

Abstract
Urinary voiding dysfunction in childhood, manifesting as incontinence, dysuria, and urinary frequency, is a common condition. Urofacial syndrome (UFS) is a rare autosomal recessive disease characterized by facial grimacing when attempting to smile and failure of the urinary bladder to void completely despite a lack of anatomical bladder outflow obstruction or overt neurological damage. UFS individuals often have reflux of infected urine from the bladder to the upper renal tract, with a risk of kidney damage and renal failure. Whole-genome SNP mapping in one affected individual defined an autozygous region of 16 Mb on chromosome 10q23-q24, within which a 10 kb deletion encompassing exons 8 and 9 of HPSE2 was identified. Homozygous exonic deletions, nonsense mutations, and frameshift mutations in five further unrelated families confirmed HPSE2 as the causative gene for UFS. Mutations were not identified in four additional UFS patients, indicating genetic heterogeneity. We show that HPSE2 is expressed in the fetal and adult central nervous system, where it might be implicated in controlling facial expression and urinary voiding, and also in bladder smooth muscle, consistent with a role in renal tract morphology and function. Our findings have broader implications for understanding the genetic basis of lower renal tract malformations and voiding dysfunction.
Main Text
Dysfunctional urinary voiding manifests variously as incontinence, dysuria, and urinary frequency. It can be accompanied by a failure to coordinate relaxation of the sphincter mechanism with bladder smooth muscle wall (detrusor) contraction, without overt neurological or anatomical explanation. It is common, affecting up to 15% of children at 6 years of age.1 Unrecognized and untreated, it can occasionally lead to kidney damage associated with impaired flow of urine from the upper renal tract into the bladder and/or vesico-ureteric reflux (VUR) of infected urine. The pathogenesis of dysfunctional urinary voiding is unclear and may be informed by understanding the basis of urofacial (Ochoa) syndrome (UFS [MIM 236730]), a rare autosomal recessive disease characterized by a severe and early-onset form of dysfunctional urinary voiding.2 Affected individuals usually present prenatally or in early childhood with grossly distorted renal tracts, comprising dysmorphic bladders and dilatation of the ureter and renal pelvis. They are at high risk of VUR, with ascending bacterial infection leading to kidney damage, hypertension, and renal failure. A third of UFS children experience constipation or fecal soiling, suggesting that the pathophysiology of the syndrome encompasses a broader functional impairment of elimination.3 Affected individuals also have a characteristic facial grimace when trying to smile, which both aids accurate diagnosis and differentiates the condition from other causes of neuropathic and nonneuropathic bladder.
Previous homozygosity and linkage mapping studies in consanguineous families of Columbian, American-Irish, Spanish, and French extraction were undertaken with microsatellite markers.4,5 These studies identified and then fine-mapped a locus to a 220 kb region of chromosome 10q23-q24 that was proposed to contain the causative gene.4,5 The region contained two genes, ACDP1 (MIM 607802) and GOT1 (MIM 138180), but subsequent sequence analyses failed to identify pathogenic mutations in any of the affected individuals.
Here we demonstrate that biallelic mutations in the HPSE2 gene on chromosome 10q23-q24 are responsible for some cases of UFS. Furthermore, we demonstrate that the gene is normally expressed in both the central nervous system and the bladder.
Family 1 (Figure 1) is a consanguineous British Pakistani family with three siblings affected with UFS. The parents are unaffected first cousins. The proband (IV-4) presented when 2 years old with acute renal failure and urinary sepsis. He was found to have a hypercontractile bladder, bilateral VUR, and hydronephrotic scarred kidneys. He underwent a surgical ileal loop urinary diversion procedure. When assessed at age 11, his glomerular filtration rate (GFR), a measurement of excretory kidney function, was at the lower end of the normal range, and he had modest proteinuria, a marker of kidney damage. He required pharmacological treatment for hypertension. When his sister (IV-3) was age 6, she was found to have dysfunctional voiding with a hypocontractile bladder and VUR; surgery was not undertaken, and her kidney function is normal. The index case's younger brother (IV-5) presented with renal pelvis dilatation on antenatal ultrasound screening. Postnatal investigations showed a low-capacity, trabeculated bladder with VUR, and he underwent surgical urinary diversion. Assessed at the age of 10 years, he had structurally abnormal kidneys, a GFR at the lower end of the normal range, and modest proteinuria, but he was normotensive. All three affected siblings have the UFS characteristic grimace upon smiling (Figure 1).
