

Abstract
One barrier to apply current tri-octylphosphine oxide (TOPO) based quantum dots (QDs) to biomedical imaging is that the TOPO on TOPO-QDs can be replaced by the proteins in living system, which may cause the degradation of QDs and/or deactivation of protein. In order to develop biocompatible optical imaging agents, a novel triblock copolymer, designed as a multidentate ligand, was synthesized to coat quantum dot nanocrystals (QDs). The copolymer consists of a polycarboxylic acid block at one end and a polythiol block at the other end with an intervening cross-linked poly(styrene-co-divinylbenzene) block bridging the ends. The multiple mercapto groups from the polythiol block act as multidentate ligands to stabilize QDs, while the polycarboxylic acid block improves the water solubility of QDs and offers reaction sites for surface modification or conjugation with bimolecules. The cross-linked poly(styrene-co-divinylbenzene) block provides a densely compacted hydrophobic shell. This shell will act as a barrier to inhibit the degradation of QDs by preventing the diffusion of ions and small molecules into the core of QDs. This new multidentate polymer coating facilitates the transfer of QDs from organic solvent into aqueous phase. The QDs directly bound to multidentate mercapto groups instead of TOPO are less likely to be affected by the mercapto or disulfide groups within proteins or other biomolecules. Therefore, this research will provide an alternative coating material instead of TOPO to produce QDs which could be more suitable for in vivo use under complex physiological conditions.
Keywords: Quantum dot, block copolymer, nanoparticle, biocompatible, coating
1. Introduction
Due to its high sensitivity, relatively inexpensive cost and lack of any risk of exposure to ionizing radiation, optical imaging has become one of the attractive non-invasive diagnostic technologies for early detection of tumors, particularly for those situated near the body surface[1]. To improve the diagnostic accuracy of optical imaging, it is essential to develop highly sensitive, tissue and/or tumor specific contrast agents with adequate stability to withstand harsh physiological microenvironment. In comparison to conventional organic fluorescent probes, semiconductor quantum dots (QDs) based fluorescent imaging probes have several advantages, including 1) high resistance to photo-bleaching and chemical degradation; 2) size-dependent emission wavelength from ultraviolet to near infrared range and wide adsorption spectra for excitation; and 3) high quantum yield and molar extinction coefficients (∼10–100 fold higher than those for organic dyes) [2–9]. In particular, the large molar extinction coefficient and resistance to photo-bleaching make QD an ideal agent for detection of small tumors.
Since the pioneering work by Alivisatos [10] and Nie[11], QDs have been covalently linked to various biomolecules such as antibodies, peptides, nucleic acids and other ligands for biological applications [12–27]. Recently, Wu [28] described that QDs covalently linked to immunoglobulin G and streptavidin could effectively label the breast cancer marker Her2 on the surface of fixed and living cancer cells for cellular imaging. In spite of successful cellular labeling in vitro, there are few examples of the use of QDs for in vivo imaging of tumors in live animals. In a report by Chen [29], commercially available QDs conjugated with RGD (arginine-glycine-aspartic acid) were shown to specifically bind to an αvβ3 integrin positive glioblastoma to enable in vivo optical imaging. In another report of in vivo imaging by Nie [30], QD probes coated with an ABC triblock amphiphilic copolymer and tumor targeting ligands were shown to accumulate in tumors because of enhanced vascular permeability and antibody binding to cancer-specific cell surface biomarkers. The authors pointed out that the strong hydrophobic interactions between the hydrophobic segment of the coating polymer and the trioctylphosphine oxide (TOPO) provide a protective hydrophobic coating layer that resists hydrolysis and enzymatic degradation of QDs even under complex in vivo conditions.
One key for successful in vivo imaging is that QDs should be able to withstand degradation by avoiding reaction with biomolecules under hostile physiological microenvironment. In general, surface coating of QDs is an efficient way to increase their stability, water solubility and biocompatibility by reducing toxicity [31]. Based on the TOPO coated QDs created by Bawendi and Murray [32], numerous techniques for surface coating have been developed. These methods can be generally classified as: encapsulation with silica shell, ligand exchange reaction with mercapto compounds, and encapsulation through hydrophobic interaction.
