

Abstract
Congenital long QT syndrome (LQTS) is a hereditary disorder that leads to sudden cardiac death secondary to fatal cardiac arrhythmias. Although many genes for LQTS have been described, the etiology remains unknown in 30%–40% of cases. In the present study, a large Chinese family (four generations, 49 individuals) with autosomal-dominant LQTS was clinically evaluated. Genome-wide linkage analysis was performed by using polymorphic microsatellite markers to map the genetic locus, and positional candidate genes were screened by sequencing for mutations. The expression pattern and functional characteristics of the mutated protein were investigated by western blotting and patch-clamp electrophysiology. The genetic locus of the LQTS-associated gene was mapped to chromosome 11q23.3-24.3. A heterozygous mutation (Kir3.4-Gly387Arg) was identified in the G protein-coupled, inwardly rectifying potassium channel subunit Kir3.4, encoded by the KCNJ5 gene. The Kir3.4-Gly387Arg mutation was present in all nine affected family members and absent in 528 ethnically matched controls. Western blotting of human cardiac tissue demonstrated significant Kir3.4 expression levels in the cardiac ventricles. Heterologous expression studies with Kir3.4-Gly387Arg revealed a loss-of-function electrophysiological phenotype resulting from reduced plasma membrane expression. Our findings suggest a role for Kir3.4 in the etiology of LQTS.
Introduction
Congenital long QT syndrome (LQTS; MIM 192500) is an inherited cardiac disorder associated with a prolonged QT interval on surface electrocardiogram (ECG), recurrent syncope, and, in some cases, a predisposition to sudden death from life-threatening cardiac arrhythmias, including torsades de pointes and ventricular fibrillation. The onset of the deleterious arrhythmias is often associated with emotional or physical stress.1 To date, mutations in 12 genes,2–13 including the genes encoding ion channels and their associated proteins, have been identified in hereditary LQTS. Mutations in these genes have been identified in approximately 60%–70% of the individuals affected with LQTS.14 Despite considerable effort over the past 15 years, the genes responsible for the remaining 30%–40% of LQTS cases remain to be determined.
LQTS originates from prolongation of cardiac cell repolarization following depolarization of the cardiac ventricles. Three potassium currents (IK1, IKs, and IKr) are believed to be responsible for the majority of ventricular repolarization.15 However, recent studies have demonstrated that the G protein-coupled, inwardly rectifying potassium channel current IKACh is masked by the constitutively active potassium channel current IK1.16,17 Thus, the role of IKACh in ventricular repolarization may be of greater pathophysiological relevance than previously believed.17
IKACh is formed by a heteromultimeric or homomultimeric complex of Kir3.1 and 3.4 subunits.18–22 Kir3.4 (also called GIRK4), encoded by the KCNJ5 gene (MIM 600734), assembles into functional homomeric channels with low channel activity, but Kir3.1 must coassemble with Kir3.2, Kir3.3, or Kir3.4 to form a functional heteromeric channel.21,23–25 Interestingly, Kir3.4 knockout mice have a modest resting tachycardia, an inability to regulate heart rate in response to parasympathetic stimulation, and a longer ventricular effective refractory period than wild-type mice.26–28
In a large Chinese family with LQTS, we have identified a mutation in the potassium channel Kir3.4 that results in a defect in channel trafficking and a reduction in the IKACh current. The data suggest that the loss-of-function mutation in Kir3.4 is associated with LQTS.
Material and Methods
Identification and Phenotypic Determination of LQTS Patients
A large family with a history of LQTS was identified in the Liaoning Province of China. Written informed consent was obtained from all participants or their guardians in agreement with the standards established by local institutional review boards. Peripheral venous blood specimens were obtained and clinical data, including medical records, ECG tracings, and echocardiogram reports were collected. Control subjects consisted of 528 ethnically matched normal healthy individuals. This investigation was performed in accordance with the Helsinki Declaration and was approved by the Research Ethics Committee of Tongji University School of Medicine, Shanghai, China. The diagnosis of LQTS was made as described previously.29 The corrected QT interval (QTc) was calculated with Bazett's formula.30
Linkage Analyses
Genomic DNA was extracted with a Wizard Genomic DNA Purification Kit (Promega). Previously identified LQTS genes (KCNQ1 [MIM 607542], KCNH2 [MIM 152427], SCN5A [MIM 600163], ANK2 [MIM 106410], KCNE1 [MIM 176261], KCNE2 [MIM 603796], KCNJ2 [MIM 600681], CACNA1C [MIM 114205], CAV3 [MIM 601253], SCN4B [MIM 608256], AKAP9 [MIM 604001], and SNTA1 [MIM 601017]) were screened for mutations in the proband (II-8) and another affected family member (II-4).2–13 A genome-wide scan was performed with a set of 383 microsatellite markers (ABI Prism Linkage Mapping Sets v2.5). Additional short tandem repeat (STR) markers were used to fine map the genetic interval on chromosome 11. PCR products were pooled, and product sizes were determined with a MegaBACE 500 DNA Analysis System (Amersham Biosciences). Two-point LOD scores were calculated with the program SAGE 5.0 under an autosomal-dominant genetic model with the disease allele frequency and penetrance of LQTS set at 0.003 and 95% respectively, evenly shared allele frequency, and no sex difference.
Mutation Screening of the Positional Candidate Genes
The complete coding regions and intron/exon boundaries of the positional candidate genes were amplified with HotStarTaq DNA Polymerase (QIAGEN). Both strands of each PCR product were sequenced with a DYEnamic ET Dye Terminator Kit (Amersham Biosciences UK Limited) and the MegaBACE 500 DNA Analysis System. For the mutated Kir3.4 protein, multiple protein sequences from various species were aligned by using the software program ClustalW.
