

Abstract
Previously, we localized the defective gene for the urofacial syndrome (UFS) to a region on chromosome 10q24 by homozygosity mapping. We now report evidence that Heparanse 2 (HPSE2) is the culprit gene for the syndrome. Mutations with a loss of function in the Heparanase 2 (HPSE2) gene were identified in all UFS patients originating from Colombia, the United States, and France. HPSE2 encodes a 592 aa protein that contains a domain showing sequence homology to the glycosyl hydrolase motif in the heparanase (HPSE) gene, but its exact biological function has not yet been characterized. Complete loss of HPSE2 function in UFS patients suggests that HPSE2 may be important for the synergic action of muscles implicated in facial expression and urine voiding.
Main Text
The urofacial syndrome (UFS [MIM 236730]) is an autosomal recessive disease characterized by urological and facial abnormalities.1,2 The typical symptom for UFS patients is a distorted face, as if in pain or sadness, when they smile or laugh. However, dysfunctional voiding, such as repeated episodes of urinary tract infection, dysuria or incontinence, frequency, urgency, or enuresis, is the life-threatening component of the syndrome. This gradually progresses to severe upper tract damage and the development of bladder trabeculation, vesicoureteral reflux (VUR), hydroureternephrosis, and subsequent renal failure.1–3 Nevertheless, no apparent neurological or urinary obstructive pathology has been noticed in any UFS patients.
Previous studies in patients originating from Colombia localized the disease gene to a region on chromosome 10q24 by homozygosity mapping.4,5 Subsequent fine mapping provided preliminary evidence that the disease gene could be located in a genomic interval of approximately 250 kb DNA between markers D10S2500 and D10S2511.6 Mutation screening in UFS patients was also carried out in two candidate genes, GOT1 (MIM 138180) and CNNM1 (ancient conserved domain protein 1, ACDP1) (MIM 607802).5–7 GOT1 contains 9 exons and spans 33.7 kb of genomic DNA. It is a ubiquitous pyridoxal phosphate-dependent enzyme, and it plays an important role in amino acid metabolism and in the urea/tricarboxylic acid cycles. CNNM1 contains 11 exons and spans a genomic region of 67.56 kb DNA sequence. CNNM1 is a novel gene belonging to a new gene family that could be implicated in divalent ion transport.8–10 Unexpectedly, no pathological mutation was discovered in these two plausible candidate genes, raising the possibility that the disease gene may be outside of the refined interval. This was a distinct possibility, because it is sometimes difficult to distinguish between recombination events and point mutations in homozygosity mapping.
To accurately redefine the disease interval, we selected multiple families with recombination events on both ends of the disease interval, which includes patient II.1 of UFS-30 collected in the United States and patients II.1 of UFS-2, II.1 of UFS-12, II.1 of UFS-13, IV.1 of UFS-14, V.1 of UFS-15, and II.1 of UFS-18, recruited from Colombia (Figure 1). Because these patients carry two identical disease chromosomes inherited from their parents, they were homozygous for the markers in the region without recombination. Haplotype analysis was then carried out to determine the disease interval. As shown in Figure 2, chromosomal regions for each patient without recombination (markers with homozygous genotypes) were boxed. Heterozygous genotypes were characterized in patients II.1 of UFS-2, II.1 of UFS-13, II.1 of UFS-18, and II.1 of UFS-30 at marker D10S1433 and its adjacent telomeric markers, indicating that D10S1433 defines the telomeric boundary for the disease interval. Similarly, patients II.1 of UFS-3, II.1 of UFS-12, IV.1 of UFS-14, and V.1 of UFS-15 showed heterozygous genotypes at marker D10S603 and other centromeric markers, demonstrating that D10S603 could be the breakpoint for centromeric recombination. Therefore, the disease region was placed at an expanded interval between markers D10S1433 and D10S603. Seventeen known genes and two unnamed transcripts (LOC100289312 and NCRNA00093) were characterized within this newly defined disease interval (Figure 3).
Figure 1.