Encapsulation of QDs within silica shells is currently seldom used because of the relatively complicated procedure and the particularly large size increase of resulting QDs [10,33,34]. In contrast, coating of QDs with amphiphilic molecules through the hydrophobic interaction between TOPO and the polymers appears to be a promising approach for stabilizing QDs [12,35,36]. Its obvious advantage is the preservation of the native surface structure as well as the original quantum efficiency of QDs. This hydrophobic shell protects QDs against hydrolysis and enzymatic degradation [30]. Although these QDs are stable under usual chemical conditions, i.e. in water or buffer, they might not have sufficient biological stability, especially under the complex in vivo conditions in living systems. One major problem could arise from the surface exchange reaction between thiols from proteins and TOPO on the surface of QDs. There are ample mercapto groups in certain proteins, which can replace the TOPO on the surface of QDs. Such ligand exchange reactions may lead to the degradation of QDs under complex in vivo imaging conditions, resulting in the release of highly toxic Cd2+ ions. On the other hand, the direct binding of proteins to QDs may result in the alteration of the chemical conformation of proteins and loss of their bioactivities, as the mercapto and disulfide groups usually play critical roles in the conformation of proteins [37].
In order to develop physiologically stable QDs for in vivo imaging studies in live animals, coating of QDs with mercapto groups instead of TOPO may be an alternative strategy. Indeed, replacement of TOPO with mercapto compounds containing carboxylic acid or amino groups has previously been used as the representative ligand exchange reaction to prepare water-soluble QDs [11,38,39]. However, one of the major obstacles for this strategy is the insufficient stability of the resulting QDs due to the oxidation of the low molecular weight mono-thiols into disulfides. Recent studies show that the stability and resistance of QDs to chemical degradation can be improved through modification with bidentate ligand [40,41]. Additionally, multidentate ligands based on polyamines, such as polyethyleneimine or poly(2-N,N-dimethylaminoethyl methacrylate), have been used to coat and improve the stability of QDs [42–47].
Therefore, in this study, we took advantage of mercapto groups, multidentate effect and hydrophobic shell to design a novel triblock copolymer for surface coating of quantum dots (CdSe-ZnS). The triblock copolymer, synthesized by successive atom transfer radical polymerization (ATRP), consists of a polycarboxylic acid at one end, a polythiol block at the other end and an intermediate poly(styrene/divinylbenezene) block, which is densely compacted and strongly hydrophobic. The polythiol block is derived from bis[2-(2-bromoisobutyryloxy)ethyl] disulfide initiators. The multiple mercapto groups of the polythiol block serve as multidentate ligands not only to bind QDs but also to prevent the exchange reaction between TOPO on the surface of QDs and biomolecules such as proteins in living systems. The polycarboxylic acid block, derived from t-butyl acrylate, will improve the water solubility of QDs and provide reaction sites for bioconjugation. In addition to the multidentate effect, divinylbenzene monomers were copolymerized with styrene to form a cross-linked hydrophobic segment separating the polycarboxylic block from the polythiol block. The densely compacted hydrophobic shell formed from the cross-linked poly(styrene/divinylbenzene) block will prohibit the diffusion of other molecules or ions through the hydrophobic shell so as to protect QDs from hydrolytic and enzymatic degradation.
2. Experimental Section
2.1 Materials
The monomers for successive ATRP polymerization, including styrene (ST, 99+%, Sigma-Aldrich), divinylbenzene (DVB, 80%, Polysciences) and t-butyl acrylate (tBA, 99%, Aldrich) were purified by passing through short columns filled with basic alumina to remove radical inhibitors prior to use. CuBr (98%, Acros) was purified by washing repeatedly with glacial acetic acid followed by ether, and then stored under argon after drying. All other reagents, including 2-mercaptoethanol (Sigma), hydrogen peroxide (30% aqueous solution, Fisher), triethylamine (99%, Fisher), 2-bromoisobutyryl bromide (98%, Aldrich) and 2,2′-bispyridine (98+%, Acros) were used without further purification.
All chemicals for preparation of QDs, including cadmium oxide (99.998%, Alfa Aesar), stearic acid (99%, Alfa Aesar), tri-n-octylphosphine oxide (TOPO, 90%, Aldrich), 1-hexadecylamine (90%, Alfa Aesar), trioctylphosphine (TOP, 90%, Aldrich), dimethylzinc (1.2 mol/L solution in toluene, Acros), hexamethyldisilathiane (Fluka) were used as received.