Figure 1.
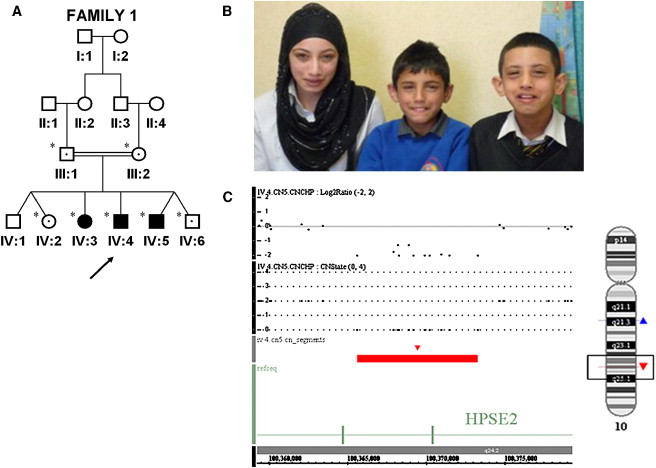
Identification of Intragenic Deletion in HPSE2 in an Affected UFS Patient
(A) The pedigree of family 1 indicates the individuals who were screened by PCR for a deletion breakpoint, indicated by ∗. Filled-in symbols represent affected individuals, and symbols containing a dot indicate heterozygote. SNP6.0 array performed on IV:4 is indicated by an arrow.
(B) Photograph of affected siblings demonstrating characteristic grimace upon smiling.
(C) SNP6.0 array copy number analysis illustrating 11 probe deletions in HPSE2. Log2ratio, copy number state (CNState), and red bar all indicate the deleted region, and an ideogram of chromosome 10 with the position of the deletion is indicated by a red triangle.
Ethical approval for this study was obtained from the University of Manchester (06138) and NHS ethics committees (06/Q1406/52). Informed consent was obtained from all participating subjects or, in the case of children, from their parents. Affymetrix SNP6.0 array genotyping in individual IV-4 via AutoSNPa6 analysis revealed a homozygous region of 16.65 Mb (flanked by rs17346680 and rs10786808) that encompassed the critical interval previously defined on 10q23-q24.5 Copy number analysis via the CN5 algorithm from Affymetrix defined an intragenic homozygous deletion of a maximum 11.25 kb (from rs12240940 to rs658053) encompassing exons 8 and 9 in HPSE2 that encodes heparanase 2 (Figure 1). HPSE2 is situated at 100,208,867−100,985,609 bp (National Center for Biotechnology Information [NCBI] build 36) and therefore lies immediately centromeric to the previously identified critical region.5 PCR analysis and DNA sequencing across the breakpoint defined a complex rearrangement encompassing a 10.81 kb deletion and a 23 bp insertion at the breakpoints. The rearrangement is proposed to result in an in-frame deletion of exons 8 and 9 (c.1099-4166_1320+840delins23, p.V367_P440del) and in a removal of 74 amino acids. The mutation segregated with the disorder within the family, and the mutation was not present in a panel of ethnically matched controls or in the Database of Genomic Variation.
Mutation screening of HPSE2 led to the identification of mutations in five previously reported unrelated families (Table 1; Figure 2) with UFS.7–9 Primers for amplification of exons and exon-intron boundaries of HPSE2 were designed with Primer3Plus by using NCBI reference sequence NM_021828.4 for exons 1 to 12 and NM_001166246.1 for transcript variant 4 alternative exon 12 (exon12b) (see Table S1 available online for primer sequences and PCR conditions). A homozygous nonsense mutation in exon 10 (c.1414C>T, p.R472X) was identified in a 16-year-old Turkish female with a history of dysfunctional voiding, a dysmorphic, poorly emptying bladder, and hydronephrosis (family 2).7
Table 1.