Molecular Cloning and Transfection
Complementary DNAs encoding human Kir3.1 (GenBank accession number NM_002239), human Kir3.4 (GenBank accession number NM_000890), and human type 2 muscarinic acetylcholine (ACh) receptor (M2, GenBank accession number NM_001006630) were kind gifts from D.E. Logothetis. The cDNAs were subcloned into pcDNA3, and the hKir3.4-Gly387Arg mutation was introduced via site-directed mutagenesis (Pfu Turbo DNA Polymerase, Stratagene). All constructs were verified by DNA sequencing.
Human embryonic kidney cells (HEK293) were transiently cotransfected with 0.6 μg pcDNA3-hM2, 0.6 μg pcDNA3-hKir3.1, and 0.2 μg pcDNA3-eGFP (reporter gene), plus 0.6 μg wild-type pcDNA3-hKir3.4, 0.6 μg pcDNA3-hKir3.4-Gly387Arg, or 0.3 μg wild-type pcDNA3-hKir3.4 and 0.3 μg pcDNA3-hKir3.4-Gly387Arg. Transfections were performed with Lipofectamine and Plus reagent (Invitrogen) according to the manufacturer's instructions.
Electrophysiology
Patch-clamp experiments were performed 2 to 3 days after transfection. Whole-cell currents were measured in symmetrical Ringer solution at room temperature. The internal pipette solution consisted of (in mM) KCl 141, EGTA 10, CaCl2 5.2, MgCl2 1.42, and HEPES 10 (pH 7.4 with KOH); the external solution consisted of (in mM) KCl 140, CaCl2 2.6, MgCl2 1.2, and HEPES 5 (pH 7.4 with KOH). 10 μM ACh was freshly prepared prior to each experiment and added to the external solution. Cells were constantly perfused with 10 μM Ach. Measurements were made with PULSE software (HEKA Elektronik) and an EPC-9 amplifier sampling at 2.5 kHz and filtering at 1.25 kHz (HEKA Elektronik). Borosilicate glass pipettes were pulled on a DPZ-Universal puller (Zeit Instrumente, Munich). The pipettes had a resistance of 1.5–2.5 MΩ when filled with intracellular solution. The series resistance recorded in the whole-cell configuration was 2–4 MΩ and was compensated (80%). The seal resistance in all experiments was greater than 1.0 GΩ.
Protein Extraction and Western Blotting
Right atrium lateral wall (RA) and left ventricular endocardial free wall (LV) specimens (n = 4) were obtained from patients undergoing valve replacement surgery at Rigshospitalet, University of Copenhagen, Denmark. Total membrane proteins were purified via a method modified from Han et al.31 Because surface membrane-associated proteins were primarily harvested by this extraction method, we ascertained that neither cytochrome c nor β-tubulin or actin were suitable as loading controls. Great care was taken to load exactly the same amount of total membrane protein onto gels, and western blotting experiments were repeated several times. The cell surface biotinylation procedure was performed as previously described.32
Total lysate (20 μg per lane human tissue extract) and HEK293 biotinylated and unbound fractions (15 μg per sample in the biotinylation procedure) were separated on 4%–15% SDS-PAGE via a minigel system (Bio-Rad). Proteins were transferred onto Hybond-P polyvinylidene difluoride transfer membranes (Amersham Biosciences, 0.45 μm), and membranes were blocked in 5% nonfat milk in TBST (0.1% Tween 20 in Tris-buffered saline solution) for 2 hr at room temperature and then incubated with primary antibody against Kir3.1 (1:1,000; Alomone Labs), Kir3.4 (1:1,000; Alomone Labs), calnexin (1:2,000; Nordic Biosite), or Na+/K+-ATPase α1 (1:500; Santa Cruz Biotechnology) overnight. Proteins were detected with HRP-conjugated donkey anti-mouse and anti-rabbit secondary antibodies (1:10,000; Jackson ImmunoResearch Laboratories). Enhanced chemiluminescence (ECL) incubation was performed via a standard procedure (SuperSignal West Pico Chemiluminescent detection system, Pierce), and immunoblots were exposed on Hyperfilm ECL (Amersham Biosciences). Band density was quantified with Quantity One software as a Gaussian trace quantity on the exposed films; quantified band densities were processed equally.
Statistical Analyses
All data are presented as mean ± standard error of the mean. For statistical analyses, paired and unpaired t tests and two-way analysis of variance combined with a Bonferroni post hoc test were used. p < 0.05 was considered statistically significant.
Results
Clinical Characteristics
The proband is a 64-year-old woman (II-8, Figure 1) referred for a cardiology consultation following recurrent syncopal episodes that began when she was 22 years old. Since that time, she had experienced 32 syncopal episodes, the majority of which were preceded by emotional or physical stress. She therefore underwent a comprehensive clinical evaluation including echocardiography and ECG recordings. The echocardiogram did not uncover any structural or functional cardiac abnormalities. However, the ECG revealed a markedly prolonged QT interval (QTc = 520 ms). A representative 12-lead ECG is shown in Figure 2A. She was treated with metoprolol 50–75 mg PO twice daily. Subsequent evaluation of 49 family members identified nine patients with LQTS characterized by similar clinical features. The participant pedigree is shown in Figure 1, and a summary of clinical characteristics of the affected members is listed in Table 1. Transmission of LQTS in this family occurred in an autosomal-dominant pattern. Interestingly, the proband also developed persistent atrial fibrillation (AF; MIM 608583) (Figure 2B). Permanent AF was also observed in affected subjects I-2 and II-4; subject I-2's elder sister was also diagnosed with LQTS and permanent AF (data not shown). Subject II-13, from whom blood samples were not obtained, suffered from a series of seizures and syncopal episodes and died suddenly 7 days after birth.
Figure 1.
