

Abstract
To implement Quality by Design (QbD) in drug development, scientists need tools that link drug products properties to in vivo performance. Physiologically based absorption models are potentially useful tools; yet, their utility of QbD implementation has not been discussed or explored much in the literature. We simulated pharmacokinetics (PK) of carbamazepine (CBZ) after administration of four oral formulations, immediate-release (IR) suspension, IR tablet, extended-release (XR) tablet and capsule, under fasted and fed conditions and presented a general diagram of a modeling and simulation strategy integrated with pharmaceutical development. We obtained PK parameters and absorption scale factors (ASFs) by deconvolution of the PK data for IR suspension under fasted condition. The model was validated for other PK profiles of IR formulations and used to predict PK for XR formulations. We explored three key areas where a modeling and simulation approach impacts QbD. First, the model was used to help identify optimal in vitro dissolution conditions for XR formulations. Second, identification of critical formulations variables was illustrated by a parameter sensitivity analysis of mean particle radius for the IR tablet that showed a PK shift with decreased particle radius, Cmax was increased and Tmax was decreased. Finally, virtual trial simulations allowed incorporation of inter-subject variability in the model. Virtual bioequivalence studies performed for two test formulations suggested that an in vitro dissolution test may be a more sensitive discriminative method than in vivo PK studies. In summary, a well-validated predictive model is a potentially useful tool for QbD implementation in drug development.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-010-9250-9) contains supplementary material, which is available to authorized users.
KEY WORDS: advanced compartmental absorption and transit (ACAT) model, gastroplus™, modified release (MR), quality by design (QbD)
INTRODUCTION
Quality by Design (QbD) is a systemic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management to meet patient needs through clinical and safety targets in the Quality Target Product Profile (QTPP) (1,2). Implementing QbD in drug development will benefit the pharmaceutical industry, the Agency, and most importantly, the target patient population. Modeling and simulation can play an important role in every stage of QbD based drug development. A variety of useful models could be developed including mechanistic models such as physiologically based absorption modeling; empirical models such as conventional in vivo–in vitro correlation (IVIVC) modeling, and statistically designed experiments; and semi-empirical models such as scale-up correlations.
The role of predictive biopharmaceutic methods in QbD was emphasized in a recent biopharmaceutics and QbD conference cosponsored by the FDA and University of Wisconsin School of Pharmacy Extension Services (3). One of the main challenges of QbD is translating in vitro observations to in vivo performance. Understanding the mechanisms of in vitro release as well as physiology in relation to drug absorption, and in silico models that mimic in vivo release characteristics have been emphasized as potential tools that will facilitate implementation of QbD (3).
Physiologically based pharmacokinetic (PBPK) models are mathematical models that integrate anatomical and physiological parameters of animals or humans, physicochemical properties of drug substances, and formulation properties of drug products to predict absorption, distribution, metabolism and excretion (ADME) of compounds in vivo (4). They give quantitative predictions by integrating prior knowledge in a mechanistic network. Physiologically based absorption models emphasize understanding and predicting the drug fate in the gastrointestinal (GI) tract, such as dissolution, precipitation, and metabolism. The absorption model connected to a distribution model predicts the in vivo PK profiles. Many research efforts have been allocated to develop such predictive absorption models in various species (5–11), and many review articles have descriptions about the model development (12–17). The mechanistic and predictive properties enable such mechanism-based models to be tools for biopharmaceutics QbD implementation although this aspect has not been discussed and explored extensively and been underused. Recently physiologically based absorption modeling approach has gained interests from pharmaceutical scientists and been applied in the development of clinical formulations (18,19), assessment of active pharmaceutical ingredient properties to reduce risks of failure in the later development stage (20), prediction of food effects (21), establishment of IVIVC (22–25), and to aid justification of biowaivers (26–28). In our study, we utilized carbamazepine (CBZ), a BCS Class II drug, as an example to illustrate the general steps of applying mechanistic modeling and simulation to identify important factors in formulation design and discuss important aspects and considerations during modeling and simulation. This general strategy can also be applied to other drug products and be modified according to prior knowledge.
METHODS
General Modeling and Simulation Strategy
Figure 1 illustrates the key modeling and simulation steps to identify critical formulation factors. Usually, physiologically based predictive models require a large number of input parameters. Modeling and simulation start from data collection. It is critical to understand the underlying ADME mechanisms of the drug substance so that this information can be integrated into the mechanism-based model. For example, some drug substances undergo significant pre-systemic gut and liver metabolism, with the result that formulations with different release rates can produce different PK profiles of the same drug substance, potentially resulting in bioavailability differences. Thus, when designing new formulations to achieve target PK profiles for such drugs, it is important to integrate pre-systemic metabolism in the model. Here, we emphasize understanding the absorption process of different formulations and use conventional compartmental PK models to describe distribution and elimination. To obtain the PK model, using PK data after intravenous (i.v.) administration is ideal because the absorption model discussed below predicts fraction absorbed and can be linked to an empirical model for clearance. However, for some drugs, PK data after i.v. administration are absent. Using oral PK data after oral (p.o.) administration of the fastest dissolving formulation (such as an oral solution if available) is suggested to minimize the effects of absorption on the selection of the PK model. With initial values of all input parameters incorporated into the model, a predicted PK profile is obtained. The parameters with high uncertainty, such as in vivo solubility, in vivo release profile, permeability, etc., are optimized so that the predicted profile closely matches the observed profile. The optimized parameters are then fixed and used to predict other PK dataset(s) obtained under different conditions, such as a different dosing regimen, or another formulation with different particle size, etc. to validate the model.
Fig. 1.

The flow diagram shows a general process of using a physiologically based absorption model in QbD-based drug development
After gaining enough confidence about the model, we can do further exploration within the model, such as understanding how formulation parameters affect the PK profiles, simulating different dosing regimens, and linking to a pharmacodynamic (PD) model in order to predict formulation impact on PD response. Here, we used CBZ as a model drug to illustrate how we apply the physiologically based absorption modeling and simulation to understand critical parameters in the formulations and how the model can be used to guide formulation design. We modeled four dosage forms of CBZ under both fasted and fed state. All simulations were performed using GastroPlus™ software, version 6.1 (Simulations Plus, Inc.).
The Model Drug
CBZ has been traditionally considered as a narrow therapeutic index drug (29). The reported population therapeutic plasma concentration range is 4–12 μg/mL (29). However, the therapeutic index is different for different subjects, leading to individual therapeutic indices which could be narrower than the indicated population therapeutic index. Thus, a small fluctuation of plasma concentration may cause less efficacy or more toxicity in some subjects (29). CBZ is a substrate of CYP3A4 and it is well-established that CBZ autoinduces its own metabolism (30). Autoinduction of CBZ metabolism can be modeled by varying systemic clearance as a function of time and dose using mechanism-based in vitro–in vivo extrapolation (31). As the major goal of the current study is to evaluate the formulation impact on PK, autoinduction was not considered in modeling and simulation. Compared to the hepatic blood flow rate, CBZ clearance is much lower after single dose administration; thus, first pass extraction by the liver was neglected in modeling and simulation. It is unlikely that CBZ is metabolized in the intestine since a CBZ perfusion study performed in rabbit small and large intestine did not show detectable metabolites in the perfusate samples (32). Thus, gut extraction was also neglected in the simulations.