Mutations in HPSE2 Identified in UFS Patients
| Family | Ethnicity | Nucleotide Change | Exons | Amino Acid Change |
|---|---|---|---|---|
| 1 | Asian | c.1099-4166_1320+840delins23 | 8, 9 | p.V367_P440del |
| 2 | Turkish | c.1414C>T | 10 | p.R472X |
| 3 | Turkish | c.457C>T | 3 | p.R153X |
| 4 | Turkish | c.57dupC | 1 | p.A20RfsX45 |
| 5 | Spanish | c.449-?_610+? | 3 | p.D150_T203del |
| 6 | Irish | c.1465_1466del | 10 | p.N489PfsX126 |
Nomenclature is based on NCBI reference sequence NM_021828.4.
Figure 2.
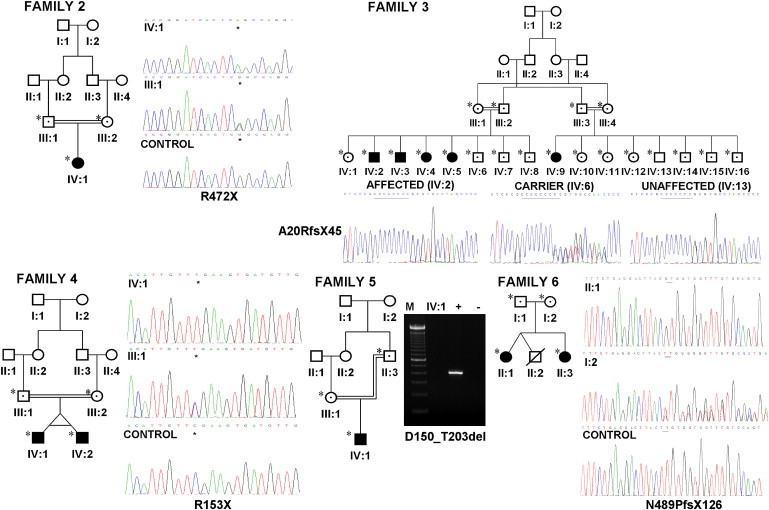
Identification of HPSE2 Mutations in UFS Families
Pedigrees and genomic sequence chromatograms of families 2, 3, 4, and 6. In family 2, a homozygous nonsense mutation in exon 10 (p.R472X) was identified; in family 3, a frameshift mutation as a result of a homozygous single base pair insertion in exon 1 (p.A20RfsX45) was identified; in family 4, a homozygous nonsense mutation in exon 3 (p.R153X) was identified; and in family 6, a 2 bp deletion at the end of exon 10 resulting in a frameshift (p.N489PfsX126) was identified. Pedigree of family 5 and agarose gel reveal no amplification of exon 3 in the affected individual. M represents the 100 bp DNA ladder, + represents the positive control, and – represents the negative control. All individuals screened are indicated by ∗, filled-in symbols represent affected individuals, and symbols containing a dot indicate heterozygote.
In family 3, five affected members of another Turkish family all carried two copies of a frameshift mutation because of a homozygous single base pair insertion of a cytosine in exon 1 resulting in a premature stop codon at position 64 (c.dup57, p.A20RfsX45).8 A homozygous nonsense mutation in exon 3 (c.457C>T, p.R153X) was identified in 18-month-old Turkish twin brothers affected by UFS (family 4).8 Both had presented with urosepsis and were found to have dysmorphic bladders.