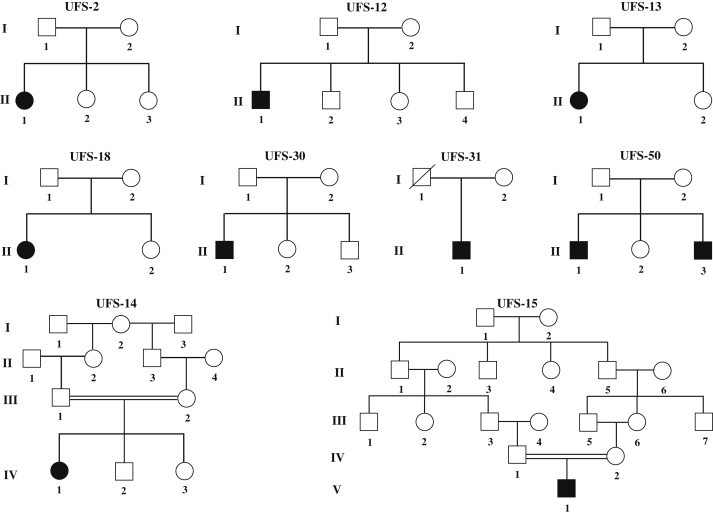
Pedigrees Used for Redefining UFS Interval and Candidate Gene Mutation Screenings
Pedigrees of UFS-2, -12, -13, -14, -15, and -18 were collected from Colombia, pedigrees UFS-30 and UFS-31 were recruited from the United States, and pedigree UFS-50 was obtained from France. Patients of UFS-30 and UFS-31 share a common Irish heritage, and patients in pedigree UFS-50 are of European descent. Of note, patients in pedigrees UFS-2, -12, -13, -18, -30, -31, and -50 were from unrelated marriages, whereas patients in pedigrees UFS-14 and -15 were from consanguineous marriages.
Figure 2.
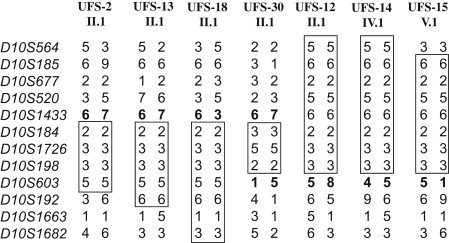
Redefined UFS Interval Based on Haplotype Analysis of Patients with Recombination Events
Patients II.1 of UFS-2, II.1 of UFS-13, II.1 of UFS-18, II.1 of UFS-12, IV.1 of UFS-14, and V.1 of UFS-15 were Colombian, whereas II.1 of UFS-30 was a United States patient. Markers with the homozygous genotype were boxed to define the region without recombination. Alleles for markers D10S1443 and D10S603 in patients with recombination events are in bold to show the telomeric and centromeric breakpoints, respectively. The disease interval was placed between markers D10S1433 and D10S603.
Figure 3.
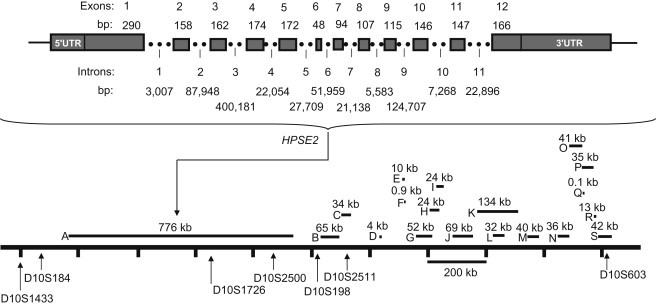
A Transcriptional Map for the Newly Defined UFS Disease Interval
Top: a detailed genomic structure for the HPSE2 gene. Bottom: all candidate genes located between D10S1433 and D10S603. The size in genomic DNA (kb) for each transcript is shown within the figure. The order and genomic location of each microsatellite marker within the map was defined based on the genomic sequence information from the Ensemble database. The letters represent the following: A, HPSE2 (NM_021828); B, CNNM1 (NM_020348); C, GOT1 (NM_002079); D, NKX2-3 (NM_145285); E, SLC25A28 (NM_031212); F, LOC100289312 (NM_002343029); G, ENTPD7 (NM_020354); H, COX15 (NM-078470); I, CUTC (NM-015960); J, ABCC2 (NM_000392); K, DNMBP (NM_015221); L, NCRNA00093 (NR_024130); M, CPN1 (NM_001308); N, ERLIN1 (NM_006459); O, CHUK (NM_001278); P, CWF19L1 (NM_018294); Q, SNORA12 (NR_002954); R, BLOC1S2 (NM_0173809); S, PKD2L1 (NM_016112).