2.2 Synthesis of disulfide (2)
Hydrogen peroxide (8.5 g of 30% aqueous solution, 75 mmol) was diluted with 10 mL of water. The diluted hydrogen peroxide was added dropwise into a solution of 2-mercaptoethanol (7.8 g, 100 mmol) in 25 mL of water. After 1 h of stirring at room temperature, the reaction mixture was extracted with 50 mL ethyl acetate three times. The ethyl acetate extract was dried over anhydrous sodium sulfate. After removal of sodium sulfate by filtering, the organic solvent was evaporated off under vacuum to obtain the disulfide 2 (figure 1) as a viscous oil in 92% yield. 1H NMR (CDCl3; δ, ppm): 2.56 (t, 2H, CH2S), 3.58 (t, 2H, CH2O). The scheme for chemical synthesis is shown in figure 1.
Figure 1.

Synthesis of disulfide initiator 4 and successive ATRP (atom transfer radical polymerization) to generate cross-linked multidentate triblock copolymer 6.
2.3 Synthesis of disulfide initiator (4)
The initiator 4 (figure 1) was prepared by a modified literature method [48,49]. Disulfide 2 (1.54 g, 10 mmol) and triethylamine (3.03 g, 30 mmol) were dissolved in dry THF (30 mL). After bubbling argon through the reaction mixture for 30 min, a solution of 2-bromoisobutyryl bromide 3 (9 g, 30 mmol in 10 mL of dry THF) was added under vigorous stirring within an ice bath. The reaction mixture was kept stirred in the ice bath for 10 min and then at room temperature for 2 h. 200 mL of water were added to quench the reaction. The aqueous solution was extracted with three portions of 50 mL ethyl acetate. The combined organic solvent was thoroughly washed first with aqueous sodium bicarbonate solution (1%, 50 mL) and then with water (50mL), respectively. The organic extract was dried over anhydrous sodium sulfate. Sodium sulfate was removed by filtering and then ethyl acetate was evaporated by using a rotary evaporator to obtain crude product 4 (viscous brown oil, 89% yield). A light yellow oil was obtained after further purification by column chromatography (silica gel 60, dichloromethane as eluent). 1H NMR (CDCl3; δ, ppm): 4.44 (t, 2H, CH2OOC), 2.98 (t, 2H, CH2S) and 1.94 (s, 6H, (CH3)2CBr).
2.4 Preparation of the cross-linked multidentate triblock copolymer (5) by successive ATRP from ST, DVB and tBA
Polymerization of ST and DVB was initiated using the disulfide initiator 4 (figure 1). Prior to polymerization experiments, the initiator was deoxygenated by bubbling with argon for 3 h. The effect of varying molar ratio of monomers, concentration, reaction time and temperature on polymerization as well as molecular weight was examined (table 1). Typically, 650 mg of ST (6.25 mmol) and 406 mg of DVB (3.125 mmol) were added into 2 mL of diphenyl ether. The mixture was degassed by 4 freeze-pump-thaw cycles. 71.5 mg of CuBr (0.5 mmol) and 156.2 mg of 2,2′-bispyridine (1.0 mmol) were added into the frozen mixture. The flask was backfilled with argon and the mixture was warmed to room temperature. Then, 113 mg of the degassed disulfide initiator 4 (0.25 mmol, molar ratio of 1/25 vs ST monomer) was injected through a syringe. The mixture was immediately heated to 65-75 °C. 100 μL of sample were taken out every 30 min and the polymeric product was precipitated by addition of 1 mL methanol to monitor the polymerization.
Table 1.
Experimental conditions, feeding ratio of reactants, and results for polymerization of styrene (ST), divinylbenzene (DVB), t-butyl acrylate (tBA) by ATRP.