An in-frame deletion of exon 3 (c.449-?_610+?, p.D150_T203del) removing 54 amino acids was identified in a 12-year-old Spanish boy who presented at the age of 3 years with urosepsis and was found to have dysfunctional and dysmorphic bladder and upper renal tract dilatation (family 5).9 A homozygous 2 base pair deletion at the end of exon 10 resulting in a frameshift (c.1465_1466del, p.N489PfsX126) was determined in two Irish sisters with UFS, with strikingly different presentations (family 6).9 The first sister (II-1) was diagnosed at age 3 with bilateral upper tract dilatation and a nonfunctioning left kidney. Her sister (II-3) experienced recurrent urinary tract infections but was not diagnosed until her third decade. The parents were both confirmed as heterozygotes for the mutation and were not knowingly related to each other.
The mutations segregated with the disease phenotype in all families and were not identified on polymorphism databases or in panels of ethnically matched controls (93 Pakistani, 96 Turkish, and 96 European). Importantly, we did not identify HPSE2 mutations in four additional families, two published cases8 and two other families with UFS that we have ascertained. Three of these additional UFS families were consanguineous, and heterozygosity for polymorphic variants at the HPSE2 locus suggests that UFS does not map to this locus and is likely to be genetically heterogeneous. This contrasts with previous genetic mapping reports that defined a single UFS locus at 10q23-q24.5,10
The nonsense mutations identified in two families, p.R153X and p.R472X, are likely to lead to nonsense-mediated decay of the transcript and absent protein. Likewise, the frameshift mutations p.A20RfsX45 and p.N489PfsX126 are predicted to result in either nonsense-mediated decay or in unfolded proteins that are readily degraded.
Therefore, the effects of the in-frame whole exonic deletions of exons 8 and 9 (family 1) and exon 3 (family 5) were considered. The Phyre method for protein fold recognition by using one- and three-dimensional sequence profiles coupled with secondary structure and solvation potential information was used to predict the functional consequence of the exon 8 and 9 deletion (Figure 3).11 Residues 195–592 of the heparanase 2 sequence were aligned to the family 51 α-L-arabinofuranosidase with ClustalW,12 and the structural model was generated by using the SWISS-MODEL automated modeling server,13 with the alignment between heparanase 2 and the family 51 α-L-arabinofuranosidase sequences as an input and the crystal structure of the family 51 α-L-arabinofuranosidase14 (Protein Data Bank code 1PZ2) chain A as a template. This predicted that the deletion most likely results in an unstructured protein that will be degraded. Further, the deletion of these two exons should remove part of the active site (the residue corresponding to Glu343 in heparanase 1, which is potentially equivalent to Glu380 in heparanase 2).
Figure 3.
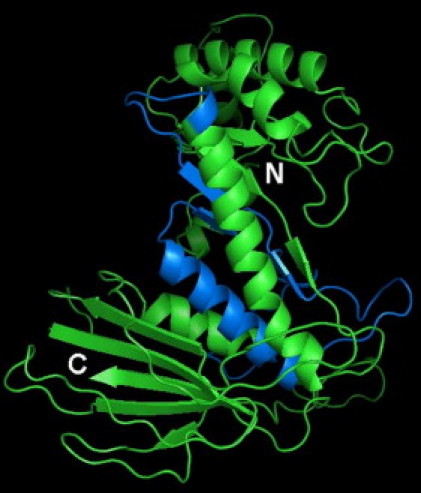
Protein Structure of Heparanase 2
Model of the predicted 50 kDa subunit of human heparanase 2 (residues 195–592) based on the crystal structure of the family 51 α-L-arabinofuranosidase13 (Protein Data Bank code 1PZ2). The part of the structure that is encoded by exons 8 and 9 is shown in marine blue. The N and C termini are indicated. This figure was prepared with the program PyMOL (DeLano Scientific LLC).
In contrast, the exon 3 deletion was not modeled because this sequence corresponds to the linker sequence, which is cleaved off in heparanase 1 to yield an active enzyme. The heparanase 2 exon 3 deletion mutant could potentially form a properly folded and active protein. It is noteworthy that this sequence is missing in isoform 4 (HPSE2a), which lacks exons 3 and 4.