The ACAT Model Description
The advanced compartmental absorption and transit model (ACAT) model implemented in GastroPlus™ was developed based on Yu and Amidon’s compartmental absorption and transit (CAT) model (5–7,33) and was used for all the simulations in the current study. Briefly, the ACAT model consists of nine compartments (stomach, duodenum, jejunum 1, jejunum 2, ileum 1, ileum 2, ileum 3, caecum, and ascending colon) to mimic the human GI tract (14,15,17,19,21,34,35). Mass balance equations described the transport of drug along the GI tract as well as through the enterocytes. In each compartment, drug exists as unreleased, undissolved, and dissolved forms. All three forms can transit along the GI tract by first order processes. But only dissolved drug can transport through enterocytes and serve as substrate for metabolic enzymes and transporters. Default physiology parameters under fasted and fed states were population mean values obtained from published data including pH, volume, length, radii, and transit time. The dissolution rate constant was calculated by a modified Noyes–Whitney dissolution equation, in which diffusion coefficient, drug particle density, particle radius, particle shape factor, and solubility are input parameters. Once the drug transports through the basolateral membrane of enterocytes, it reaches the portal vein and then the liver. Liver blood flow consists of about 75% of portal vein blood flow and 25% of hepatic arterial blood flow. After drug exits the liver, it reaches the systemic circulation from where the ACAT model is connected to either a conventional PK compartment model or a PBPK model that describes drug distribution in major tissues. The link between formulation performance and pharmacokinetics of drug products is the key feature of the ACAT model. Thus, it could be a powerful tool to guide formulation design to achieve target PK profiles.
Data Collection
Input parameters in the ACAT model can be categorized into three classes: formulation properties (such as drug particle size distribution and density, controlled release profiles for modified release products), physicochemical properties of drug substance (such as diffusion coefficient, lipophilicity, pKa, solubility, and permeability), and pharmacokinetic parameters (such as clearance, volume of distribution, and the disposition model). Input parameters for baseline simulations of four CBZ formulations are summarized in Table I. All data in Table I were collected from literature, in silico predictions, multiple New Drug Applications (NDAs), or Abbreviated New Drug Applications (ANDAs) except the particle density for IR tablet which was an optimized value. PK data after oral administration of the four CBZ dosage forms of interest were collected from multiple NDAs and ANDAs. In the current study, all simulations were performed for the reference listed drug (RLD) of each dosage form. If more than one study under the same conditions with electronic data was available, then pooled mean PK data and pooled mean body weight were used. This selection of data represents that which could be available to a formulator developing either a generic version of an existing modified release product or a new modified release dosage form.
Table I.
Summary of Input Parameters for CBZ ACAT Model Baseline Simulation
| Parameters | Suspension | IR tablet | XR tablet | XR capsule |
|---|---|---|---|---|
| Dosage forma | IR: Suspension | IR: Tablet | CR: Integral Tablet | CR: Dispersed |
| MW | 236.3 | 236.3 | 236.3 | 236.3 |
| logPb | 2.38 | 2.38 | 2.38 | 2.38 |
| Solubilityc (mg/mL at pH = 6.8) | 0.12 (fasted) | 0.12 (fasted) | 0.12 (fasted) | 0.12 (fasted) |
| 0.32 (fed) | 0.32 (fed) | 0.32 (fed) | 0.32 (fed) | |
| pK da | 12.01 (acidic) | 12.01 (acidic) | 12.01 (acidic) | 12.01 (acidic) |
| 0.26 (basic) | 0.26 (basic) | 0.26 (basic) | 0.26 (basic) | |
| Dose (mg) | 200 | 400 | 400 | 300 |
| Dose volume (mL) | 240 | 240 | 240 | 240 |
| Particle density (g/mL) | 1.2d | 1.5e | 1.2d | 1.2d |
| Mean particle radius (μm) | 5f | 75 g | 100f | 50f |
| Particle radius standard deviation | 0 | 20 | 10 | 20 |
| Particle radius bin # | 1 | 5 | 3 | 3 |
| Precipitation time (sec)d | 900 | 900 | 900 | 900 |
| Diffusion coefficient (cm2/s)d | 9.72 × 10−4 | 9.72 × 10−4 | 9.72 × 10−4 | 9.72 × 10−4 |
| f up (%)h | 22 | 22 | 22 | 22 |
| Permeability (cm/s)i | 4.30 × 10−4 | 4.30 × 10−4 | 4.30 × 10−4 | 4.30 × 10−4 |
| Simulation time (h) | 168 | 168 | 240 | 192 |
| Body weight (kg) | 75 (fasted) | 75 (fasted) | 70 (fasted) j | 75 (fasted) |
| 80 (fed) | 70 (fed) j | 70 (fed) j | 75 (fed) |
aDosage forms selected in GastroPlus™ for simulations for different CBZ dosage forms
bFrom reference (50)
cFrom reference (24,42). Aqueous solubility was used in simulations under fasted sate and solubility measured in phosphate buffer (pH 6.8) containing 0.1% sodium lauryl sulfate (SLS) was used in simulations under fed state
dPredicted by ADMET predictor, or default values in GastroPlus™
eOptimized values
fApproximated from multiple ANDA reviews
gFrom reference (48)
hFrom reference (51)
iFrom reference (52)
jBody weight data were missing and an average value of 70 kg was assumed
Dosage Forms
Four marketed dosage forms of CBZ were studied, for which the RLD products are as follows: IR suspension (Tegretol®), IR tablet (Tegretol®), XR tablet (Tegretol® XR), and XR capsule (Carbatrol®). For the IR suspension and tablet, the corresponding dosage forms in GastroPlus™ were selected for simulations. IR formulations begin dissolving in stomach as soon as the dose is administered. In GastroPlus™, particle dissolution equations for IR suspensions, capsules, and tablets are identical except that when simulating solution, suspension, and capsule dosage forms, a shorter stomach transit time is suggested than if the same simulations were performed for a tablet dosage form. The dissolution was predicted according to the Nernst–Brunner modification of the Noyes–Whitney equation. For XR tablet and capsule, control release (CR): Integrated Tablet and CR: Dispersed were selected for simulations, respectively. Controlled release profiles described by a four-parameter Weibull function (Eq. 1) were used for both extended release formulations.
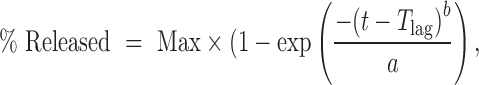 |
1 |
where Max is the percentage of maximum release, Tlag is the lag time, a is the scale factor, and b is the shape factor.
The PK Model
PK data after oral administration of the IR suspension under a fasted state were fit in PKPlus™ to obtain the systemic clearance, volume of distribution, and distribution constants between central and peripheral compartments. Two methods were used to obtain the PK parameters: (1) Empirical PK models were fitted to the data obtained after oral administration of IR suspension under fasted state to get the PK parameters (Vc, CL, K12, K21, Ka, and tlag). Then, we fixed the PK parameters and optimized the ASF values to get the physiology model described in detail in the following section. Fitting of empirical PK models were performed in PKPlus™, which fits all three models (1-, 2-, and 3-compartment) at once and treats the absorption rate as a constant. (2) We also performed fitting in the ACAT model. To accomplish this, an ‘optimization’ module was used. Nine parameters were selected for optimization simultaneously: systemic clearance, central volume of distribution, distribution constant from central compartment to peripheral compartment, distribution constant from peripheral compartment to central compartment, mean particle radius, drug particle density, solubility, and C1 and C2 constants used in calculation of ASFs in the physiology model. The Hooke & Jeeves pattern search method was used in both fittings, and weighting was equal to 1/Yhat2. Akaike information criterion (AIC) was used to select the best fitted compartment model. After the PK parameters were obtained, they were fixed and used in simulations for other dosage forms.
The Physiology Model
The physiology parameters used for CBZ simulations were summarized in Tables S1 and S2 in Supporting Information I for fasted and fed state, respectively. The ASF in GastroPlus™ is a multiplier used to scale the effective permeability to account for variations in surface-to-volume ratio, pH effects, influx, or efflux transporter differences, and other absorption-rate-determining effects that differ from one compartment to another (36). There are eight basic ASF models in version 6.1. ASFs are calculated from four coefficients C1–C4 in all ASF models except in the “User-Defined” model. The ASFs were optimized using a PK dataset after single dose oral administration of IR suspension in the fasted state by changing constants C1 and C2. Coefficient C3 and C4 were kept as default values C4 (0.0734 and 0.32, respectively) since they are used to calculate the ASFs of colon and had less impact on the absorption of a fast dissolving formulation. For the IR suspension under the fasted state, a stomach transit time of 0.1 h was used while a stomach transit time of 0.25 h was used for the tablet and capsule. Under fed conditions, a stomach transit time of 1 h was used for all dosage forms. A colon transit time of 36 h was used for all dosage forms under both fasted and fed conditions. Other parameters were default values in GastroPlus™.
