenotypic effect and the allele frequency of a mutation within an evolutionary model that incorporates purifying selection. These simulations highlighted the potential value of a statistical approach that uses a variable allele-frequency threshold instead of a fixed threshold. We have implemented such an approach, assessing statistical significance by permutation testing with variable thresholds, and we show that this approach indeed improves statistical power in both simulated and empirical data sets. In particular, this approach does not make implicit assumptions about the relationship between allele frequency and odds ratio.
Next, we have incorporated computational predictions of the functional effect of amino acid changes17,18 in the statistical test. The test gives higher weight to allelic variants predicted to be functionally significant and lower weight to variants predicted to be functionally insignificant. We show that incorporating computational predictions of functional importance further boosts power.
Material and Methods
Simulation Framework
The demographic history and distribution of selection coefficients associated with de novo missense mutations were determined by analyzing resequencing data from individuals of European ancestry, as described previously.4 The parameters of the demographic-history model included ancestral population size, bottleneck population size, final population size, and duration of exponential growth. The missense-to-synonymous ratio for de novo mutations was assumed to equal 2.3 (see 4). The model was shown to recapitulate the site-frequency spectrum of nonsynonymous human SNPs. We note that exponential population growth and purifying selection are critical features of the model for the analysis of rare alleles. We used this population-genetics model to generate 9 kb of sequence data in each of 10,000 individuals. We considered only missense mutations; nonsense, frameshift, and splice-site mutations were not simulated. Corresponding phenotypes were generated as follows: individuals that harbored a missense mutation with an associated selection coefficient greater than s had a normal distribution of quantitative trait values, with the same variance as noncarriers but with a mean shifted by δ standard deviations. Although the use of a discrete cutoff on selection coefficients for assigning function is clearly an oversimplification, we believe that this will not affect our results because selection coefficients do not explicitly enter into any of our association tests, which consider only weakly related variables. We generated data for 10,000 independent simulations for each of s = 0.0001, s = 0.001, and s = 0.01. (We note that the choice of s affects not only the average selection coefficient associated with damaging missense mutations but also the fraction of missense mutations that affect phenotypes.) In each case, we simulated 3 kb of sequence for each of δ = 0.125, δ = 0.25, and δ = 0.5. Our main results (Tables 1 and Table 2) focus on δ = 0.25, which is intermediate between values characteristic for relatively common SNPs segregating in the population (< 0.05 standard deviations [SD]) and mutations associated to Mendelian syndromes (> 1 SD), and on s = 0.001, which is close to the median value for s estimated from empirical data.4 We note that values of s and δ, rather than average values for segregating alleles, were specifically chosen to model new missense mutations.
Table 1.
Power of Various Approaches Using Quantitative Phenotypes
| T1 | T5 | WE | VT | VTP | |
|---|---|---|---|---|---|
| α = 0.001 | 0.137 | 0.182 | 0.098 | 0.204 | 0.259 |
| α = 0.05 | 0.547 | 0.503 | 0.543 | 0.600 | 0.686 |
We display results for T1 (1% allele-frequency threshold), T5 (5% allele-frequency threshold), WE (weighted), VT (variable threshold), and VTP (VT plus Polyphen) analyses for α = 0.001 and α = 0.05 significance levels, based on 10,000 independent simulations.
Table 2.
Power of Various Approaches Using Dichotomous Phenotypes
| T1 | T5 | WE | VT | VTP | |
|---|---|---|---|---|---|
| α = 0.001 | 0.089 | 0.150 | 0.078 | 0.161 | 0.213 |
| α = 0.05 | 0.482 | 0.458 | 0.488 | 0.533 | 0.625 |
As in Table 1, we display results for T1, T5, WE, VT, and VTP analyses for α = 0.001 and α = 0.05 significance levels, based on 10,000 independent simulations.