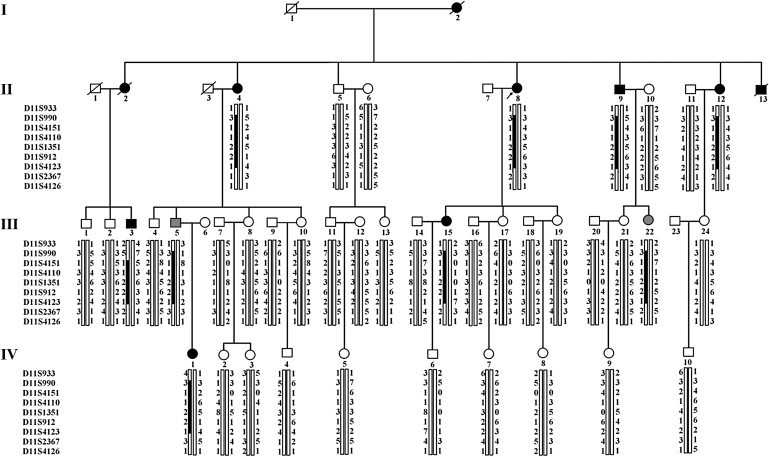
Pedigree and Mapping Analysis of a Family with Congenital Long QT Syndrome
Black squares and circles represent male and female affected subjects, respectively; white squares and circles represent unaffected subjects; gray symbols represent subjects with clinical status undetermined. Slashes through symbols indicate deceased individuals; the arrow denotes the proband. A vertical bar beneath a family member indicates the presence of the chromosomal segment as determined by genetic evaluation. Kir3.4 is located at 11q24.3, and the markers tested in this region of chromosome 11 are listed to the left of the pedigree, with each individual's allele (represented by numbers) for each marker shown next to the chromosome bar. The chromosomal segment originating from the mutation-carrying chromosome of the affected individual in the first generation (I-2) is represented by the black bar.
Figure 2.
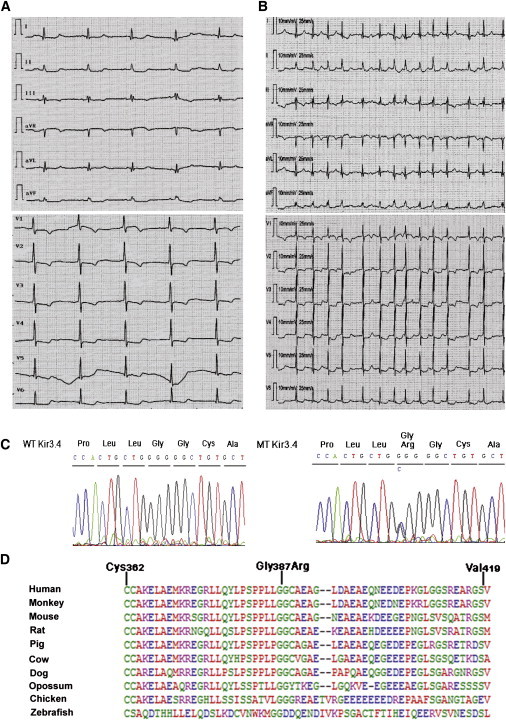
The Kir3.4-Gly387Arg Mutation Is Associated with Congenital Long QT Syndrome
(A and B) Representative 12-lead surface electrocardiograms from the proband, demonstrating a prolonged QT interval (A) and atrial fibrillation (B).
(C) Sequencing of genomic DNA from the proband revealed a heterozygous mutation in Kir3.4. The G-to-C substitution results in the introduction of an arginine (R) instead of the normal glycine (G) at codon 387 (Gly387Arg).
(D) Alignment of Kir3.4 protein sequences across species demonstrating that Gly387 is conserved throughout evolution.
Table 1.
Clinical Characteristics of the Pedigree Members with Kir3.4-Gly387Arg or Sudden Death
|
Subject Information |
Symptom |
Electrocardiogram |
Echocardiogram |
|||||
|---|---|---|---|---|---|---|---|---|
| Identity | Gender | Age (Years) | Recurrent Syncope | Recurrent Palpitation | QTc (ms) | Other Findings | LVEDD (mm) | LVEF (%) |
| I-2 | F | 72a | + | + | NA | Permanent AF | NA | NA |
| II-2 | F | 67a | + | + | 490 | AT, TdP | NA | NA |
| II-4 | F | 66 | + | + | 560 | Permanent AF | 60 | 50 |
| II-8 | F | 64 | + | + | 520 | Persistent AF | 50 | 54 |
| II-9 | M | 60 | + | + | 450 | AT | 40 | 57 |
| II-12 | F | 48 | + | + | 470 | − | 49 | 64 |
| II-13 | M | 7 daysa | + | NA | NA | NA | NA | NA |
| III-3 | M | 48 | + | + | 450 | − | 49 | 71 |
| III-5 | M | 40 | − | + | 430 | − | 52 | 64 |
| III-15 | F | 38 | + | + | 470 | − | 40 | 61 |
| III-22 | F | 35 | − | + | 460 | − | 39 | 65 |
| IV-1 | F | 13 | + | + | 460 | − | 36 | 72 |
The following abbreviations are used: QTc, QT interval corrected for heart rate; LVEDD, left ventricular end-diastolic diameter; LVEF, left ventricular ejection fraction; +, present; −, absent; NA, not available or not applicable; AF, atrial fibrillation; AT, atrial tachycardia; TdP, torsades de pointes.
Age at death.
Chromosomal Location
Sequencing of 12 known LQTS genes did not reveal any mutational change in two affected family members, excluding the possibility of their causative effect. Thus, we assumed that the observed LQTS syndrome in the current pedigree could be caused by an unknown gene that was expected to be identified by performing a genome-wide linkage analyses. Initial evidence of linkage was obtained with marker D11S4151 on chromosome 11q23 with a two-point LOD score of 1.74 (at recombination fraction [θ] = 0.00). Fine mapping of the region resulted in positive two-point LOD scores for sequential markers from D11S4110 to D11S4123 with a peak LOD score of 5.12 (at θ = 0.00) at D11S4123 on chromosome 11q24.3 (Table 2). Haplotype analyses indicated a shared region (11q23.3-24.3) among affected family members flanked by markers D11S4151 and D11S4123, corresponding to a genetic distance of approximately 4.78 cM.