Parameter Sensitivity Analysis
Parameter sensitivity analysis (PSA) was performed for four dosage forms under both fasted and fed conditions. When performing PSA, one parameter was changed gradually within a realistic span based on prior knowledge while keeping the other parameters at baseline levels. PSA was performed for parameters with high uncertainty and key parameters in formulations, including diffusion coefficient, dose volume, mean particle radius, particle radius standard deviation, drug particle density, permeability, solubility, precipitation time, and four Weibull parameters. For the IR suspension, in order to capture the early time points and get an accurate estimation of AUCt and AUCinf, simulation times of 12 and 168 h were used.
Virtual Bioequivalence Studies
Virtual bioequivalence (BE) studies were performed for a hypothetical XR tablet under fasted and fed conditions using a Weibull function fitted to the in vitro dissolution profile of RLD. A conventional 2 × 2 crossover design was applied. Physiological and PK parameters were randomly sampled from log-normal distributions defined in Table S3 in Supporting Information I for 25 subjects. PK profiles for the 25 subjects were predicted after a single dose administration of RLD under the fasted state. Two test formulations (Tests 1 and 2) were given to the same 25 subjects and PK profiles were predicted from hypothetical in vitro dissolution profiles described by Weibull function given to the two test formulations. A random sequence was assigned to the test formulations for 90% confidence intervals (CI) calculation of Cmax, AUCt, and AUCinf. F2 values were calculated to compare the similarity of dissolution profiles between RLD and test formulations. The same procedure was repeated for a fed virtual BE study on another 25 virtual subjects.
RESULTS
PK Profiles after Oral Administration of Four Dosage Forms Were Closely Predicted by the ACAT Model under both Fasted and Fed State
AIC indicated that a two-compartment model best described CBZ pharmacokinetics following administration of the IR formulation. A conventional compartmental model with first-order absorption and elimination in the presence of a lag time was fit to mean PK data after oral administration of IR suspension under the fasted state. Meanwhile, we also fit an ACAT model to the same dataset. Fitted PK parameters were summarized in Table II. Comparing PK parameters obtained in the conventional PK model versus ACAT model revealed no significant differences. This gave us confidence about the PK parameters. Furthermore, the half life and volume of distribution values determined by fitting the models were within the reported ranges (37–40). The optimized solubility, particle radius, and particle density were also close to the values used in baseline simulations (Table I). The parameter values obtained by fitting the conventional PK model were used for further simulations for clearance, central volume of distribution, distribution constant from central compartment to peripheral compartment, and distribution constant from peripheral compartment to central compartment (Table II). For body weight, mean values from multiple studies under the same conditions (dosage form, dosing regimen, and food condition) were used. Parameters in Table I were used for baseline simulations for four dosage forms.
Table II.
PK Parameters Deconvoluted from PK Data After Single Dose Oral Administration of CBZ IR Suspension Under Fasted Condition in Conventional PK Model and ACAT Model
| Parameters | Conventional PK model | ACAT model |
|---|---|---|
| Systemic clearance, CL/F (L/h) | 1.127 | 1.127 |
| Central compartment volume, V c/F (L) | 63.06 | 60.68 |
| Half life, T 1/2 (h) | 44.65 | 43.38 |
| Peripheral compartment volume, V 2/F (L) | 8.939 | 9.079 |
| Absorption rate constant, K a (1/h) a | 6.025 | N.A. |
| Lag time, t lag (h) * | 0.309 | N.A. |
| Distribution constant central to peripheral, K 12 (1/h) | 0.034 | 0.036 |
| Distribution constant peripheral to central, K 21 (1/h) | 0.241 | 0.240 |
| Mean particle radius (μm) | N.A. | 2.94 |
| Drug particle density (g/mL) | N.A. | 0.9873 |
| Solubility | N.A. | 0.1135 |
| Mean body weight (kg) | 75 | 75 |
a K a and t lag were not used in ACAT modeling
N.A. not available
Obtaining the systemic clearance using the PK datasets after i.v. or p.o. administration of the most rapidly dissolving formulation in conventional PK models is critical because, for many modified release oral products, bioavailability may contribute significantly to apparent oral clearance and volume of distribution. The ACAT model predicts the fraction absorbed mechanistically and calculates the bioavailability as a function of liver extraction, gut extraction, and fraction absorbed. Thus, if an underestimated systemic clearance was used, it is possible that the terminal elimination phase will be over-predicted. When fitting the ACAT model to PK data, it is important to minimize the number of parameters to be optimized and keep them in a realistic range in order to avoid overfitting. This process needs a good understanding of the formulation (such as particle size and density) as well as the physicochemical properties of the drug substance (such as pKa, logP, diffusion coefficient, etc.).
When we began ACAT modeling of the CBZ PK profile after oral administration of the IR suspension under fasted conditions, all ASF models in GastroPlus™ predicted Tmax values later than those observed. We studied the effects of particle size and solubility on the Tmax prediction under fasted state after administration of IR suspension. The results showed that even though the particle radius was decreased by tenfold to 0.5 μm, Tmax was over predicted by 1.5 h. If the solubility was increased by fivefold, the Tmax was over-predicted by 0.95 h, and if the solubility was increased by tenfold, the Tmax was over predicted by 0.88 h. Tmax was not sensitive to the change of drug particle density either. In order to capture the early Tmax after oral administration of IR suspension while keeping the particle size and solubility in a realistic range, we optimized the constants C1 and C2 of Opt logD Model to obtain optimized ASF for the small intestine. Coefficients C1 and C2 were 0.6 and 4.3, respectively, obtained by optimizing another independent PK dataset after p.o. administration of 100 mg IR suspension (data not shown). The optimized ASF values (Tables S1 and S2 in Supporting Information I) were about 10 times higher than the values in default Opt logD Model, indicating a fast absorption of CBZ in the small intestine.
Predicted mean PK profiles after p.o. administration of IR suspension and IR tablet were close to the observed mean PK profiles (Table III, Fig. 2a and b) using parameters in Table I, PK parameters in Table II, and physiology parameters in Tables S1 and S2 in Supporting Information I. The ratios of predicted values to observed mean values were between 0.85 and 1.14 for Cmax, AUCt, and AUCinf, under both fasted and fed conditions. An absorption plateau exists after oral administration of the CBZ IR tablet under the fasted condition (Fig. 2b). The POT20 (peak occupancy time, time span over which the concentration is within 20% of Cmax) ranged from 3.7 to 41 h under fasted conditions. The model also captured this property well and the predicted POT20 ranged from 2.9 to 40 h (Table III).
Table III.