Weighted Approaches Correspond to Implicit Assumptions about Log Odds Ratios
We derive the result that a log likelihood ratio for a causal model with specified odds ratios will weight counts of variants in proportion to their log odds ratios. For SNP i (i = 1 to m), suppose that the allele frequency pi in controls is known, let Ri be the odds ratio in the causal model, and suppose that observed counts in cases are Ci copies of the reference allele and Ni − Ci copies of the variant allele. It follows that the allele frequency qi in cases under the causal model is qi = Ripi / [1 + (R − 1)pi], so that the likelihood ratio for the causal model versus the null model is
Thus, conditional on the total counts Ni, the log likelihood ratio is proportional to . It follows that the weights for Ci proposed by 16 correspond to the implicit assumption that . (We note that this relationship between log odds ratio and allele frequency was also assumed in the simulations of 16.)
Fixed-Threshold Approach
For dichotomous phenotypes, we computed the score , in which i indexes SNPs, Ci is the reference allele count of SNP i in cases, and ξi is an indicator variable that is equal to 1 if the frequency of SNP i is below a specified threshold (1% or 5%) and is equal to 0 otherwise. Statistical significance was assessed by permutations on phenotypes. We generalize this to quantitative phenotypes by computing , in which j indexes samples, Cij is the reference allele count of SNP i in sample j, and πj is the phenotype of sample j.
Weighted Approach
For dichotomous phenotypes, following 16 we computed the score , in which is the inverse square root of expected variance based on allele frequencies pi computed from controls only, with pseudocounts (see Equation 1 of 16). Statistical significance was assessed by permutations on phenotypes. We generalize this to quantitative phenotypes by computing , in which ξi is now based on allele frequencies pi computed from all samples, with pseudocounts.
Variable-Threshold Approach
We propose a variable-threshold approach. The intuition behind this approach is that there exists some (unknown) threshold T for which variants with a minor allele frequency (MAF) below T are substantially more likely to be functional than are variants with an MAF above T. Thus, we compute a z-score z(T) for each allele-frequency threshold T, define zmax as the maximum z-score across values of T, and assess statistical significance of zmax by permutations on phenotypes, allowing zmax in permuted data to be attained at values of T different than those in unpermuted data to ensure the validity of the permutation test. In contrast to the fixed-threshold approach, it is necessary for z(T) to account for the fact that the variances of relevant sums vary with different values of T, which will be compared when computing zmax. Thus, for either dichotomous or quantitative phenotypes, we compute z(T) as the z-score of a regression across samples of phenotypes versus counts of mutations meeting the allele-frequency threshold T. For computational speedup, we use linear regression instead of logistic regression, letting be an indicator variable that is equal to 1 if the frequency of SNP i is below the threshold T and equal to 0 otherwise, letting be the mean value of across samples j, and defining , which is proportional to a standard normal variable.
We used 1000 permutations in all of our simulations. In the case of dichotomous phenotypes, ties between zmax in permuted and unpermuted data may arise. These ties were broken randomly to ensure an appropriate uniform distribution for permutation statistics under null data. We define a p value as (x + 1) / (P + 1), in which P is the total number of permutations and x is the number of permutations for which zmax is higher in unpermuted data than in permuted data.
The use of multiple allele-frequency thresholds in a permutation-testing framework raises questions as to the computational complexity of this approach. However, by aggregating partial sums for increasing values of allele frequency T, starting with singleton mutations and continuing with only those values of T corresponding to allele frequencies of actual SNPs, the computational cost for analyzing a single gene is proportional only to the total number of minor alleles observed times the number of permutations tested. For each of our simulations involving one gene, 10,000 individuals, and 1000 permutations, running time was roughly 1 s, which is scalable to genome-wide studies of 20,000 genes. Although a larger number of permutations may be desired for the achievement of genome-wide significance, these can be limited to genes with suggestive evidence of association on the basis of 1,000 permutations, a standard approach to permutation testing in GWAS.
Cheating Approach to Incorporating ϕ(p)
We investigated the potential advantage of explicitly capturing the relationship between allele frequency and functional effect, via a “cheating” approach that weights variants according to the probability ϕ(p) that an allele of frequency p is functional, as inferred by using the same simulated data used to evaluate power. We implemented the cheating approach for quantitative phenotypes, using the weighted score , in which was computed with the use of binned values from simulations. Statistical significance was assessed by permutations on phenotypes.