A human multiple-tissue RNA dot blot (RNA Masterblot; CLONTECH) hybridized with a full-length 32P-labeled HPSE2b probe showed widespread HPSE2 expression in the adult central nervous system, including the spinal cord, caudate nucleus, thalamus, substantia nigra, medulla oblongata, putamen, and pons (Figure S1).
Furthermore, as assessed by RT-PCR of a human embryonic brain panel (MRC Newcastle Brain Tissue Resource) from Carnegie stage 16 (week 6) and stage 21 (week 8) human embryos, HPSE2 was expressed in the developing forebrain, telencephalon, diencephalon, midbrain, hindbrain, and spinal cord (Figure 4). Dot-blot analysis also showed prominent HPSE2 expression in the adult urinary bladder, and in situ hybridization, as previously described,15 demonstrated that transcripts localized within tissue sections to longitudinal and circular layers of detrusor muscle (Figure 4). On dot blots, it was apparent that HPSE2 was also expressed in the adult fore-, mid-, and lower gut and in both the adult and fetal kidney. As assessed by RT-PCR of wild-type mouse bladder, brain, kidney, and ureter,16 Hpse2 expression is widespread, with all organs positive at E15 and in the adult samples (Figure 4). In contrast, for Hpse1 at E15, the bladder, kidney, and ureter are positive, with no expression in the brain. In the adult samples, however, Hpse1 is also expressed in the bladder, but there is no expression in the brain; in addition, the kidney expression is positive, but very weak, and the ureter is negative.
Figure 4.
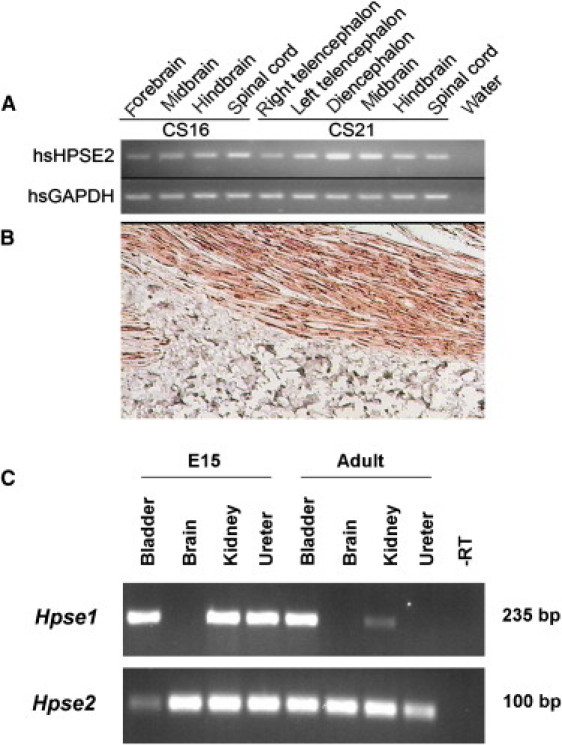
Expression of HPSE2 in Human and Murine Tissues
(A) RT-PCR demonstrating expression of HPSE2 in embryonic brain tissue at Carnegie stages 16 and 21.
(B) Direct in situ RT-PCR detection of HPSE2 mRNA expression human bladder tissue. Strong brown staining demonstrates expression in longitudinal smooth muscle tissue.
(C) RT-PCR demonstrating expression of Hpse2 in E15 and adult mouse bladder, brain, kidney, and ureter. Hpse1 expression at E15 in the murine bladder, kidney, and ureter are positive, with no expression in the brain. In adult mouse tissue, Hpse1 is expressed in the bladder, but not in the brain. Hpse1 expression in the adult kidney is positive, but very weak, and the ureter is negative.