Comparison of Predicted vs. Observed Mean Plasma PK Parameters
| Parameters | Suspension | IR tablet | XR tablet | XR capsule | |||||
|---|---|---|---|---|---|---|---|---|---|
| Obs. | Pred. | Obs. | Pred. | Obs. | Pred. | Obs. | Pred. | ||
| Dose (mg) | 200 | 400 | 400 | 300 | |||||
| C max (ng/mL) | Fasted | 3066.7 | 2914.6 | 3610.1 | 3713.7 | 3005.2 | 3105.2 | 2066.1 | 2120.8 |
| Fed | 2580.0 | 2506.9 | 5920.0 | 5501.2 | 3329.9 | 3786.6 | 2661.9 | 2798.5 | |
| AUCt (μg × h/mL) | Fasted | 166.6 | 163.3 | 279.8 | 301.6 | 270.4 | 263.7 | 194.3 | 190.2 |
| Fed | 165.2 | 152.9 | 401.3 | 348.6 | 286.1 | 288.6 | 221.1 | 222.2 | |
| AUCinf (μg × h/mL) | Fasted | 179.8 | 177.3 | 298.6 | 330.0 | 285.8 | 272.2 | 226.4 | 203.8 |
| Fed | 180.7 | 166.2 | 444.9 | 379.3 | 304.7 | 297.5 | 246.7 | 237.0 | |
| T max (hr) | Fasted | 1 | 1.45 | 24 | 16 | 24 | 28.9 | 26 | 20.98 |
| Fed | 4 | 3.66 | 12 | 4.8 | 24 | 16.4 | 15 | 17.8 | |
| POT20 (hr) | Fasted | [0.6,8.5] | [0.8,10] | [3.7,41] | [2.9,40] | [10,42] | [13,44] | [8.2,54] | [8.9,48] |
| Fed | [1.1,16] | [1.4,16] | [3.5,28] | [2.3,19] | [8.1,42] | [10,34] | [7.3,44] | [8.8, 39] | |
| F a (%) | Fasted | N.A. | 99.9 | N.A. | 93.0 | N.A. | 71.6 | N.A. | 76.6 |
| Fed | N.A. | 99.9 | N.A. | 99.8 | N.A. | 78.2 | N.A. | 89.0 | |
| Correlation coefficient (R 2) | Fasted | 0.956 | 0.975 | 0.974 | 0.977 | ||||
| Fed | 0.940 | 0.876 | 0.954 | 0.991 | |||||
POT 20 peak occupancy time, time span over which the concentration is within 20% of C max; F a fraction absorbed; N.A. not available
Fig. 2.

Predictions of PK profiles for different dosage forms using the ACAT Model. a IR Suspension; b IR tablet; c XR tablet; d XR capsule. Legend in (a) can be applied to (b), (c), and (d) Observed values were expressed as pooled mean and standard deviation
To predict the PK profiles after oral administration of XR products, Weibull controlled-release functions were used as inputs. We first deconvoluted mean PK profiles after p.o. administration of XR tablets and capsules under fasted and fed conditions to obtain the four sets of Weibull parameters. However, for XR tablet simulations, the Weibull function parameters obtained by fitting an in vitro dissolution profile gave slightly closer predictions to the observed mean PK profiles judging by correlation coefficient (R2), visual inspection, as well as by comparing predicted and observed mean PK parameters (Cmax, AUCt, and AUCinf). The deconvolution algorithm favored release profiles that provided complete release and thus did not locate the in vitro curve. Thus, Weibull parameters used for XR tablet baseline simulations were Max = 79.46%, Tlag = 0 h, scale factor a = 20.61, and shape factor b = 1.528. The same Weibull function was used for both fasted and fed conditions. The predicted profiles were compared with observed data in Fig. 2c. The corresponding Weibull release profile was shown in Fig. 3c.
Fig. 3.

Comparison of in vitro dissolution, Weibull controlled release profiles, and in vivo dissolution profiles for different dosage forms: a IR Suspension; b IR tablet; c XR tablet; and d XR capsule
For XR capsule simulations, Weibull parameters of Max = 100%, Tlag = 0.7278 h, scale factor a = 5.393, and shape factor b = 0.6293 were used for fasted study simulation; and Max = 97.144%, Tlag = 1.44 h, scale factor a = 5.454, and shape factor b = 0.8985 were used for fed study simulation, both of which were obtained from optimization/deconvolution. Figure 2d shows the predicted and observed PK profiles under fasted and fed conditions. The corresponding Weibull functions were shown in Fig. 3d.
The use of the same Weibull release profile for fed and fasted states for the tablet may be related to the formulation of that product as an osmotic pump while the different fed and fasting profiles for the capsule may indicate that drug release from that formulation is more sensitive the physiological environment.
The Nature of the Food-Formulation Interactions Differ with Dosage Form
An interesting observation was that food effects on the rate of CBZ absorption varied with the formulation. Co-administration of the IR suspension with food resulted in a later Tmax and lower Cmax. Co-administration of the IR tablet or XR capsule with food, by contrast, resulted in an earlier Tmax and higher Cmax. Finally, co-administration of the XR tablet with food had no significant impact on Tmax and increased Cmax only slightly. Food affects the physiology of the gastrointestinal (GI) tract via a diverse variety of mechanisms, and some of these mechanisms may underlay the different types of food effects observed for the different dosage forms. For example, prolongation of gastric emptying time may explain why the IR suspension has a later Tmax when taken with food. On the other hand, a high-fat meal increases bile output, leading to an increase in bile salts in the GI tract, which can enhance the dissolution rate of low-solubility drugs by micellar solubilization (41). This could be a possible mechanism causing the earlier Tmax and higher Cmax for the IR tablet and XR capsule. By incorporating gastric empting time and different in vivo solubility under fasted and fed states into the model, the different food effect trends on different formulations were well-predicted (Table III and Fig. 2).
In Vitro Dissolution Profiles Correlated to in Vivo Dissolution Rate can be Identified
Finding the in vitro dissolution testing conditions that best predict in vivo performance is one of the major tasks for implementation of QbD. A review of in vitro dissolution profiles under different conditions published in the literature or submitted to the Office of Generic Drugs (OGD) for different CBZ products, we identified dissolution testing factors affecting the in vitro dissolution rate and their role in comparisons with the in vivo dissolution profiles.
As the reported aqueous solubility of CBZ ranged from 0.12 to 0.26 mg/mL (24) use of surfactant, such as SLS, is common in developing in vitro dissolution methodology for CBZ formulations. Use of surfactant increases the solubility and dissolution rate of the CBZ IR tablet and XR capsule. Dissolution rate is not affected by pH because CBZ exists mainly as a neutral form from pH 1 to 11.
For the IR suspension (strength 100 mg/5 mL), dissolution testing was conducted in 900 mL water at 37°C using USP Apparatus II with a rotation speed of 50 rpm without surfactant. In dissolution datasets submitted to OGD by multiple firms, the IR suspension was completely dissolved within 30 min although the percentage of dissolved CBZ at 15 min showed relatively high variability. Figure 3a showed ranges of percentage dissolved at each time point tested under above conditions from multiple studies. In vitro dissolution was slower than in vivo dissolution in the fed state but faster than in vivo dissolution in the fasted state.
For the IR tablet, in vitro dissolution was tested in 900 mL media containing different amounts of SLS at different pH values using USP Apparatus II with paddle speed of 75 rpm (42). Figure 3b shows representative in vitro dissolution profiles in media containing 0.1% SLS (extracted from (42)) and 1% SLS (from data available to FDA). In vivo dissolution in the fed state was close to in vitro dissolution profiles tested in media containing 0.1% SLS. The f2 value between those two profiles was 47.3. Fasted state in vivo dissolution was slower than any measured in vitro profiles. In a simulation performed for the fasted state using the same solubility as for the fed state, in vivo dissolution rate increased and was comparable with the rate under the fed state (Fig. S1 A and B in Supporting Information I). Thus, the difference of in vivo dissolution rates between fasted and fed states for IR formulations was caused by the difference of in vivo solubility under fasted and fed state. The above findings supported the higher solubility (similar to that measured in media containing 0.1% SLS) used in fed state simulations.