Incorporation of Computational Predictions of Functional Effects
We investigated whether incorporation of PolyPhen-2 scores improves our statistical test.17,18 We calculated distribution of PolyPhen-2 probabilistic scores for neutral and damaging amino acid changes. For the neutral set we used amino acid substitutions that were fixed in the human lineage after divergence from chimpanzee, and for the damaging set we used known disease-causing missense mutations that cause the same phenotype as do nonsense mutations in the same gene. From these two distributions we determined posterior probabilities p(S) of being functional for each SNP, given raw PolyPhen-2 probabilistic score S. These recalibrated posterior probabilities p(S) were applied as weights in the regression. We used the PolyPhen-2 predictions only for rare variants (MAF < 1%), applying a constant weight of 0.5 for low-frequency or common variants (1.0 for nonsense, frameshift, and splice-site variants, which are extremely likely to be functional), so that average PolyPhen-2 weight was independent of allele frequency.
To evaluate the incorporation of computational predictions within our simulation framework, we needed to generate simulated PolyPhen-2 predictions. We did this by sampling PolyPhen-2 scores from their known distributions for functional and neutral variants (which are defined in our simulations according to selection coefficient threshold s). The functional and neutral distributions of PolyPhen-2 scores for damaging and neutral mutations provide a reasonable approximation to PolyPhen-2 scores that would be generated from empirical sequence data.
Application to Empirical Data Sets
We analyzed resequencing data providing established associations between multiple rare or low-frequency variants and triglyceride levels, type 1 diabetes, and obesity, as described in previous work.11,13,14 Data for triglyceride level were split into six groups according gender and ethnicity (Non-Hispanic Whites, Non-Hispanic Blacks, and Hispanics), and normalized rank within the group was used as a quantitative trait value. For this data set, following 14 we also considered a discrete phenotype defined by membership in top or bottom quartiles, with data from remaining samples ignored for the discrete phenotype analysis. Functional predictions for each missense variant were calculated with PolyPhen-2.17,18 PolyPhen-2 scores were used for computing Bayesian posterior probabilities, and weights were applied as described above. Statistical tests were applied to previously associated gene sets: ANGPTL3 (MIM 604774), ANGPTL4 (MIM 605910), ANGPTL5 (MIM 607666) for triglyceride levels, IFIH1 (MIM 606951) for type 1 diabetes, and a set of 21 genes for obesity.11,13,14 For each statistical test, the number of permutations was increased to 100,000, 10,000,000, and 100,000 for the analyses of triglyceride level, type 1 diabetes, and obesity, respectively, for increased precision in p values.
Results
Simulation Framework
We used population-genetics simulations to characterize the allele-frequency spectrum of rare variants involved in complex traits and to motivate statistical strategies to identify such alleles (see Material and Methods). We primarily focus on the parameter values δ = 0.25 and s = 0.001, the most realistic values, but we also consider other values to explore robustness of the model characteristics with respect to these parameters (see Material and Methods).4 All simulations involved a sample size of 10,000 individuals.
We first investigated how phenotypic effects vary with allele frequency for nonsynonymous variants in this simulation framework. In Figure 1A, we plot the probability ϕ(p) that a nonsynonymous variant is functional (i.e., has a selection coefficient greater than s) as a function of the allele frequency p, normalized by the value ϕ(1/2N) for singleton variants (N = 10,000). Because our model assumes that the phenotypic distribution is shifted for these variants, ϕ(p) is directly related to the expected phenotypic effect of an allele with frequency p (the distinction between quantitative and dichotomous phenotypes is addressed below). For comparison, we also plot the value ϕ(p) ∼, as implicitly assumed by the approach of 16 (see Material and Methods). Figure 1A indicates that ϕ(p) is a decreasing function of p, but decreasing at a rate that may differ substantially from, depending on the value of s. Furthermore, ϕ(p) is not robust with respect to model parameters and is likely to vary across genes and phenotypes.