Table 2.
Two-Point LOD Scores
| Marker |
Recombination Fraction |
||||||
|---|---|---|---|---|---|---|---|
| 0.00 | 0.01 | 0.05 | 0.10 | 0.20 | 0.30 | 0.40 | |
| D11S933 | −1.08 | −0.91 | −0.06 | 0.31 | 0.48 | 0.39 | 0.20 |
| D11S990 | (−) | 0.57 | 1.07 | 1.14 | 0.94 | 0.58 | 0.20 |
| D11S4151 | 1.74 | 1.70 | 1.56 | 1.37 | 0.98 | 0.56 | 0.19 |
| D11S4110 | 2.05 | 2.01 | 1.84 | 1.63 | 1.18 | 0.71 | 0.25 |
| D11S1351 | 3.74 | 3.67 | 3.38 | 3.02 | 2.24 | 1.40 | 0.55 |
| D11S912 | 4.34 | 4.26 | 3.94 | 3.52 | 2.64 | 1.69 | 0.67 |
| D11S4123 | 5.12 | 5.03 | 4.67 | 4.20 | 3.18 | 2.04 | 0.81 |
| D11S2367 | (−) | −2.95 | −0.44 | 0.39 | 0.78 | 0.60 | 0.23 |
| D11S4126 | −0.05 | −0.04 | 0.00 | 0.02 | 0.03 | 0.02 | 0.01 |
Identification of a Mutation in Kir3.4
A bioinformatic analysis revealed 15 defined genes in the above chromosomal region. Among these genes, two ion channel genes were considered strong candidate genes for LQTS: KCNJ1 (MIM 600359), encoding Kir1.1, and KCNJ5, encoding Kir3.4. Upon sequencing of each candidate, a heterozygous mutation was identified in Kir3.4. A cytosine-to-guanine substitution at nucleotide 1473 (c.1473C>G) was identified in KCNJ5 that resulted in a change in the Kir3.4 amino acid residue 387 from glycine to arginine (Gly387Arg) (Figure 2C). A cross-species alignment of the Kir3.4 protein sequence demonstrated that Gly387Arg is highly conserved across species (Figure 2D). The Gly387Arg mutation was present in all nine affected family members and absent in 528 ethnically matched controls. Interestingly, LQTS is incompletely penetrant in subject III-5 and III-22, pointing toward the need for long-term monitoring of asymptomatic subjects carrying the Gly387 allele in order to confirm its clinical significance.
Electrophysiological Analysis of Mutated Kir3.4
The functional effects of the Kir3.4-Gly387Arg mutation were examined by patch-clamp analyses on HEK293 cells cotransfected with Kir3.1 plus Kir3.4, Kir3.4 and Kir3.4-Gly387Arg, or Kir3.4-Gly387Arg. Coexpression of Kir3.1/Kir3.4-Gly387Arg or Kir3.1/Kir3.4/Kir3.4-Gly387Arg in HEK293 cells resulted in a drastic reduction of inward currents compared to wild-type Kir3.1/Kir3.4 (Figure 3). The fact that heterozygous expression of wild-type and mutant Kir3.4 gives low current amplitude indicates that Kir3.4-Gly387Arg has a dominant-negative effect.
Figure 3.
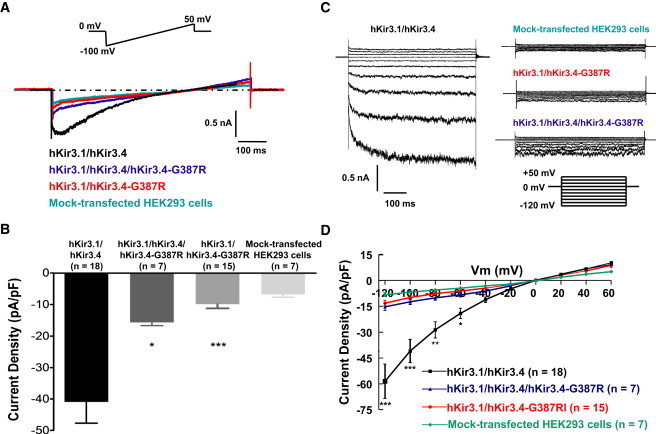
Electrophysiological Analyses of Kir3.4-Gly387Arg
Whole-cell patch clamping of HEK293 cells transiently transfected with hKir3.1/hKir3.4, hKir3.1/hKir3.4/hKir3.4-Gly387Arg, or hKir3.1/hKir3.4-Gly387Arg. Note: G387R in this figure is equivalent to Gly387Arg.
(A) Representative traces. Current was recorded in response to a 1000 ms voltage ramp from −100 to +50 mV from a holding potential of 0 mV (inset at top). Symmetrical potassium ion solutions with 10 μM acetylcholine (ACh) were used.
(B) Normalized average ± standard error of the mean (SEM) current density values measured at −100 mV (current was elicited by the ramp protocol described above). hKir3.1/hKir3.4: −40.96 ± 6.75 pA/pF; hKir3.1/hKir3.4/hKir3.4-Gly387Arg: −15.69 ± 0.95 pA/pF; hKir3.1/hKir3.4-Gly387Arg: −9.92 ± 1.32 pA/pF; mock-transfected cells: −6.83 ± 0.86 pA/pF. ∗p < 0.05, ∗∗∗p < 0.001 versus hKir3.1/hKir3.4.
(C) Representative traces. Current was recorded during 1000 ms voltage-clamp pulses from −120 to +60 mV in 20 mV steps from a holding potential of 0 mV (inset at lower right).