The CBZ XR tablet was developed using an osmotic release delivery system designed to release CBZ at a constant rate up to certain percentage of total drug release. This tablet has a solid core, which contains CBZ, an osmotic agent, and water soluble viscosity generating agents (43). A water permeable membrane surrounds the core through which a single opening has been made on one side of the tablet. The tablet absorbs water from gastric contents through the semi-permeable membrane and a homogeneous solution of the drug is formed. Osmotic pressure differences then drive the CBZ solution through the opening in the membrane (43,44). Thus, the release rate is determined by the permeability of the water permeable membrane. CBZ XR tablet dissolution testing was conducted in 900 mL buffer of different pH (1.1, 4.5, and 6.8) using USP Apparatus I at 100 rpm. No significant difference was observed under different conditions. Figure 3c shows the range of in vitro dissolution conducted for 200 mg XR tablets in water and the fitted Weibull function used for baseline simulation. When these parameters were used as simulation inputs the PK profile was well predicted, and the observed in vivo release rate was similar in the fed state but slower in the fasted state. Thus, the difference found between the in vivo dissolution under the fasted versus the fed state was caused by different solubility. Performing simulation using the same solubility for the fasted state as the solubility in the fed state gave an almost identical in vivo dissolution profile (Figure S1 C in Supporting Information I).
CBZ XR capsules (RLD Carbetrol®) contain three different types of beads: IR, XR, and enteric- coated, to achieve extended release in vivo (44). Some factors affecting in vitro dissolution may have counteracting effects (Table IV). For example the f2 value was 50.2 comparing a dissolution profile of the 100 mg capsule in 1,000 mL of 0.1 N HCl containing 0.1% SLS at 75 rpm with a dissolution profile conducted in 900 mL of 0.1 N HCl (0–2 h) + phosphate buffer (pH 7.5, 2–12 h) containing 0.9% SLS at 50 rpm. In vitro dissolution ranges plotted in Fig. 3d were for 300 mg capsules in 900 mL of media (pH 1.2, 4.5, and 6.8) with 0.1% SLS using USP Apparatus II under different rotation speed (50 vs. 75 rpm). For the XR capsule, the best relationship between in vivo and in vitro data was at 50 rpm and 0.1% SLS.
Table IV.
Summary of Dissolution Testing Conditions that affect CBZ XR capsule in vitro Dissolution Rate
| Parameters | Effects |
|---|---|
| Dose strength | Dissolution rate is slightly higher at lower strength suggesting non-sink condition. |
| pH | Dissolution rate is not sensitive to pH because CBZ exists mainly as a neutral form from pH 1 to 11. |
| %SLS or other surfactant | Increasing surfactant concentration increases dissolution rate and solubility. |
| Speed | Increasing paddle speed increases dissolution rate. |
| Paddle type | Paddle type affects initial dissolution rate presumable due to the change of hydrodynamics. |
Tested in USP Apparatus II, 900 mL Volume, 100, 200, and 300 mg
CBZ was Mainly Absorbed in Small Intestine for IR Formulation, but in Colon for XR Formulation
The ACAT model also predicts the regional absorption. Formulations may have significant impact on regional absorption as indicated in Fig. 4. For both fasted and fed conditions, CBZ was mainly absorbed in small intestine if it was formulated as IR formulation but was mainly absorbed in caecum and colon if it was formulated as XR formulation. Compared with XR formulations, CBZ IR tablet has a rapid absorption followed by a plateau absorption phase (Fig. 2b) under the fasted condition. Thus, we observed about 25% of CBZ in IR tablet formulation was absorbed in colon under the fasted state (Fig. 4a). Food had the greatest effects on the rate of absorption from the IR suspension and tablet and increased CBZ absorption in duodenum (Fig. 4b).
Fig. 4.

Effects of dosage forms on regional absorption. a Fasted; b Fed
Dosage Form Selection May have Significant Impact on Simulation Results, Suggesting Understanding the Formulation Design is Critical
GastroPlus™ provides a variety of dosage forms. It is important to understand the formulations and then chose the right dosage form for simulation. Otherwise, the simulation may lead to misleading conclusions. We chose CR for both XR formulation simulations. According to the formulation design, the XR tablet (osmotic pump) releases saturated solutions. There are some uncertainties in the XR capsule since it consists of three types of beads. IR beads could release dissolved or undissolved drug and enteric-coated beads could release dissolved drug or undissolved particles depending on the formulation design and drug properties. Thus, the XR capsule could be a mixture of dissolved drugs and undissolved particles. We compared the simulation results of using different dosage forms while keeping all other parameters the same. The largest sensitivity was observed for XR tablet under fasted conditions (Fig. 5a). The XR tablet simulations under the fed condition and the XR capsule simulations under both fasted and fed conditions were less sensitive to the changes (Fig. 5b–d).
Fig. 5.

Effect of dosage form selection on simulation results. a XR tablet (fasted state); b XR tablet (fed state); c XR capsule (fasted state); d XR capsule (fed state)
We also compared whether it was better to model the XR tablet using unit transit or dispersed transit. Although it is obvious that an osmotic tablet actually transits as a discrete unit within an individual subject, the ACAT model is intended to provide a prediction of population mean absorption and pharmacokinetics. In this context, dispersed transit of the osmotic pump formulation reflects variations in transit between individuals. For this reason, we found that modeling the XR tablet as a dispersed dosage form provided better predictions of the observed data in terms of Tmax under fed condition (Table S4 and Figure S2 in Supporting Information I).
PK Profile Sensitivity to the same Parameters Could be Different for Different Formulations
We further performed PSA for the four dosage forms under both fasted and fed conditions for important formulation parameters and physiological parameters with high uncertainty. PSA is potentially a powerful tool for QbD implementation once it is validated for key formulation factors. Figure 6 shows sensitivity of PK parameters to effective permeability under the fasted condition. If CBZ is formulated as a suspension, permeability has less effect on PK parameters. Particle size and density had a significant effect on CBZ PK from IR formulations but no effect on PK from XR formulations (Figures in Supporting Information II). That was because in XR formulations the particle size effects were integrated in the dissolution profiles, which were translated into Weibull functions for input to the ACAT model. Comparing the two IR formulations, particle size has less impact on CBZ AUC and Cmax from the IR suspension (Page 6 of Supporting Information II) than from the IR tablet (Page 38 of Supporting Information II). In general for all formulations, in vivo solubility had significant impact on PK profiles when it was less than 0.2 mg/mL under the fasted state (Figures in Supporting Information II). This is within the reported range of aqueous solubility of CBZ (24), suggesting that CBZ absorption is dissolution rate-limited rather than solubility-limited, which is also consistent with literature reports (37).
Fig. 6.

PK parameters sensitivity to effective permeability in different dosage forms. a AUCt, b C max, c T max. Legend in (c) is also applied to (a) and (b). Simulations were performed under fasted condition. Other PSA results were presented in Supporting Information II
PK Profile Sensitivity to the same Parameters Could be Different Under Fasted Versus Fed States
Another interesting phenomenon observed during PSA was that PK profiles had different sensitivity to the same parameters depending upon whether under the fasted or fed states existed. Figure 7 presented the CBZ PK profiles changing with the change of particle radius in IR suspension. When the particle radius decreased from 48.62 to 10.28 μm, Cmax increased 1.2-fold under the fasted state but did not change much under fed conditions. This observation suggests that when conducting pilot PK studies to evaluate a new formulation, if the study is not properly designed, it may not be possible to detect the formulation-related differences in drug absorption. Evaluating all the PSA results in the Supporting Information II shows that in general, PK profiles are more sensitive to parameters changing under fasted state than under fed state for CBZ.
Fig. 7.