Figure 1.
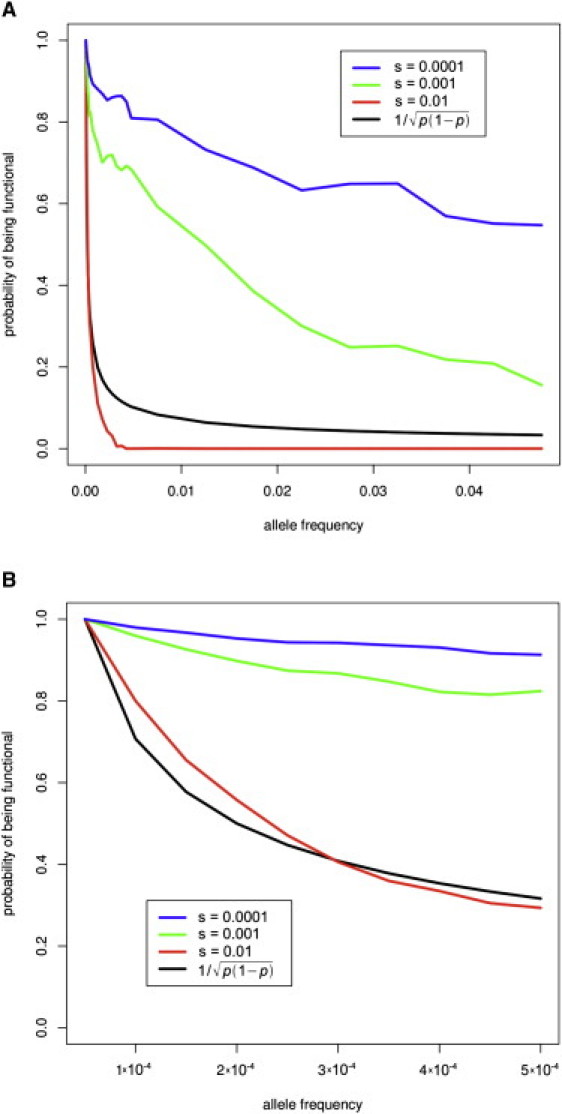
Probability ϕ(p) that a Nonsynonymous Variant Is Functional as a Function of Allele Frequency p
Results are based on 10,000 independent simulations in which we vary the selection parameter s: s = 0.01 (log10s = −2), s = 0.001 (log10s = −3), or s = 0.0001 (log10s = −4). We also plot the value ϕ(p) ∼ (as implicitly assumed by the weighted approach). All probabilities are normalized by the corresponding probability for singleton mutations. Panels (A) and (B) are identical except for the different range of the allele frequency p, with panel (B) restricted to very rare alleles.
Of particular interest are variants whose individual phenotypic association can be confirmed in a well-powered follow-up genotypic study in a larger sample. These variants should be of strong phenotypic effect and sufficient population frequency (even though still can be classified as low-frequency variants). We refer to these variants as “goldilocks” variants. For example, an allele with a frequency of 0.5% could be easily detected by resequencing of 1000 samples. Assuming an effect size of δ = 0.25, follow-up genotyping of 10,000 samples has 57% power to identify a significant association at p = 0.001 (92% power at p = 0.05). Our simulations suggest that goldilocks alleles can be quite common under our simple mutation-selection-drift model. For δ = 0.25 and s = 0.001, we find that about one-eighth of genes affecting the trait are expected to have at least one functional allele in the frequency range between 0.5% and 2%. However, this depends on the strength of selection: functional alleles in this frequency range are expected to be present in a substantial fraction of genes with variants under weak selection (s = 0.0001) affecting the trait, but absent from the population under strong selection (s = 0.01) (Figure S1, available online). It is important that methods for detecting association be effective either in the presence or in the absence of goldilocks alleles; otherwise, true associations may be missed.