Mutation screening was then carried out for the 14 new genes and the two above indicated unnamed transcripts located in the centromeric region of the interval between D10S198 and D10S603. We selected three patients, originating from Colombia (II.1 of UFS-2), the United States (II.1 of UFS-30), and France (II.1 of UFS-50), with each representing a disease haplotype (mutation), except for II.1 of UFS-50, who inherited two different disease haplotypes from his parents. We also included a pooled control DNA (equal amount of DNA pooled from eight normal individuals) to evaluate normal polymorphisms. We designed a PCR walking strategy, with each PCR flanking a 600–800 bp of genomic region. The screenings have covered 4 kb of genomic DNA flanking the promoter region and 5′ UTR region, entire exons, 120 bp of exon/intron junctions, and the 3′ UTR region of each candidate gene. Unfortunately, we were not able to identify any apparent pathological mutation in these genes (data not shown).
Finally, we turned our attention to the region telomeric to CNNM1 (ACDP1), which contains a single gene (HPSE2) that spans 776 kb of genomic DNA (Figure 3). A homozygous nonsense mutation (c.1516C>T) was first identified in exon 11 of the HPSE2 gene in the Colombian patient (Figure 4A). This mutation, designated R506X (c.1516C>T), produces a truncated protein that misses 86 amino acids at the C terminus of the HPSE2 protein. The mutation abolishes the TaqI restriction site that is present in the wild-type DNA. PCR amplification followed by TaqI digestion was used for rapid genotyping of the 31 other UFS patients in our collection. As expected from the haplotype data on these patients, all UFS patients from Colombia are homozygous for the R506X (c.1516C>T) mutation (data not shown).
Figure 4.
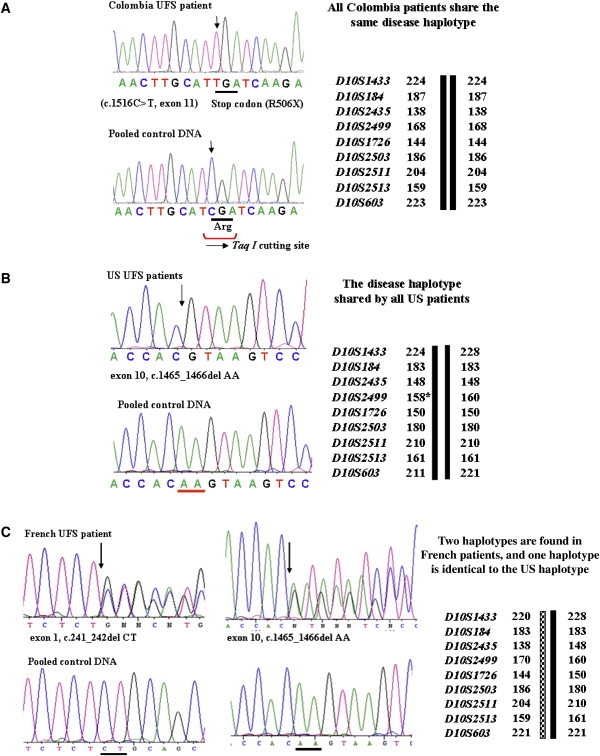
Mutations Identified in UFS Patients
(A) All Colombian patients share one identical disease haplotype and carry a homozygous nonsense mutation c.1516C>T (R506X) in exon 11.