PK profiles sensitivity to the mean particle radius is different under fasted and fed state. a IR suspension, fasted state; b IR suspension, fed state. Legend in (b) is also applied to (a)
Virtual BE Studies Indicate that Dissolution is a Sensitive Discrimination Method
PSA is a one-dimensional simulation to evaluate the trend and range of PK changes with the change of certain parameters. In reality, many sensitive parameters have counteracting effects that may interact together and result in no apparent effect. To incorporate all possible effects simultaneously, we performed virtual BE studies on XR tablet. The XR tablet was selected as an example since its in vitro dissolution gave good predictions in vivo. Virtual trial is a stochastic simulation that randomly samples parameters from predefined distributions. Two virtual test formulations were examined. Test 1 had an f2 value of 67.4 compared with RLD and test 2 had an f2 value of 38.2 compared with RLD (Table V). Dissolution profiles were shown in Fig. 8a. Although test 2 and RLD showed large difference in in vitro dissolution, the virtual BE study showed that test 2 was still bioequivalent to the RLD using the 80–125% criteria (Table V and Fig. 8). The result was consistent with the general conclusion that in vitro dissolution is a more sensitive method than an in vivo study. We further noticed that the 90% CIs calculated for the test/reference ratios after logarithm transformation from virtual crossover BE studies were narrower than observed CIs. That was because in the simulation the physiological and pharmacokinetic parameters of the same subjects were identical when they were administered test versus reference formulations. However, in reality, physiological and pharmacokinetic parameters could fluctuate within the same subjects if they were given the different formulations on different occasions. By incorporating such variability, test 2 formulation might be not be bioequivalent to the RLD in an in vivo study using a relatively modest number of subjects since its 90% CI is already close to the edge of the BE limits.
Table V.
Summary of Virtual BE Studies
| Parameters | RLD vs. Test 1 | RLD vs. Test 2 | ||||||
|---|---|---|---|---|---|---|---|---|
| f2 | 67.4 | 38.2 | ||||||
| Fasted | Fed | Fasted | Fed | |||||
| P.E. | 90% CI | P.E. | 90% CI | P.E. | 90% CI | P.E. | 90% CI | |
| C max | 1.09 | [1.07, 1.12] | 1.00 | [0.99, 1.00] | 0.93 | [0.91, 0.96] | 0.89 | [0.88, 0.90] |
| AUCt | 1.05 | [1.04, 1.06] | 1.00 | [0.99, 1.00] | 1.09 | [1.07, 1.12] | 1.11 | [1.08, 1.14] |
| AUCinf | 1.05 | [1.03, 1.06] | 0.99 | [0.99, 1.00] | 1.10 | [1.07, 1.13] | 1.11 | [1.09, 1.14] |
P.E. point estimate
Fig. 8.

Virtual BE study. a Weibull controlled release profiles; b Fasted state; c Fed state
DISCUSSION
PBPK modeling has been widely used in environmental science and toxicology for many years to predict chemical distributions in human body due to the infeasibility of performing such studies in humans (45). Recently, it has been used in drug discovery and development focused on predicting clinical PK from preclinical studies as an alternative approach to allometric scaling (46,47). The ACAT model is one type of PBPK models which focuses on simulating oral drug products releasing, dissolving, and transport along the GI tract, as well as transport through the enterocytes from which the drug substance connects to the systemic circulation. Conceptually, this predictive model is a tool for QbD implementation as it connects the drug substance properties (pKa, solubility, permeability, lipophilicity) and formulation properties (particle size distribution, drug particle density) with drug products in vivo behavior (PK profiles). However, its utility in QbD-based drug development has not been discussed and explored much in the literature.
In this study, we used CBZ, a poorly soluble, highly permeable drug, as an example to illustrate how the ACAT model could be potentially used in QbD implementation. We started modeling with an IR suspension formulation, which was the fastest dissolving formulation of CBZ, to find the appropriate physiological model. It is crucial to start with the most rapidly dissolving formulation because otherwise, the results could lead to misleading conclusions. For example, deconvolution of the PK data obtained after oral administration of XR capsules under the GastroPlus™ Opt logD Model showed that in vitro dissolution profile tested in media containing 0.1% SLS at rotation speed of 75 rpm was the closest to in vivo release (data not shown). However, using the Opt logD Model could not capture the early Tmax of PK profile after oral administration of IR suspension. The ASFs in the physiology model obtained after optimizing the PK data after administration of the IR suspension in the fasted state could predict the PK data after administration of IR suspension in the fed state, as well as IR tablet under fasted and fed conditions (Table III and Fig. 2a and b). Promisingly, the model captured the plateau of the PK profile after administration of IR tablet under fasted state (Fig. 2b).
The partially validated model was further applied to extended release formulations. Deconvolution of PK profiles after administration of XR formulations gave in vivo dissolution profiles. Comparing in vitro dissolution profiles tested under different conditions with in vivo dissolution profiles, we identified that dissolution testing conducted in 900 mL buffer using USP Apparatus I correlated well with in vivo dissolution for XR tablet under fed condition; and dissolution profile obtained in 900 mL buffer containing 0.1% SLS using USP Apparatus II at rotation speed of 50 rpm was the closest to in vivo dissolution profiles under both fasted and fed conditions. It should be noted that for controlled release simulations, Weibull function is used as a model input. But Weibull function does not always represent in vivo dissolution (Fig. 3c). That was because in vivo dissolution was the outcome of a complex interaction of dissolution, solubility, and gastric intestinal transit time.
For modified release products finding in vitro dissolution testing conditions that best represent in vivo performance is one of the important tasks in QbD. Here, we reviewed all dissolution data submitted to FDA and selected the ones that predicted PK profiles closely. In pharmaceutical development, an appropriate approach would be deconvolution of PK profiles to obtain target in vitro release profile and find the dissolution conditions that give the target dissolution profile by DOE (Design of Experiment) models. The absorption model and dissolution conditions would then be tested through the development of another formulation.
Model validation plays a key role in the modeling and simulation procedures. Model validation includes many aspects, such as predictability of Tmax, peak occupancy time, dose non-linearity caused by incomplete absorption, gut extraction, or liver extraction, trend of PK profiles changing according to the changes of certain critical parameter, trend of food effects, etc. The PSA analysis for the IR tablet showed that when mean particle radius was decreased, Cmax was increased but Tmax was decreased. The same trend was observed by M. Dam et al. (48) for CBZ IR tablet. One of the limits of using BE data available to the FDA was that they were all successful BE studies and thus difficult to evaluate the predictions by PSA that would result in BE failure. Ideally, in pharmaceutical development stage, an additional well-designed pilot PK study to confirm the trend predicted by PSA would be very informative and give more confidence to the model. For example, in order to confirm the model predictability to other extended release formulations, the appropriate steps would be designing another extended release formulation with different dissolution rate, testing the in vitro dissolution rate under the proper conditions found in relevant studies, conducting the pilot PK study, and comparing the results with model prediction. Unlike conventional IVIVC which requires three PK studies for three formulations with slow, medium, and fast releasing rate, the IVIVR (in vivo-in vitro relationship) requires less study but needs the context of a mechanistic model.
We demonstrated how to perform a 2 × 2 crossover virtual BE study in the ACAT model, which could be incorporated as part of IVIVR to validate the model. Virtual trial enables introduction of population variability to the model. However, without including intra-subject variability, the confidence intervals are narrower than observed. Nevertheless, the virtual trial for the XR tablet PK profiles after single dose administration well captured the population variability between subjects (data not shown).
One of the important model outcomes was the regional absorption of CBZ. The CBZ XR formulations model showed that about 50% of the dose was absorbed in colon (Fig. 4). A fusion study in rabbit intestine showed the CBZ could be absorbed both in duodenum and jejunum as well as colon (32), consistent with the model prediction. The colon is traditionally not considered as an important absorption site. However, today, an increasing number of modified release formulations are being developed to delay or extend drug absorption in order to achieve the optimized dose regimen. A study of colonic drug absorption for 42 drugs in humans showed that BCS Class I drugs were well absorbed in colon (49). However, there were insufficient data available to fully assess the impact of low solubility and slow dissolution rate on colon absorption (49). In addition, our understanding of the role that colon physiology plays in drug absorption is not as extensive it is for the small intestine. Combining all these factors, we believe that improving an understanding of drug absorption in colon can be of great help in developing better mechanism-based predictive absorption models.