The HPSE2 transcript comprises 12 exons and encodes a 592 amino acid polypeptide, heparanase 2 (previously HPA2). Four potential isoforms of HPSE2 have been identified, as a consequence of differential splicing.17 The four different mRNAs encode putative proteins of 480, 534, 538, and 592 amino acids and result from differential splicing of exons 3 and 4. Because we identified a whole exon pathogenic deletion of exon 3 in a family with UFS, this suggests that the isoforms containing exon 3 are critical for HPSE2 function.17 We were unable to obtain mRNA derived from these patients, which precluded an examination of the effect at the transcript level. The 2 bp deletion in family 6 predicted to create a frameshift mutation disrupts a consensus heparin-binding motif (NHHNHN) at the boundary of exons 10 and 11 in HPSE2. This may therefore produce a mutated form of the protein that is unable to bind to its glycosaminoglycan substrate. Removal of this activity would be consistent with a predicted null phenotype and consistent with the other disease-associated nonsense and frameshift mutations.
HPSE2 was identified through sequencing of expressed sequence tags homologous to HPSE1 (MIM 604724).17 HPSE1 encodes heparanase 1 (HPA1), an endoglycosidase enzyme (the active form consisting of a heterodimer of 8 kDA and 50 kDa subunits) that is known to cleave heparan sulfate glycosaminoglycans from proteoglycan core proteins. Heparanase 1 and the 2c isoform share 45% homology spanning both subunits, with higher patches of homology or identity covering critical regions, including small subunit domains and catalytic glutamic acid residues. We are currently investigating the proposal that the heparanase 2 isoforms also have carbohydrate processing activity and so play a role in extracellular matrix remodeling (E.A.M., unpublished data).
Because of the association with an abnormal facial expression and voiding dysfunction in UFS, it has been suggested that the gene or genes mutated in the syndrome may have roles within the brain and/or brainstem in areas that control both facial expression and also bladder and bowel voiding; for example, the midbrain contains the pontine micturition center and facial nerve nucleus.18 However, it could also be speculated that the renal tract dysfunction is caused by lesions in neurons within or near the bladder and/or that the renal tract dysmorphology could be caused by failure to form normally functioning smooth muscle within the walls of bladder and ureters. Indeed, neural networks are present even in developing bladders,19 whereas mutations of genes expressed in urothelium or nascent smooth muscle can cause renal tract malformations.20,21 When HPSE2 was first described, preliminary studies showed that transcripts were expressed in adult human brain, bladder, uterus, small intestine, and prostate.17 Our current data confirm and extend these observations, showing that the gene is widely expressed in both the mature and nascent human brain and spinal cord, consistent with the hypothesis that the UFS phenotype is generated by alteration of processes in the central nervous system. On the other hand, the collective data demonstrate that the gene is normally expressed within the bladder itself, where transcripts are found in smooth muscle cells. Our murine studies demonstrate that Hpse2 is also expressed in the developing bladder (as well as in the adult organ). In addition, it is expressed in developing ureters. These observations are consistent with a new hypothesis, namely that heparanase 2 has key roles in the structural and functional maturation of the lower renal tract itself. Certainly, the observation that some affected individuals present with markedly dysmorphic ureters as fetuses (e.g., family 1) is consistent with a direct role for HPSE2 in lower renal tract development. Furthermore, that HPSE2 is expressed in both the adult and developing kidney suggests that it may have intrinsic roles in this organ, leading to the consideration that not all the renal damage found in affected individuals is secondary to lower tract dysfunction. In the mouse, it is also interesting that although there is overlapping expression between Hpse1 and Hspe2 in the renal tract, only Hpse2 is expressed in the developing and mature brain, indicating distinct roles for the encoded proteins. However, it is unclear how absent HPSE2 expression in the central nervous system and/or bladder causes UFS. We speculate that lack of heparanase 2 affects the extracellular matrix composition and hence growth factor signaling and muscle differentiation; e.g., VEGF-A is normally expressed by the nascent bladder and enhances detrusor growth,22 and this factor binds to, and its activity may be modified by, matrix molecules with heparan sulfate chains, which could be disrupted by absent heparanase 2.