(D) Current-voltage relationships of hKir3.1/hKir3.4, hKir3.1/hKir3.4/hKir3.4-Gly387Arg, and hKir3.1/hKir3.4-Gly387Arg with that of mock-transfected HEK cells as control. To measure current-voltage relationships, current was elicited by the step protocol described above, and current amplitudes were measured at the end of each voltage step. Kir3.1/3.4 was significantly different from Kir3.1/Kir3.4/Kir3.4-Gly387Arg or Kir3.1/Kir3.4-Gly387Arg by two-way analysis of variance (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus hKir3.1/hKir3.4). Kir3.1/Kir3.4/Kir3.4-Gly387Arg was not significantly different from Kir3.1/Kir3.4-Gly387Arg. At −60 mV, the average current was −19.13 ± 2.96 pA/pF for Kir3.1/Kir3.4, −8.39 ± 1.14 pA/pF for Kir3.1/Kir3.4/Kir3.4-Gly387Arg, and −6.22 ± 0.84 pA/pF for Kir3.1/Kir3.4-Gly387Arg.
Kir3.1 and Kir3.4 Protein Trafficking
Because the Gly387Arg mutation resides in a region of Kir3.4 previously described to be involved in the surface targeting of the heteromeric Kir3.1/Kir3.4 complex,33 we decided to examine whether a reduced surface expression of the two subunits could explain the Kir3.4-Gly387Arg-induced current reduction. To this end, cell surface and intracellular Kir3.4 and Kir3.1 were analyzed via western blotting of cells cotransfected with Kir3.4-Gly387Arg/Kir3.1 or wild-type Kir3.4/Kir3.1 (Figure 4). In Kir3.4-Gly387Arg/Kir3.1-cotransfected cells, the plasma membrane and intracellular expression levels of Kir3.4 and Kir3.1 were markedly reduced compared to that of control (wild-type Kir3.4/Kir3.1) (Figure 4). Cotransfection of wild-type Kir3.4 together with Kir3.4-Gly387Arg and Kir3.1 completely recovered the intracellular Kir3.4 and Kir3.1 levels but failed to recover the expression of both Kir3.4 and Kir3.1 on the cell surface (Figure 4).
Figure 4.
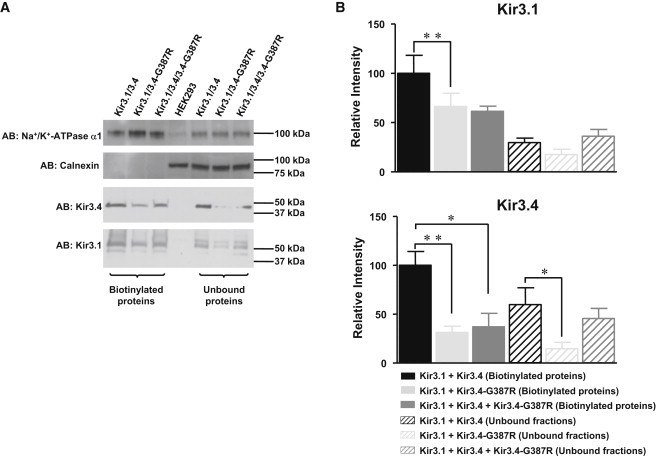
Cell Surface Expression of Kir3.4-Gly387Arg and Kir3.1
Western blotting of HEK293 cells transiently cotransfected with hKir3.1 and hKir3.4 or hKir3.4-Gly387Arg. Note: G387R in this figure is equivalent to Gly387Arg.
(A) Representative exposures of biotinylated proteins and unbound protein fractions with the indicated antibodies (AB). For mock-transfected HEK293 cells, whole-cell lysate was loaded. Na+/K+-ATPase α1 served as a loading control to make sure that equal amounts of cell lysates were loaded onto the gel. Calnexin, which is an endoplasmic-reticulum-resident protein, served as control for selective biotinylation of plasma membrane proteins.
(B) Average ± SEM values from quantification of six independent experiments. ∗p < 0.05, ∗∗p < 0.001.
Human Cardiac Kir3.1 and Kir3.4 Protein Expression
Atrial and endocardial left ventricular tissue samples were obtained from four patients undergoing cardiac valve surgery. Protein was extracted from these samples, and western blotting was performed (Figure 5). Both Kir3.1 and Kir3.4 were demonstrated to be expressed in the atria as well as the ventricles.
Figure 5.
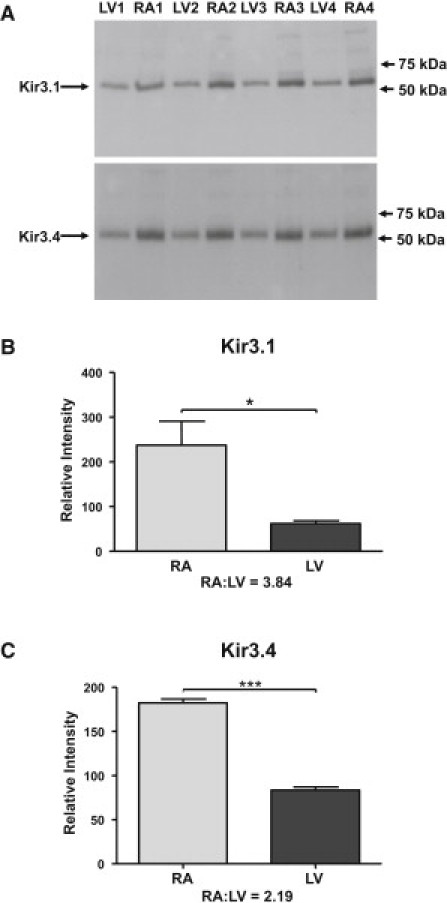
Human Atrial and Ventricular Expression of Kir3.1 and Kir3.4
(A) Western blotting of proteins extracted from tissue from the left ventricular endocardial free wall (LV) and the lateral wall of the right atrium (RA) from four patients (e.g., patient 1: LV1 and RA1).