(B) The United States patients possess one identical disease haplotype and carry a homozygous AA deletion (c.1465_1466delAA) in exon 10 at nucleotide positions 1465 and 1466 (start codon as position 1).
(C) The French patients carry two different disease haplotypes: the first is identical to the United States patients, and the second is different from other patients. They carry a 2 bp AA deletion (c.1465_1466delAA) in exon 10, like the United States patients, and a 2 bp CT deletion (c.241_242delCT) in exon 1 at positions 241 and 242 (start codon as position 1).
We then screened mutations for the rest of the UFS patients. Patients from two United States pedigrees (II.1 of UFS-30 and II.1 of UFS-31) with a common Irish heritage share one same disease haplotype. Unlike the Colombian patients, these two patients carry a homozygous 2 bp deletion (c.1465_1466delAA) in exon 10 at nucleotide positions 1465 and 1466 (start codon as position 1) (Figure 4B), resulting in a larger protein of 613 aa, with a completely different sequence for the last 125 aa because of reading frame shift. Of note, these patients carry a point mutation at marker D10S2499 that was previously used to define the centromeric boundary.6
Unlike the homozygous mutations characterized in the above patients, two French patients (II.1 and II.3 of UFS-50) from the same family of European decent carry two different disease haplotypes originating from their nonconsanguineous parents (Figure 1): the first is identical to the United States haplotype, and the second is unique to the French patients. As expected, compound heterozygous loss-of-function mutations were characterized in these two patients. One is the same 2 bp deletion mutation found in the United States patients (c.1465_1466delAA), whereas the other is a 2 bp deletion mutation (c.241_242delCT) in exon 1 at nucleotide positions 241 and 242 (start codon as position 1) (Figure 4C).
The full-length HPSE2 gene consists of 2353 bp of nucleotides encoding a protein of 592 aa that contains a region showing sequence homology to the glycosyl hydrolase motif in the heparanase (HPSE) gene,11 but its exact biological function has not yet been characterized. Interestingly, phylogenetic analysis revealed that no HPSE2 orthologs exist in species of nonvertebrates such as Drosophila melanogaster (see Figure S1 available online). HPSE2 is evolutionarily conserved among diverse vertebrates. For example, the similarity between mouse and human is 93% for the DNA coding sequence and 97% at the protein sequence level (Figure S2).
Because of the involvement of both the urinary system and the facial muscles for the UFS phenotype, it has been thought that UFS may result from defects in a region of the brain that controls micturation and facial muscle.1,3,12 Contrary to this hypothesis, cranial MRI analysis of UFS patients showed negative results for lesions in the brain, and MRI analysis further demonstrated a normal spine and conus medularis in the UFS patients, suggesting a nonneurological origin for the disease pathogenesis.3,13 Furthermore, RT-PCR analysis failed to detect HPSE2 expression in the brain and spinal cord. On the contrary, both the facial muscle and urinary bladder showed high levels of HPSE2 expression. However, unlike urinary bladder, very low levels of HPSE2 mRNA were detected in the stomach and intestine, and, unlike facial muscle, HPSE2 was almost completely absent in the skeletal muscle (data not shown). Given the phenotypic characteristics of UFS patients (they show distorted facial expression and detrusor-sphincter dyssynergia), our results suggest that HPSE2 could regulate the coordinated action of muscles implicated in facial expression and urine voiding in the periphery. Moreover, the characterization of the HPSE2 gene for the syndrome has now paved the way to fully dissect the underlying mechanisms for the puzzling observations on the clinical phenotype of patients with this devastating disease.
In summary, this study identified three loss-of-function mutations in the HPSE2 gene in UFS patients from multiple countries, which provides evidence for the conclusion that HPSE2 is the culprit gene responsible for the syndrome. Voiding disorders such as frequency, urgency, enuresis, dysuria or incontinence without apparent neurological abnormality, and urinary obstruction are highly prevalent in the general population but are greatly underrecognized and underreported.12,14–19 Because UFS patients share clinical, radiological, and urodynamic features with those patients with dysfunctional voiding in the general population,3,13,20–22 it will be interesting to determine whether altered HPSE2 function is implicated in the pathogenesis of general voiding disorders.