In summary, we have demonstrated that biallelic mutations in HPSE2, predicted to abolish activity of heparanase 2, cause UFS in families from different ethnic groups. Because of the variable phenotype of this condition, it is likely that UFS is underrecognized as a cause of nonneuropathic bladder. Further work to understand the biochemical and genetic role of HPSE2 in normal urogenital development will provide greater insight into urinary voiding dysfunction and may be relevant in unraveling the genetics of primary, nonsyndromic VUR, a common familial condition,23 which can be associated with bladder dysfunction and which, in the UK, causes up to 10% of kidney failure, requiring treatment with renal transplantation and/or life-long dialysis.24
Acknowledgments
Genetic Medicine and A.S.W. are supported by the National Institute of Health Research Manchester Biomedical Research Centre, University of Manchester, and the MAHSC. D.A.L is supported by a Senior Non-Clinical Fellowship from Kidney Research UK. Thanks to Tarkan Soygur, Fatos Yalcınkaya, and Zeynep Birsin Ozcakar for their valuable contributions in gathering the data. Thanks to Cathy Merry for helpful discussions.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
AutoSNPa, http://dna.leeds.ac.uk/autosnpa/
Database of Genomic Variation, http://projects.tcag.ca/variation/
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/Omim/
Primer3Plus, http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi
References
- 1.Hoebeke P., van der Walle J. Voiding dysfunction, recurrent urinary infection, constipation, and vesicoureteric reflux: A common disease complex. Dialog. Pediatr. Urol. 2002;25:2–3. [Google Scholar]
- 2.Ochoa B., Gorlin R.J. Urofacial (ochoa) syndrome. Am. J. Med. Genet. 1987;27:661–667. doi: 10.1002/ajmg.1320270320. [DOI] [PubMed] [Google Scholar]
- 3.Ochoa B. The urofacial (Ochoa) syndrome revisited. J. Urol. 1992;148:580–583. doi: 10.1016/s0022-5347(17)36659-4. [DOI] [PubMed] [Google Scholar]
- 4.Wang C.Y., Huang Y.Q., Shi J.D., Marron M.P., Ruan Q.G., Hawkins-Lee B., Ochoa B., She J.X. Genetic homogeneity, high-resolution mapping, and mutation analysis of the urofacial (Ochoa) syndrome and exclusion of the glutamate oxaloacetate transaminase gene (GOT1) in the critical region as the disease gene. Am. J. Med. Genet. 1999;84:454–459. [PubMed] [Google Scholar]
- 5.Wang C.Y., Davoodi-Semiromi A., Shi J.D., Yang P., Huang Y.Q., Agundez J.A., Moran J.M., Ochoa B., Hawkins-Lee B., She J.X. High resolution mapping and mutation analyses of candidate genes in the urofacial syndrome (UFS) critical region. Am. J. Med. Genet. A. 2003;119A:9–14. doi: 10.1002/ajmg.a.20042. [DOI] [PubMed] [Google Scholar]
- 6.Carr I.M., Flintoff K.J., Taylor G.R., Markham A.F., Bonthron D.T. Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum. Mutat. 2006;27:1041–1046. doi: 10.1002/humu.20383. [DOI] [PubMed] [Google Scholar]
- 7.Derbent M., Melek E., Arman A., Uçkan S., Baskin E. Urofacial (ochoa) syndrome: Can a facial gestalt represent severe voiding dysfunction? Ren. Fail. 2009;31:589–592. doi: 10.1080/08860220903003370. [DOI] [PubMed] [Google Scholar]