(B and C) Quantification of expression levels of Kir3.1 (B) and Kir3.4 (C) in the atria and ventricles. Average ± SEM arbitrary quantification values for atrial versus ventricular expression were as follows: Kir3.1, 237 ± 54 versus 62 ± 7; Kir3.4, 182 ± 5 versus 82 ± 4. ∗p < 0.05, ∗∗∗p < 0.01.
Discussion
We have identified KCNJ5, encoding Kir3.4, as a causative gene in LQTS. In a large Chinese family with LQTS, we detected a mutation, Gly387Arg, in a highly conserved residue in Kir3.4. The Kir3.4-Gly387Arg mutation resulted in a loss of IKACh channel function by disrupting membrane targeting and stability of Kir3.4.
In the heart, the functional IKACh channels are formed largely by heteromeric Kir3.4 and Kir3.1 subunits.26 In this study, we determined that, when cotransfected in HEK293 cells, the expression of Kir3.4-Gly387Arg led to reduced levels of Kir3.4 and Kir3.1 in both the plasma membrane and cytoplasmic fractions, indicating that this mutation in Kir3.4 leads to defects in Kir3.4 and Kir3.1 membrane targeting and stability. Interestingly, the coexpression of wild-type Kir3.4, Kir3.4-Gly387Arg, and Kir3.1, a situation mimicking the in vivo heterozygous expression of Kir3.4 in patients with LQTS as described in this study, rescued the cytoplasmic expression levels of Kir3.4 and Kir3.1 but failed to rescue defects in membrane targeting of these proteins. This suggests that the Kir3.4-Gly387Arg mutation interferes with the formation of functional channels containing Kir3.4. As a consequence, Gly387Arg mutant channels exhibited a near complete loss of channel activity and failed to respond to ACh stimulation.
IKACh is predominantly expressed in the sinoatrial node, atria, and atrioventricular node.34 Biochemical and genetic studies have demonstrated that IKACh plays an important role in both parasympathetic slowing of the heart rate and repolarization of atrial action potentials.34 The mechanism of function of Kir3.4 in the context of Kir3.1 is also well established in the atria. Parasympathetic release of ACh leads to activation of the muscarinic M2 receptor, which in turn activates Kir3.1/Kir3.4 channels via G protein βγ subunits.33 However, the role of Kir3.1/Kir3.4 channels in ventricular function is less clear. IKACh has been recorded in the ventricular myocytes of multiple species including human,35 ferret,36 and dog.37 Other studies have shown that adenosine or sphingosine-1-phosphate receptor stimulation may activate an IKACh-like current in ferret and mouse ventricular myocytes,38,39 suggesting that the activation of IKACh in ventricles may be distinct from the predominantly muscarinic activation in atria. The relatively uncharacterized role of Kir3.1/Kir3.4 channels in the ventricles could also be due to their resemblance to IK1.16,17
Genetic studies have demonstrated that Kir3.1 or Kir3.4 knockout mice exhibit a modest resting tachycardia and inability to regulate heart rate changes secondary to parasympathetic stimulation.26–28 The loss-of-function phenotype in mice is strikingly different from that seen in the human patients described in this study. The reasons behind this apparent discrepancy remain unclear, but several possible mechanisms can be hypothesized. First, the differences observed between mice and humans may simply reflect differences between species, inasmuch as other Kir3 family members in mice may compensate for the loss of Kir3.4 in the ventricle. Second, the phenotype of the Gly387Arg mutation observed in humans may be attributable to a loss of function of both Kir3.4 and Kir3.1, because the mutant protein appears to affect targeting and stability of both Kir3.4 and Kir3.1. Interestingly, normal Kir3.1 membrane location is impaired in the myocytes of Kir3.4 knockout mice, which supports our finding of a Kir3.4-Gly387Arg mutation-induced Kir3.1 membrane targeting defect.28 Nevertheless, our analyses have revealed a physiological and pathophysiological function for the Kir3.4 protein in human ventricular myocytes.
Although AF is significantly more common in patients with LQTS compared to the general population,40 little is known about the molecular mechanisms underlying LQTS-coexistent arrhythmias. In the present study, three of the affected individuals presented with permanent or persistent AF, and an additional two affected individuals experienced atrial tachycardia (Table 1), suggesting that Kir3.4-Gly387Arg may affect both the atria and the ventricles. In fact, upregulation of IKACh has been reported in AF and has long been speculated to play a role in the incidence and persistence of AF.41 It has also become increasingly clear that heterogeneity in repolarization may promote arrhythmias such as AF.42,43 Thus, we hypothesize that the Gly387Arg mutation in Kir3.4 may create sufficient atrial repolarization heterogeneity to contribute to AF or atrial tachycardia in affected patients.
In conclusion, a mutation in Kir3.4 exerts dominant-negative effects on Kir3.1/Kir3.4 channel complexes, linking it to congenital LQTS. This suggests a potential role for these channels in ventricular repolarization.