Acknowledgments
We are grateful to the affected subjects and their family members for participating in this study. We are also grateful to Leszek Ignatowicz for his great suggestion for manuscript preparation and Yan Jiao for her advice in high-throughput mutation screening. This work was supported by grants from the National Institutes of Health (DK074957 to C.-Y.W. and DK53266 to J.-X.S.).
Contributor Information
Jin-Xiong She, Email: jshe@mcg.edu.
Cong-Yi Wang, Email: cwang@mcg.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
NCBI Protein Database, http://www.ncbi.nlm.nih.gov/protein/
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/Omim/
The Ensembl Project genome database, http://uswest.ensembl.org/
References
- 1.Ochoa B. The urofacial (Ochoa) syndrome revisited. J. Urol. 1992;148:580–583. doi: 10.1016/s0022-5347(17)36659-4. [DOI] [PubMed] [Google Scholar]
- 2.Ochoa B., Gorlin R.J. Urofacial (ochoa) syndrome. Am. J. Med. Genet. 1987;27:661–667. doi: 10.1002/ajmg.1320270320. [DOI] [PubMed] [Google Scholar]
- 3.Ochoa B. Can a congenital dysfunctional bladder be diagnosed from a smile? The Ochoa syndrome updated. Pediatr. Nephrol. 2004;19:6–12. doi: 10.1007/s00467-003-1291-1. [DOI] [PubMed] [Google Scholar]
- 4.Wang C.Y., Hawkins-Lee B., Ochoa B., Walker R.D., She J.X. Homozygosity and linkage-disequilibrium mapping of the urofacial (Ochoa) syndrome gene to a 1-cM interval on chromosome 10q23-q24. Am. J. Hum. Genet. 1997;60:1461–1467. doi: 10.1086/515469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang C.Y., Huang Y.Q., Shi J.D., Marron M.P., Ruan Q.G., Hawkins-Lee B., Ochoa B., She J.X. Genetic homogeneity, high-resolution mapping, and mutation analysis of the urofacial (Ochoa) syndrome and exclusion of the glutamate oxaloacetate transaminase gene (GOT1) in the critical region as the disease gene. Am. J. Med. Genet. 1999;84:454–459. [PubMed] [Google Scholar]
- 6.Wang C.Y., Shi J.D., Huang Y.Q., Cruz P.E., Ochoa B., Hawkins-Lee B., Davoodi-Semiromi A., She J.X. Construction of a physical and transcript map for a 1-Mb genomic region containing the urofacial (Ochoa) syndrome gene on 10q23-q24 and localization of the disease gene within two overlapping BAC clones (<360 kb) Genomics. 1999;60:12–19. doi: 10.1006/geno.1999.5908. [DOI] [PubMed] [Google Scholar]
- 7.Wang C.Y., Davoodi-Semiromi A., Shi J.D., Yang P., Huang Y.Q., Agundez J.A., Moran J.M., Ochoa B., Hawkins-Lee B., She J.X. High resolution mapping and mutation analyses of candidate genes in the urofacial syndrome (UFS) critical region. Am. J. Med. Genet. A. 2003;119A:9–14. doi: 10.1002/ajmg.a.20042. [DOI] [PubMed] [Google Scholar]
- 8.Wang C.Y., Shi J.D., Yang P., Kumar P.G., Li Q.Z., Run Q.G., Su Y.C., Scott H.S., Kao K.J., She J.X. Molecular cloning and characterization of a novel gene family of four ancient conserved domain proteins (ACDP) Gene. 2003;306:37–44. doi: 10.1016/s0378-1119(02)01210-6. [DOI] [PubMed] [Google Scholar]