Other functions could be developed beyond the studies we have shown here. For example, we only performed one-dimensional PSA in which only one parameter changed while the other parameters were fixed. By performing multi-dimensional PSA, an optimized design space can be constructed as a function of important parameters. A further step from the physiologically based absorption modeling toward clinical and safety target is to link the PK model to a PD model (mechanistic or empirical). Thus the starting point (pharmaceutical design) and the end point (clinical performance) of the drug development chain will be connected through modeling and simulation.
To summarize, in this study we simulated four formulations of CBZ in a physiologically based absorption model under both fasted and fed conditions. We demonstrated what the general steps would be in establishing a model based on data from different dosage forms and how the model could be potentially used in QbD-based drug development. Model validation is important and can be evaluated from different aspects at different points in the process. The key to optimal implementation of mechanistic models in QbD is to understand the drug substance as well as the formulations, including the release mechanisms. We conclude that well-validated predictive models incorporating aspects of both in vivo and in vitro dosage form performance can be used to optimize drug product formulation and strengthen existing QbD programs.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
(DOC 392 kb)
(PDF 1275 kb)
ACKNOWLEDGEMENTS
The authors thank the Science Team Staff in the Office of Generic Drugs for excellent scientific discussion and debates, and Dr. John Chung for his help with virtual BE simulations.
Footnotes
The views presented in this article by the authors do not necessarily reflect those of the Food and Drug Administration (FDA).
REFERENCES
- 1.van Baarsen LG, Bos CL, van der Pouw Kraan TC, Verweij CL. Transcription profiling of rheumatic diseases. Arthritis Res Ther. 2009;11(1):207. doi: 10.1186/ar2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cox MA, Gawlitta D, Driessen NJ, Oomens CW, Baaijens FP. The non-linear mechanical properties of soft engineered biological tissues determined by finite spherical indentation. Comput Methods Biomech Biomed Eng. 2008;11(5):585–592. doi: 10.1080/10255840701771768. [DOI] [PubMed] [Google Scholar]
- 3.Gawlitta D, Oomens CW, Baaijens FP, Bouten CV. Evaluation of a continuous quantification method of apoptosis and necrosis in tissue cultures. Cytotechnology. 2004;46(2–3):139–150. doi: 10.1007/s10616-005-2551-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones HM, Gardner IB, Watson KJ. Modelling and PBPK Simulation in Drug Discovery. AAPS J. 2009;11(1):155–166. doi: 10.1208/s12248-009-9088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu LX. An integrated model for determining causes of poor oral drug absorption. Pharm Res. 1999;16(12):1883–1887. doi: 10.1023/A:1018911728161. [DOI] [PubMed] [Google Scholar]
- 6.Yu LX, Amidon GL. A compartmental absorption and transit model for estimating oral drug absorption. Int J Pharm. 1999;186(2):119–125. doi: 10.1016/S0378-5173(99)00147-7. [DOI] [PubMed] [Google Scholar]
- 7.Yu LX, Crison JR, Amidon GL. Compartmental transit and dispersion model analysis of small intestinal transit flow in humans. Int J Pharm. 1996;140(1):111–118. doi: 10.1016/0378-5173(96)04592-9. [DOI] [Google Scholar]
- 8.Willmann S, Hohn K, Edginton A, Sevestre M, Solodenko J, Weiss W, et al. Development of a physiology-based whole-body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. J Pharmacokinet Pharmacodyn. 2007;34(3):401–431. doi: 10.1007/s10928-007-9053-5. [DOI] [PubMed] [Google Scholar]
- 9.Willmann S, Schmitt W, Keldenich J, Dressman JB. A physiologic model for simulating gastrointestinal flow and drug absorption in rats. Pharm Res. 2003;20(11):1766–1771. doi: 10.1023/B:PHAM.0000003373.72652.c0. [DOI] [PubMed] [Google Scholar]
- 10.Willmann S, Schmitt W, Keldenich J, Lippert J, Dressman JB. A physiological model for the estimation of the fraction dose absorbed in humans. J Med Chem. 2004;47(16):4022–4031. doi: 10.1021/jm030999b. [DOI] [PubMed] [Google Scholar]
- 11.Willmann S, Thelen K, Becker C, Dressman JB, Lippert J. Mechanism-based prediction of particle size-dependent dissolution and absorption: cilostazol pharmacokinetics in dogs. Eur J Pharm Biopharm. 2010;76(1):83–94. doi: 10.1016/j.ejpb.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 12.Grass GM. Simulation models to predict oral drug absorption from in vitro data. Adv Drug Deliv Rev. 1997;23(1–3):199–219. doi: 10.1016/S0169-409X(96)00436-X. [DOI] [Google Scholar]
- 13.Norris DA, Leesman GD, Sinko PJ, Grass GM. Development of predictive pharmacokinetic simulation models for drug discovery. J Control Release. 2000;65(1–2):55–62. doi: 10.1016/S0168-3659(99)00232-1. [DOI] [PubMed] [Google Scholar]
- 14.Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Deliv Rev. 2001;50(Suppl 1):S41–67. doi: 10.1016/S0169-409X(01)00179-X. [DOI] [PubMed] [Google Scholar]
- 15.Parrott N, Lave T. Prediction of intestinal absorption: comparative assessment of GASTROPLUS and IDEA. Eur J Pharm Sci. 2002;17(1–2):51–61. doi: 10.1016/S0928-0987(02)00132-X. [DOI] [PubMed] [Google Scholar]
- 16.Jamei M, Turner D, Yang J, Neuhoff S, Polak S, Rostami-Hodjegan A, et al. Population-based mechanistic prediction of oral drug absorption. AAPS J. 2009;11(2):225–237. doi: 10.1208/s12248-009-9099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang W, Lee SL, Yu LX. Mechanistic approaches to predicting oral drug absorption. AAPS J. 2009;11(2):217–224. doi: 10.1208/s12248-009-9098-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dannenfelser R-M, He H, Joshi Y, Bateman S, Serajuddin ATM. Development of clinical dosage forms for a poorly water soluble drug I: application of polyethylene glycol-polysorbate 80 solid dispersion carrier system. J Pharm Sci. 2004;93(5):1165–1175. doi: 10.1002/jps.20044. [DOI] [PubMed] [Google Scholar]
- 19.Kuentz M, Nick S, Parrott N, Rothlisberger D. A strategy for preclinical formulation development using GastroPlus as pharmacokinetic simulation tool and a statistical screening design applied to a dog study. Eur J Pharm Sci. 2006;27(1):91–99. doi: 10.1016/j.ejps.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 20.Kesisoglou F, Wu Y. Understanding the effect of API properties on bioavailability through absorption modeling. AAPS J. 2008;10(4):516–525. doi: 10.1208/s12248-008-9061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones HM, Parrott N, Ohlenbusch G, Lave T. Predicting pharmacokinetic food effects using biorelevant solubility media and physiologically based modelling. Clin Pharmacokinet. 2006;45(12):1213–1226. doi: 10.2165/00003088-200645120-00006. [DOI] [PubMed] [Google Scholar]