Acknowledgments
We thank Yunfu Sun, Emelia J. Benjamin, and Kui Hong for their helpful comments. This work was supported by the “973” Program Fund of China (2007CB512100), the “863” Program Fund of China (2007AA02Z438), the Program Fund for Shanghai Subject Chief Scientists, the Program Fund for Innovative Research Teams by the Ministry of Education of China, the National Science Fund of China (30425016 and 30330290 to Y.-H.C.), the Shanghai Pujiang Program Fund (07PJ14058 to L.P.), the Danish National Research Foundation (to S.-P.O.), the Danish National Research Council (to B.L. and M.G.), the Lundbeck Foundation (to T.J.), and the National Institutes of Health (HL092577 and DA027021 to P.T.E.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GenBank, http://www.ncbi.nlm.nih.gov/genbank/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Keating M.T., Sanguinetti M.C. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569–580. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- 2.Wang Q., Curran M.E., Splawski I., Burn T.C., Millholland J.M., VanRaay T.J., Shen J., Timothy K.W., Vincent G.M., de Jager T. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 3.Curran M.E., Splawski I., Timothy K.W., Vincent G.M., Green E.D., Keating M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 4.Wang Q., Shen J., Splawski I., Atkinson D., Li Z., Robinson J.L., Moss A.J., Towbin J.A., Keating M.T. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 5.Mohler P.J., Schott J.J., Gramolini A.O., Dilly K.W., Guatimosim S., duBell W.H., Song L.S., Haurogné K., Kyndt F., Ali M.E. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 6.Splawski I., Tristani-Firouzi M., Lehmann M.H., Sanguinetti M.C., Keating M.T. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat. Genet. 1997;17:338–340. doi: 10.1038/ng1197-338. [DOI] [PubMed] [Google Scholar]
- 7.Abbott G.W., Sesti F., Splawski I., Buck M.E., Lehmann M.H., Timothy K.W., Keating M.T., Goldstein S.A. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- 8.Plaster N.M., Tawil R., Tristani-Firouzi M., Canún S., Bendahhou S., Tsunoda A., Donaldson M.R., Iannaccone S.T., Brunt E., Barohn R. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell. 2001;105:511–519. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 9.Splawski I., Timothy K.W., Sharpe L.M., Decher N., Kumar P., Bloise R., Napolitano C., Schwartz P.J., Joseph R.M., Condouris K. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 10.Vatta M., Ackerman M.J., Ye B., Makielski J.C., Ughanze E.E., Taylor E.W., Tester D.J., Balijepalli R.C., Foell J.D., Li Z. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–2112. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- 11.Medeiros-Domingo A., Kaku T., Tester D.J., Iturralde-Torres P., Itty A., Ye B., Valdivia C., Ueda K., Canizales-Quinteros S., Tusié-Luna M.T. SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation. 2007;116:134–142. doi: 10.1161/CIRCULATIONAHA.106.659086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L., Marquardt M.L., Tester D.J., Sampson K.J., Ackerman M.J., Kass R.S. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc. Natl. Acad. Sci. USA. 2007;104:20990–20995. doi: 10.1073/pnas.0710527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ueda K., Valdivia C., Medeiros-Domingo A., Tester D.J., Vatta M., Farrugia G., Ackerman M.J., Makielski J.C. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc. Natl. Acad. Sci. USA. 2008;105:9355–9360. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zareba W., Cygankiewicz I. Long QT syndrome and short QT syndrome. Prog. Cardiovasc. Dis. 2008;51:264–278. doi: 10.1016/j.pcad.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 15.Nerbonne J.M., Kass R.S. Molecular physiology of cardiac repolarization. Physiol. Rev. 2005;85:1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 16.Wickman K., Clapham D.E. Ion channel regulation by G proteins. Physiol. Rev. 1995;75:865–885. doi: 10.1152/physrev.1995.75.4.865. [DOI] [PubMed] [Google Scholar]
- 17.Beckmann C., Rinne A., Littwitz C., Mintert E., Bosche L.I., Kienitz M.C., Pott L., Bender K. G protein-activated (GIRK) current in rat ventricular myocytes is masked by constitutive inward rectifier current (I(K1)) Cell. Physiol. Biochem. 2008;21:259–268. doi: 10.1159/000129381. [DOI] [PubMed] [Google Scholar]
- 18.Mark M.D., Herlitze S. G-protein mediated gating of inward-rectifier K+ channels. Eur. J. Biochem. 2000;267:5830–5836. doi: 10.1046/j.1432-1327.2000.01670.x. [DOI] [PubMed] [Google Scholar]
- 19.Wickman K.D., Iñiguez-Lluhl J.A., Davenport P.A., Taussig R., Krapivinsky G.B., Linder M.E., Gilman A.G., Clapham D.E. Recombinant G-protein beta gamma-subunits activate the muscarinic-gated atrial potassium channel. Nature. 1994;368:255–257. doi: 10.1038/368255a0. [DOI] [PubMed] [Google Scholar]
- 20.Dascal N., Schreibmayer W., Lim N.F., Wang W., Chavkin C., DiMagno L., Labarca C., Kieffer B.L., Gaveriaux-Ruff C., Trollinger D. Atrial G protein-activated K+ channel: Expression cloning and molecular properties. Proc. Natl. Acad. Sci. USA. 1993;90:10235–10239. doi: 10.1073/pnas.90.21.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krapivinsky G., Gordon E.A., Wickman K., Velimirović B., Krapivinsky L., Clapham D.E. The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K(+)-channel proteins. Nature. 1995;374:135–141. doi: 10.1038/374135a0. [DOI] [PubMed] [Google Scholar]
- 22.Kubo Y., Reuveny E., Slesinger P.A., Jan Y.N., Jan L.Y. Primary structure and functional expression of a rat G-protein-coupled muscarinic potassium channel. Nature. 1993;364:802–806. doi: 10.1038/364802a0. [DOI] [PubMed] [Google Scholar]