A Variable-Threshold Approach for Pooling Multiple Rare Variants
On the basis of these results, we were motivated to develop a statistical strategy for associating multiple rare variants to a quantitative trait that can adapt to properties of individual genes. Different genes may have very different relationships between allele frequency and functional effect (Figure 1). In addition, some genes may harbor functional alleles at higher frequencies, whereas other genes may have only private functional variants (Figure S1). In our variable-threshold approach, we group rare alleles together by optimizing an allele-frequency threshold that maximizes the difference (as quantified by a z-score; see Material and Methods) between distributions of trait values for individuals with and without rare alleles. The value of the optimal allele-frequency threshold often varies considerably, even for fixed simulation parameter values, but the optimal score is robust to the shape of the z-score versus frequency-threshold curve. To control type I error, we apply the same optimization procedure to permuted data to obtain an exact p value for association (see Material and Methods and Web Resources).
Incorporation of Computational Predictions of Functional Effects
A major limitation of statistical tests combining multiple variants is that, without prior functional information about individual alleles, nonfunctional alleles are combined together with functional alleles. Statistical power can in principle be improved by the incorporation of functional predictions,11,19 and this motivates a systematic effort to evaluate formal methods for incorporating these predictions. We previously developed the PolyPhen software for predicting the effect of amino acid changes.17,18 We incorporated PolyPhen-2 scores into our statistical method as described above (see Material and Methods). We used the PolyPhen-2 predictions only for rare variants (MAF < 1%) so as not to exclude the signals of low-frequency or common variants (see Material and Methods). (PolyPhen-2 is most effective in predicting the functional effect of rare variants, which are more likely to be deleterious than are low-frequency variants, and the cost of misprediction becomes too high and may reduce power if the test is dominated by a few low-frequency variants.)
Evaluation of Statistical Tests on Simulated Data Sets
We evaluated several approaches: a fixed allele-frequency threshold approach (1% or 5%), a weighted approach generalizing 16, and our variable-threshold approach with or without incorporation of simulated PolyPhen predictions (see Material and Methods). We first applied these approaches to quantitative phenotypes, using the simulation framework described above. In each case we computed statistical significance for each of 10,000 independent simulations by applying the same test to 1000 different sets of permuted phenotypes, ensuring a properly controlled type I error rate. We computed power to detect associations at a significance level of either α = 0.05 or α = 0.001. (These significance levels correspond to candidate-gene studies that are currently being carried out; whole-exome studies would require much larger sample sizes to be well-powered at these effect sizes4). Results are displayed in Table 1. We see that the variable-threshold approach outperforms the fixed-threshold approach and the weighted approach; the improvement is highly statistically significant on the basis of 10,000 independent simulations (p << 10−12). Our results indicate a further increase in power for incorporating PolyPhen-2 predictions, which is generally similar to or greater than the benefit of the variable-threshold approach (Table 1). These results are based on δ = 0.25 and s = 0.001 (values considered most realistic4), but a wider range of simulations using δ = 0.125, 0.25, or 0.50 and s = 0.0001, 0.001, or 0.01 yielded similar relative results (Table S1). We also obtained similar results in a simulation in which phenotypes are independent of selection coefficient (Table S2a), and we verified that results are improved by incorporation of PolyPhen-2 predictions arising from either high-quality or low-quality sequence alignments (Table S2b).
We repeated the comparison using a dichotomous case-control phenotype defined by the > 75% and < 25% percentiles of our simulated quantitative phenotype. We again see that the variable-threshold approach outperforms the weighted approach and that incorporating PolyPhen-2 predictions further improves power (Table 2). As above, all of these results are based on δ = 0.25 and s = 0.001, but a wider range of simulations using δ = 0.125, 0.25, or 0.50 and s = 0.0001, 0.001, or 0.01 yielded similar relative results (Table S3). We also see that the power that can be attained by using sequence data from all individuals (Table 1) is higher than the power attained by using sequence data from the top and bottom phenotype quartiles only (Table 2). We finally note that all approaches (not just the variable-threshold approach) benefited from the incorporation of PolyPhen-2 predictions (Table S4).