- 22.Wei H, Lobenberg R. Biorelevant dissolution media as a predictive tool for glyburide a class II drug. Eur J Pharm Sci. 2006;29(1):45–52. doi: 10.1016/j.ejps.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Okumu A, DiMaso M, Lobenberg R. Computer simulations using GastroPlus to justify a biowaiver for etoricoxib solid oral drug products. Eur J Pharm Biopharm. 2009;72(1):91–98. doi: 10.1016/j.ejpb.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 24.Kovacevic I, Parojcic J, Homsek I, Tubic-Grozdanis M, Langguth P. Justification of biowaiver for carbamazepine, a low soluble high permeable compound, in solid dosage forms based on IVIVC and gastrointestinal simulation. Mol Pharm. 2009;6(1):40–47. doi: 10.1021/mp800128y. [DOI] [PubMed] [Google Scholar]
- 25.Okumu A, DiMaso M, Lobenberg R. Dynamic dissolution testing to establish in vitro/in vivo correlations for montelukast sodium, a poorly soluble drug. Pharm Res. 2008;25(12):2778–2785. doi: 10.1007/s11095-008-9642-z. [DOI] [PubMed] [Google Scholar]
- 26.Jantratid E, Prakongpan S, Amidon GL, Dressman JB. Feasibility of biowaiver extension to biopharmaceutics classification system class III drug products: cimetidine. Clin Pharmacokinet. 2006;45(4):385–399. doi: 10.2165/00003088-200645040-00004. [DOI] [PubMed] [Google Scholar]
- 27.Tsume Y, Amidon GL. The biowaiver extension for BCS class III drugs: the effect of dissolution rate on the bioequivalence of BCS class III immediate-release drugs predicted by computer simulation. Mol Pharm. 2010;7(4):1235–1243. doi: 10.1021/mp100053q. [DOI] [PubMed] [Google Scholar]
- 28.Kortejarvi H, Urtti A, Yliperttula M. Pharmacokinetic simulation of biowaiver criteria: the effects of gastric emptying, dissolution, absorption and elimination rates. Eur J Pharm Sci. 2007;30(2):155–166. doi: 10.1016/j.ejps.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 29.Bialer M, Levy RH, Perucca E. Does carbamazepine have a narrow therapeutic plasma concentration range? Ther Drug Monit. 1998;20(1):56–59. doi: 10.1097/00007691-199802000-00010. [DOI] [PubMed] [Google Scholar]
- 30.Kudriakova TB, Sirota LA, Rozova GI, Gorkov VA. Autoinduction and steady-state pharmacokinetics of carbamazepine and its major metabolites. Br J Clin Pharmacol. 1992;33(6):611–615. doi: 10.1111/j.1365-2125.1992.tb04089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shou M, Hayashi M, Pan Y, Xu Y, Morrissey K, Xu L, et al. Modeling, prediction, and in vitro in vivo correlation of CYP3A4 induction. Drug Metab Dispos. 2008;36(11):2355–2370. doi: 10.1124/dmd.108.020602. [DOI] [PubMed] [Google Scholar]
- 32.Riad LE, Sawchuk RJ. Absorptive clearance of carbamazepine and selected metabolites in rabbit intestine. Pharm Res. 1991;8(8):1050–1055. doi: 10.1023/A:1015817426713. [DOI] [PubMed] [Google Scholar]
- 33.Yu LX, Lipka E, Crison JR, Amidon GL. Transport approaches to the biopharmaceutical design of oral drug delivery systems: prediction of intestinal absorption. Adv Drug Deliv Rev. 1996;19(3):359–376. doi: 10.1016/0169-409X(96)00009-9. [DOI] [PubMed] [Google Scholar]
- 34.Jones HM, Parrott N, Jorga K, Lave T. A novel strategy for physiologically based predictions of human pharmacokinetics. Clin Pharmacokinet. 2006;45(5):511–542. doi: 10.2165/00003088-200645050-00006. [DOI] [PubMed] [Google Scholar]
- 35.Parrott N, Lukacova V, Fraczkiewicz G, Bolger MB. Predicting pharmacokinetics of drugs using physiologically based modeling–application to food effects. AAPS J. 2009;11(1):45–53. doi: 10.1208/s12248-008-9079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hendriksen BA, Felix MV, Bolger MB. The composite solubility versus pH profile and its role in intestinal absorption prediction. AAPS PharmSci. 2003;5(1):E4. doi: 10.1208/ps050104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balguid A, Driessen NJ, Mol A, Schmitz JP, Verheyen F, Bouten CV, et al. Stress related collagen ultrastructure in human aortic valves–implications for tissue engineering. J Biomech. 2008;41(12):2612–2617. doi: 10.1016/j.jbiomech.2008.06.031. [DOI] [PubMed] [Google Scholar]
- 38.Wangemann M, Retzow A, Evers G, Mazur D, Schug B, Blume H. Bioavailability study of two carbamazepine containing sustained release formulations after multiple oral dose administration. Arzneimittelforschung. 1998;48(12):1131–1137. [PubMed] [Google Scholar]
- 39.Garnett WR, McLean AM, Zhang Y, Clausen S, Tulloch SJ. Simulation of the effect of patient nonadherence on plasma concentrations of carbamazepine from twice-daily extended-release capsules. Curr Med Res Opin. 2003;19(6):519–525. doi: 10.1185/030079903125002144. [DOI] [PubMed] [Google Scholar]
- 40.Bastings MM, van Baal I, Meijer EW, Merkx M. One-step refolding and purification of disulfide-containing proteins with a C-terminal MESNA thioester. BMC Biotechnol. 2008;8:76. doi: 10.1186/1472-6750-8-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kraakman EM, Bontrop RE, Groenestein R, Jonker M, Haaijman JJ, t Hart BA. Characterization of the natural immune response of rhesus monkey CD4 + ve T cells to the bacterial antigen streptolysin O (SLO) J Med Primatol. 1995;24(4):306–312. doi: 10.1111/j.1600-0684.1995.tb00183.x. [DOI] [PubMed] [Google Scholar]
- 42.Lee H, Park SA, Sah H. Surfactant effects upon dissolution patterns of carbamazepine immediate release tablet. Arch Pharm Res. 2005;28(1):120–126. doi: 10.1007/BF02975147. [DOI] [PubMed] [Google Scholar]
- 43.Schroeder Y, Elliott DM, Wilson W, Baaijens FP, Huyghe JM. Experimental and model determination of human intervertebral disc osmoviscoelasticity. J Orthop Res. 2008;26(8):1141–1146. doi: 10.1002/jor.20632. [DOI] [PubMed] [Google Scholar]
- 44.Stevens RE, Limsakun T, Evans G, Mason DH., Jr Controlled, multidose, pharmacokinetic evaluation of two extended-release carbamazepine formulations (Carbatrol and Tegretol-XR) J Pharm Sci. 1998;87(12):1531–1534. doi: 10.1021/js980203+. [DOI] [PubMed] [Google Scholar]
- 45.Nestorov I. Whole-body physiologically based pharmacokinetic models. Expert Opin Drug Metab Toxicol. 2007;3(2):235–249. doi: 10.1517/17425255.3.2.235. [DOI] [PubMed] [Google Scholar]
- 46.Rostami-Hodjegan A, Tucker GT. Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat Rev Drug Discov. 2007;6(2):140–148. doi: 10.1038/nrd2173. [DOI] [PubMed] [Google Scholar]
- 47.Rodgers T, Rowland M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm Res. 2007;24(5):918–933. doi: 10.1007/s11095-006-9210-3. [DOI] [PubMed] [Google Scholar]
- 48.Dam M, Christiansen J, Kristensen CB, Helles A, Jaegerskou A, Schmiegelow M. Carbamazepine: a clinical biopharmaceutical study. Eur J Clin Pharmacol. 1981;20(1):59–64. doi: 10.1007/BF00554668. [DOI] [PubMed] [Google Scholar]
- 49.Sato S, Nakamura Y, Kaneko T, Asamizu E, Kato T, Nakao M, et al. Genome structure of the legume, Lotus japonicus. DNA Res. 2008;15(4):227–239. doi: 10.1093/dnares/dsn008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen ML, Yu L. The use of drug metabolism for prediction of intestinal permeability. Mol Pharm. 2009;6(1):74–781. doi: 10.1021/mp8001864. [DOI] [PubMed] [Google Scholar]
- 51.Johannessen SI, Strandjord RE. The concentration of carbamazepine (Tegretol R) in serum and in cerebrospinal fluid in patients with epilepsy. Acta Neurol Scand Suppl. 1972;51:445–446. [PubMed] [Google Scholar]
- 52.Lennernas H. Intestinal permeability and its relevance for absorption and elimination. Xenobiotica. 2007;37(10–11):1015–1051. doi: 10.1080/00498250701704819. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 392 kb)
(PDF 1275 kb)