- 9.Wang C.Y., Yang P., Shi J.D., Purohit S., Guo D., An H., Gu J.G., Ling J., Dong Z., She J.X. Molecular cloning and characterization of the mouse Acdp gene family. BMC Genomics. 2004;5:7. doi: 10.1186/1471-2164-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo D., Ling J., Wang M.H., She J.X., Gu J., Wang C.Y. Physical interaction and functional coupling between ACDP4 and the intracellular ion chaperone COX11, an implication of the role of ACDP4 in essential metal ion transport and homeostasis. Mol. Pain. 2005;1:15. doi: 10.1186/1744-8069-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKenzie E., Tyson K., Stamps A., Smith P., Turner P., Barry R., Hircock M., Patel S., Barry E., Stubberfield C. Cloning and expression profiling of Hpa2, a novel mammalian heparanase family member. Biochem. Biophys. Res. Commun. 2000;276:1170–1177. doi: 10.1006/bbrc.2000.3586. [DOI] [PubMed] [Google Scholar]
- 12.Schulman S.L. Voiding dysfunction in children. Urol. Clin. North Am. 2004;31:481–490. doi: 10.1016/j.ucl.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 13.Nicanor F.A., Cook A., Pippi-Salle J.L. Early diagnosis of the urofacial syndrome is essential to prevent irreversible renal failure. Int. Braz. J. Urol. 2005;31:477–481. doi: 10.1590/s1677-55382005000500012. [DOI] [PubMed] [Google Scholar]
- 14.Austin P.F., Ritchey M.L. Dysfunctional voiding. Pediatr. Rev. 2000;21:336–341. doi: 10.1542/pir.21-10-336. [DOI] [PubMed] [Google Scholar]
- 15.Bauer S.B., Retik A.B., Colodny A.H., Hallett M., Khoshbin S., Dyro F.M. The unstable bladder in childhood. Urol. Clin. North Am. 1980;7:321–336. [PubMed] [Google Scholar]
- 16.Feldman A.S., Bauer S.B. Diagnosis and management of dysfunctional voiding. Curr. Opin. Pediatr. 2006;18:139–147. doi: 10.1097/01.mop.0000193289.64151.49. [DOI] [PubMed] [Google Scholar]
- 17.Nowara A., Witek A., Wilk K. Overactive bladder—definition, epidemiology, pathogenesis. Ginekol. Pol. 2007;78:484–487. [PubMed] [Google Scholar]
- 18.Stewart W.F., Van Rooyen J.B., Cundiff G.W., Abrams P., Herzog A.R., Corey R., Hunt T.L., Wein A.J. Prevalence and burden of overactive bladder in the United States. World J. Urol. 2003;20:327–336. doi: 10.1007/s00345-002-0301-4. [DOI] [PubMed] [Google Scholar]
- 19.Tyagi S., Thomas C.A., Hayashi Y., Chancellor M.B. The overactive bladder: Epidemiology and morbidity. Urol. Clin. North Am. 2006;33:433–438. doi: 10.1016/j.ucl.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 20.Aydogdu O., Burgu B., Demirel F., Soygur T., Ozcakar Z.B., Yalcinkaya F., Tekgul S. Ochoa syndrome: A spectrum of urofacial syndrome. Eur. J. Pediatr. 2010;169:431–435. doi: 10.1007/s00431-009-1042-9. [DOI] [PubMed] [Google Scholar]
- 21.Chauve X., Missirian C., Malzac P., Girardot L., Guys J.M., Louis C., Philip N., Voelckel M.A. Genetic homogeneity of the urofacial (Ochoa) syndrome confirmed in a new French family. Am. J. Med. Genet. 2000;95:10–12. doi: 10.1002/1096-8628(20001106)95:1<10::aid-ajmg3>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Minaur S., Oliver F., Yanez J.M., Soriano J.R., Quinn F., Reardon W. Three new European cases of urofacial (Ochoa) syndrome. Clin. Dysmorphol. 2001;10:165–170. doi: 10.1097/00019605-200107000-00002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.