The variable-threshold approach was motivated by the observation that the relationship between allele frequency and functional effect is not robust with respect to simulation parameters (Figure 1). However, we wondered whether it could be advantageous to explicitly capture this relationship for a fixed set of parameters. To investigate this, we implemented a “cheating” approach that weights variants according to allele frequency p on the basis of the values of ϕ(p) from our simulations (see Material and Methods). Our results should be viewed as an upper bound on the true power that could be attained by this specific approach, because we used the same data to compute ϕ(p) and to evaluate power. Nonetheless, power was no better than the variable-threshold approach: 0.187 for α = 0.001 and 0.595 for α = 0.05 (compare to Table 1). This suggests that the variable-threshold approach performs well relative to approaches that explicitly model the relationship between allele frequency and functional effect.
Evaluation of Statistical Tests on Empirical Data Sets
We analyzed data from a previous study showing that multiple rare or low-frequency variants in the ANGPTL3, ANGPTL4, and ANGPTL5 genes are collectively associated to low triglyceride levels in a multiethnic population from the Dallas Heart Study.14 Resequencing data and log triglyceride levels (adjusted for ethnicity and gender) were obtained for 3476 samples (see Material and Methods). We evaluated all statistical approaches as described above. Although the relative results should be viewed with caution in light of the small size of these and other empirical data sets, the variable threshold with PolyPhen-2 approach performed well (Table 3). Consistent with our simulations, statistical significance was reduced when using a discrete phenotype defined by top and bottom quartiles only (p value increases from 0.002 to 0.009 for the weighted approach, from 0.0004 to 0.005 for the variable-threshold approach).
Table 3.
Results for Three Empirical Data Sets
| T1 | T5 | WE | VT | VTP | |
|---|---|---|---|---|---|
| Triglyceride level | 0.013 | 0.00007 | 0.0020 | 0.00038 | 0.00002 |
| Type 1 diabetes | 0.001 | 0.0000002 | 0.0000004 | 0.0000008 | 0.0000002 |
| Obesity | 0.032 | 0.053 | 0.010 | 0.010 | 0.0017 |
We display p values for T1, T5, WE, VT, and VTP analyses on triglyceride level, type 1 diabetes, and obesity data sets. All p values are one-sided, reflecting the direction of the originally reported association.
We further analyzed data from a previous study showing that that multiple rare or low-frequency variants in the IFIH1 gene are associated with a lower risk of type 1 diabetes.13 Resequencing data were obtained for 480 cases and 480 controls (see Material and Methods). The variable threshold with PolyPhen approach again performed well (Table 3). This indicates that the variable-threshold approach is effective in detecting low-frequency “goldilocks” variants associated with phenotype.
Finally, we analyzed data from a previous study showing that multiple rare or low-frequency variants in 21 genes (historically associated with monogenic forms of obesity in humans or mice) are collectively associated to obesity in a cohort of extremely obese or lean individuals.11 Resequencing data were obtained for 379 extremely obese samples and 378 lean samples (see Material and Methods). Once again, the variable threshold with PolyPhen approach performed well (Table 3). The association signal in this case is driven primarily by very rare alleles. Overall, our results on empirical data sets confirm our simulation results indicating that our approach is robust in detecting a wide variety of association signals.
Discussion
The motivation for studying rare alleles in complex traits is based on the hypothesis that rare alleles may have larger phenotypic effects than common alleles as a consequence of purifying selection. Although our understanding of the genetic architecture of complex traits is far from complete, our simulations suggest that the relationship between allele frequency and effect size may vary widely with the intensity of selection, motivating our variable-threshold approach. We have shown that this approach performs well on simulated and empirical data, relative to other methods, and demonstrated that the incorporation of computational predictions of functional effects provides a further improvement in power, concordant with recent work.11,19 The variable-threshold approach is robust to a range of scenarios, and it will be particularly valuable when little is known about the likely allele frequencies and effect sizes of the causal variants. However, there will always be examples in which other methods perform better: in the type 1 diabetes example, the true signal is largely due to MAF > 1% variants, and the inclusion of lower thresholds (T < 1%) in the variable-threshold test increases noise and reduces power (Table 3). Conversely, if the true signal is largely due to singleton or extremely rare variants, then methods that explicitly give higher weights to extremely rare variants may perform better. A final caveat is that population stratification may lead to false-positive associations in this and other approaches. If sufficient data for inferring genetic ancestry are available, a solution is to analyze residual phenotypes with respect to genetic ancestry.