- 23.Bender K., Wellner-Kienitz M.C., Inanobe A., Meyer T., Kurachi Y., Pott L. Overexpression of monomeric and multimeric GIRK4 subunits in rat atrial myocytes removes fast desensitization and reduces inward rectification of muscarinic K(+) current (I(K(ACh))). Evidence for functional homomeric GIRK4 channels. J. Biol. Chem. 2001;276:28873–28880. doi: 10.1074/jbc.M102328200. [DOI] [PubMed] [Google Scholar]
- 24.Hedin K.E., Lim N.F., Clapham D.E. Cloning of a Xenopus laevis inwardly rectifying K+ channel subunit that permits GIRK1 expression of IKACh currents in oocytes. Neuron. 1996;16:423–429. doi: 10.1016/s0896-6273(00)80060-4. [DOI] [PubMed] [Google Scholar]
- 25.Krapivinsky G., Krapivinsky L., Velimirovic B., Wickman K., Navarro B., Clapham D.E. The cardiac inward rectifier K+ channel subunit, CIR, does not comprise the ATP-sensitive K+ channel, IKATP. J. Biol. Chem. 1995;270:28777–28779. doi: 10.1074/jbc.270.48.28777. [DOI] [PubMed] [Google Scholar]
- 26.Corey S., Clapham D.E. Identification of native atrial G-protein-regulated inwardly rectifying K+ (GIRK4) channel homomultimers. J. Biol. Chem. 1998;273:27499–27504. doi: 10.1074/jbc.273.42.27499. [DOI] [PubMed] [Google Scholar]
- 27.Kovoor P., Wickman K., Maguire C.T., Pu W., Gehrmann J., Berul C.I., Clapham D.E. Evaluation of the role of I(KACh) in atrial fibrillation using a mouse knockout model. J. Am. Coll. Cardiol. 2001;37:2136–2143. doi: 10.1016/s0735-1097(01)01304-3. [DOI] [PubMed] [Google Scholar]
- 28.Wickman K., Nemec J., Gendler S.J., Clapham D.E. Abnormal heart rate regulation in GIRK4 knockout mice. Neuron. 1998;20:103–114. doi: 10.1016/s0896-6273(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 29.Keating M., Atkinson D., Dunn C., Timothy K., Vincent G.M., Leppert M. Linkage of a cardiac arrhythmia, the long QT syndrome, and the Harvey ras-1 gene. Science. 1991;252:704–706. doi: 10.1126/science.1673802. [DOI] [PubMed] [Google Scholar]
- 30.Bazett H.C. An analysis of the time relations of electrocardiograms. Heart. 1920;7:353–370. [Google Scholar]
- 31.Han W., Bao W., Wang Z., Nattel S. Comparison of ion-channel subunit expression in canine cardiac Purkinje fibers and ventricular muscle. Circ. Res. 2002;91:790–797. doi: 10.1161/01.res.0000039534.18114.d9. [DOI] [PubMed] [Google Scholar]
- 32.Jespersen T., Rasmussen H.B., Grunnet M., Jensen H.S., Angelo K., Dupuis D.S., Vogel L.K., Jorgensen N.K., Klaerke D.A., Olesen S.P. Basolateral localisation of KCNQ1 potassium channels in MDCK cells: Molecular identification of an N-terminal targeting motif. J. Cell Sci. 2004;117:4517–4526. doi: 10.1242/jcs.01318. [DOI] [PubMed] [Google Scholar]
- 33.Kennedy M.E., Nemec J., Corey S., Wickman K., Clapham D.E. GIRK4 confers appropriate processing and cell surface localization to G-protein-gated potassium channels. J. Biol. Chem. 1999;274:2571–2582. doi: 10.1074/jbc.274.4.2571. [DOI] [PubMed] [Google Scholar]
- 34.Kurachi Y., Tung R.T., Ito H., Nakajima T. G protein activation of cardiac muscarinic K+ channels. Prog. Neurobiol. 1992;39:229–246. doi: 10.1016/0301-0082(92)90017-9. [DOI] [PubMed] [Google Scholar]
- 35.Koumi S., Sato R., Nagasawa K., Hayakawa H. Activation of inwardly rectifying potassium channels by muscarinic receptor-linked G protein in isolated human ventricular myocytes. J. Membr. Biol. 1997;157:71–81. doi: 10.1007/s002329900217. [DOI] [PubMed] [Google Scholar]
- 36.Ito H., Hosoya Y., Inanobe A., Tomoike H., Endoh M. Acetylcholine and adenosine activate the G protein-gated muscarinic K+ channel in ferret ventricular myocytes. Naunyn Schmiedebergs Arch. Pharmacol. 1995;351:610–617. doi: 10.1007/BF00170160. [DOI] [PubMed] [Google Scholar]
- 37.Yang Z.K., Boyett M.R., Janvier N.C., McMorn S.O., Shui Z., Karim F. Regional differences in the negative inotropic effect of acetylcholine within the canine ventricle. J. Physiol. 1996;492:789–806. doi: 10.1113/jphysiol.1996.sp021346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Landeen L.K., Dederko D.A., Kondo C.S., Hu B.S., Aroonsakool N., Haga J.H., Giles W.R. Mechanisms of the negative inotropic effects of sphingosine-1-phosphate on adult mouse ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2008;294:H736–H749. doi: 10.1152/ajpheart.00316.2007. [DOI] [PubMed] [Google Scholar]
- 39.Dobrzynski H., Janvier N.C., Leach R., Findlay J.B., Boyett M.R. Effects of ACh and adenosine mediated by Kir3.1 and Kir3.4 on ferret ventricular cells. Am. J. Physiol. Heart Circ. Physiol. 2002;283:H615–H630. doi: 10.1152/ajpheart.00130.2002. [DOI] [PubMed] [Google Scholar]
- 40.Johnson J.N., Tester D.J., Perry J., Salisbury B.A., Reed C.R., Ackerman M.J. Prevalence of early-onset atrial fibrillation in congenital long QT syndrome. Heart Rhythm. 2008;5:704–709. doi: 10.1016/j.hrthm.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ehrlich J.R. Inward rectifier potassium currents as a target for atrial fibrillation therapy. J. Cardiovasc. Pharmacol. 2008;52:129–135. doi: 10.1097/FJC.0b013e31816c4325. [DOI] [PubMed] [Google Scholar]
- 42.Fatkin D., Otway R., Vandenberg J.I. Genes and atrial fibrillation: A new look at an old problem. Circulation. 2007;116:782–792. doi: 10.1161/CIRCULATIONAHA.106.688889. [DOI] [PubMed] [Google Scholar]
- 43.Arora R., Verheule S., Scott L., Navarrete A., Katari V., Wilson E., Vaz D., Olgin J.E. Arrhythmogenic substrate of the pulmonary veins assessed by high-resolution optical mapping. Circulation. 2003;107:1816–1821. doi: 10.1161/01.CIR.0000058461.86339.7E. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.