- 8.Aydogdu O., Burgu B., Demirel F., Soygur T., Ozcakar Z.B., Yalcınkaya F., Tekgul S. Ochoa syndrome: A spectrum of urofacial syndrome. Eur. J. Pediatr. 2010;169:431–435. doi: 10.1007/s00431-009-1042-9. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Minaur S., Oliver F., Yanez J.M., Soriano J.R., Quinn F., Reardon W. Three new European cases of urofacial (Ochoa) syndrome. Clin. Dysmorphol. 2001;10:165–170. doi: 10.1097/00019605-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Chauve X., Missirian C., Malzac P., Girardot L., Guys J.M., Louis C., Philip N., Voelckel M.A. Genetic homogeneity of the urofacial (Ochoa) syndrome confirmed in a new French family. Am. J. Med. Genet. 2000;95:10–12. doi: 10.1002/1096-8628(20001106)95:1<10::aid-ajmg3>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 11.Kelley L.A., Sternberg M.J.E. Protein structure prediction on the Web: A case study using the Phyre server. Nat. Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 12.Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwede T., Kopp J., Guex N., Peitsch M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hövel K., Shallom D., Niefind K., Belakhov V., Shoham G., Baasov T., Shoham Y., Schomburg D. Crystal structure and snapshots along the reaction pathway of a family 51 alpha-L-arabinofuranosidase. EMBO J. 2003;22:4922–4932. doi: 10.1093/emboj/cdg494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stamps A.C., Terrett J.A., Adam P.J. Application of in situ reverse trancriptase-polymerase chain reaction (RT-PCR) to tissue microarrays. J. Nanobiotechnology. 2003;1:3. doi: 10.1186/1477-3155-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Price K.L., Woolf A.S., Long D.A. Unraveling the genetic landscape of bladder development in mice. J. Urol. 2009;181:2366–2374. doi: 10.1016/j.juro.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 17.McKenzie E., Tyson K., Stamps A., Smith P., Turner P., Barry R., Hircock M., Patel S., Barry E., Stubberfield C. Cloning and expression profiling of Hpa2, a novel mammalian heparanase family member. Biochem. Biophys. Res. Commun. 2000;276:1170–1177. doi: 10.1006/bbrc.2000.3586. [DOI] [PubMed] [Google Scholar]
- 18.Ochoa B. Can a congenital dysfunctional bladder be diagnosed from a smile? The Ochoa syndrome updated. Pediatr. Nephrol. 2004;19:6–12. doi: 10.1007/s00467-003-1291-1. [DOI] [PubMed] [Google Scholar]
- 19.Thiruchelvam N., Wu C., David A., Woolf A.S., Cuckow P.M., Fry C.H. Neurotransmission and viscoelasticity in the ovine fetal bladder after in utero bladder outflow obstruction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003;284:R1296–R1305. doi: 10.1152/ajpregu.00688.2002. [DOI] [PubMed] [Google Scholar]
- 20.Jenkins D., Bitner-Glindzicz M., Malcolm S., Allison J., Hu C.C., Winyard P.J., Gullett A.M., Thomas D.F., Belk R.A., Feather S.A. De novo Uroplakin IIIa heterozygous mutations cause human renal adysplasia leading to severe kidney failure. J. Am. Soc. Nephrol. 2005;16:2141–2149. doi: 10.1681/ASN.2004090776. [DOI] [PubMed] [Google Scholar]
- 21.Caubit X., Lye C.M., Martin E., Coré N., Long D.A., Vola C., Jenkins D., Garratt A.N., Skaer H., Woolf A.S., Fasano L. Teashirt 3 is necessary for ureteral smooth muscle differentiation downstream of SHH and BMP4. Development. 2008;135:3301–3310. doi: 10.1242/dev.022442. [DOI] [PubMed] [Google Scholar]
- 22.Burgu B., Medina Ortiz W.E., Pitera J.E., Woolf A.S., Wilcox D.T. Vascular endothelial growth factor mediates hypoxic stimulated embryonic bladder growth in organ culture. J. Urol. 2007;177:1552–1557. doi: 10.1016/j.juro.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 23.Cordell H.J., Darlay R., Charoen P., Stewart A., Gullett A.M., Lambert H.J., Malcolm S., Feather S.A., Goodship T.H., Woolf A.S., UK VUR Study Group Whole-genome linkage and association scan in primary, nonsyndromic vesicoureteric reflux. J. Am. Soc. Nephrol. 2010;21:113–123. doi: 10.1681/ASN.2009060624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.The Renal Association, UK Renal Registry, The Ninth Annual Report. Available at: http://www.renalreg.com/Reports/2006.html. Accessed March 5, 2010.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.