We have focused here on a simplified scenario involving individual-level resequencing data and an excess of rare variants at one phenotypic extreme. However, allele counts obtained from the resequencing of pools of individuals (with concordant phenotypes within each pool) could be analyzed in a similar fashion, though variation in the depth of coverage between pools warrants careful statistical treatment. In addition, if functional rare variants are expected to affect phenotype in either direction, a straightforward extension of our method to capture this signal is to scale the allele counts (in both original and permuted data) by the direction of each variant's association to phenotype, effectively searching for an excess of rare variants of large absolute effect. Going forward, the wealth of resequencing data yet to be generated will shed further light on the true contribution of rare variants to disease risk.
Acknowledgments
We are grateful to J. Cohen for sharing data from 14 and to L. Pennacchio for sharing data from 11. This work was funded by NIH grants R01 MH084676 and R01 GM078598.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM) http://www.ncbi.nlm.nih.gov/omim/
VT Test Software for implementing the methods described, http://genetics.bwh.harvard.edu/rare_variants
References
- 1.Altshuler D., Daly M.J., Lander E.S. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., McCarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turner E.H., Lee C., Ng S.B., Nickerson D.A., Shendure J. Massively parallel exon capture and library-free resequencing across 16 genomes. Nat. Methods. 2009;6:315–316. doi: 10.1038/nmeth.f.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kryukov G.V., Shpunt A., Stamatoyannopoulos J.A., Sunyaev S.R. Power of deep, all-exon resequencing for discovery of human trait genes. Proc. Natl. Acad. Sci. USA. 2009;106:3871–3876. doi: 10.1073/pnas.0812824106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pritchard J.K. Are rare variants responsible for susceptibility to complex diseases? Am. J. Hum. Genet. 2001;69:124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kryukov G.V., Pennacchio L.A., Sunyaev S.R. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am. J. Hum. Genet. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frazer K.A., Murray S.S., Schork N.J., Topol E.J. Human genetic variation and its contribution to complex traits. Nat. Rev. Genet. 2009;10:241–251. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]
- 8.Cohen J.C., Kiss R.S., Pertsemlidis A., Marcel Y.L., McPherson R., Hobbs H.H. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–872. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- 9.Cohen J.C., Boerwinkle E., Mosley T.H., Jr., Hobbs H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006;354:1264–1272. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 10.Romeo S., Pennacchio L.A., Fu Y., Boerwinkle E., Tybjaerg-Hansen A., Hobbs H.H., Cohen J.C. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat. Genet. 2007;39:513–516. doi: 10.1038/ng1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahituv N., Kavaslar N., Schackwitz W., Ustaszewska A., Martin J., Hebert S., Doelle H., Ersoy B., Kryukov G., Schmidt S. Medical sequencing at the extremes of human body mass. Am. J. Hum. Genet. 2007;80:779–791. doi: 10.1086/513471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji W., Foo J.N., O'Roak B.J., Zhao H., Larson M.G., Simon D.B., Newton-Cheh C., State M.W., Levy D., Lifton R.P. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 2008;40:592–599. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nejentsev S., Walker N., Riches D., Egholm M., Todd J.A. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romeo S., Yin W., Kozlitina J., Pennacchio L.A., Boerwinkle E., Hobbs H.H., Cohen J.C. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J. Clin. Invest. 2009;119:70–79. doi: 10.1172/JCI37118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li B., Leal S.M. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am. J. Hum. Genet. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madsen B.E., Browning S.R. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5:e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramensky V., Bork P., Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.