| No. of Experiment | Molar ratio of I/ST/DVB/tBA | Temperature | Volume of solvent (mL) | Results of polymerization |
|---|---|---|---|---|
| 1 | 1 : 25 : 125 : 0 | 90 °C | 2 mL | Gelation |
| 2 | 1 : 25 : 125 : 0 | 70 °C | 2 mL | Controlled |
| 3 | 1 : 25 : 125 : 0 | 60 °C | 2 mL | No polymer |
| 5 | 1 : 25 : 125 : 0 | 70 °C | 4 mL | Controlled |
| 6 | 1 : 25 : 125 : 0 | 70 °C | 0.5 mL | Gelation |
| 7 | 1 :25 : 0 : 0 | 70 °C | 2 mL | Controlled |
| 8 | 1 : 0 : 25 : 0 | 70 °C | 2 mL | Gelation |
| 9 | 1 : 25 : 25 : 0 | 70 °C | 2 mL | Gelation |
| 10 | 1 : 125 :25 : 0 | 70 °C | 2 mL | Gelation |
| 11 | 1 : 25 : 125: 125 | 70 °C | 2 mL | Controlled |
a) The feeding amount of disulfide initiator (I), CuBr or 2,2′-bispyridine is fixed at 0.25, 0.5 and 1.0 mmol, respectively;
b) The meaning of ‘controlled’ is that the amount or molecular weight of polymer can be controlled by the temperature, feeding ratio or time of polymerization.
After the polymerization of ST and DVB had progressed for 2.5 h, 400 mg of tBA (3.125 mmol) was added immediately. The reaction continued for another 1 h to allow polymerization of tBA to proceed. The cooled mixture was purified by reprecipitation with methanol/dichloromethane. Briefly, 1 mL of the reaction mixture was added into 5 mL of methanol and then centrifuged at 8000 rpm for 5 min. After washing with 1 mL methanol three times, the precipitated polymer was redissolved in 0.25 mL dichloromethane, and the resulting suspension was centrifuged at 8000 rpm for 5 min to remove the insoluble copper catalyst. The polymer was reprecipitated by addition of 1 mL of methanol and separated by centrifugation. The procedures were repeated three times to thoroughly remove the unpolymerized monomers and insoluble catalyst. The triblock copolymer 5 (figure 1) was then dried overnight under vacuum at room temperature. 1HNMR (CDCI3; δ, ppm): 6-8 (br), 3-4 (br) and 1-2 (br). Molecular weight distribution of polymer was determined on the basis of gel permeation chromatography (GPC).
2.5 Reductive cleavage of disulfide bonds within polymer 5 into mercapto groups
100 mg of block copolymer 5 (figure 1) was dissolved in 1 mL of DMF followed by addition of 2-mercaptoethanol (0.1 mL). After stirring overnight under argon protection, the mixture was poured into 10 mL of water, and then extracted three times with 20 mL of dichloromethane. After evaporation of organic solvent, the polymer 6 (figure 1) was reprecipitated three times using dichloromethane-methanol mixture to completely remove the residual 2-mercaptoethanol, and kept in argon before ligand exchange with TOPO/QDs.
2.6 Synthesis of TOPO stabilized quantum dot nanocrystals (TOPO/QDs)
TOPO/QDs in this paper represent CdSe-ZnS core-shell nanocrystals stabilized with TOPO. TOPO/QDs were synthesized according to the method described in the literature [30]. Cadmium oxide (25.6 mg, 0.2 mmol), stearic acid (0.5 g) and TOPO (2.0 g) were mixed and flushed with argon flow. Then, the mixture was heated to 250 °C until a clear solution was formed. Two grams of hexadecylamine were added to the clear solution after it had cooled down to room temperature. The mixture was heated back to 250 °C for 10 min and then raised to 300 °C. Selenium (15.8 mg, 0.2 mmol) in 2 mL of trioctyl phosphine was quickly injected into the hot solution. The mixture immediately changed colour to orange red, indicating formation of quantum dots. The reaction mixture was refluxed for 30 min, and then cooled down to 220 °C. Dimethylzinc (83.3 μL of 1.2 mol/L in toluene, 0.1 mmol) and hexamethyldisilathiane (17.8 mg, 0.1 mmol) in 5 mL of trioctyl phosphine were slowly added dropwise (over 10 min) to the reaction mixture at 220 °C. The mixture was refluxed for 30 min and then cooled to room temperature. The TOPO/QDs were extracted with toluene or hexane and reprecipitated with methanol for 5 times. Size selective precipitation was performed using methanol and hexane. The TOPO/QDs were redispersed in toluene or chloroform for measuring dynamic light scattering (DLS) and recording UV-vis and fluorescent spectra.
2.7 Surface ligand exchange of TOPO/QDs with multimercapto polymers and formation of water-soluble QDs
5 mg of the multimercapto triblock copolymer 6 (figure 1) were dissolved in 1 mL of toluene. 2.0 mg of TOPO/QDs in toluene (1 mL) were added into the polymer solution under vigorous stirring in an argon atmosphere. The mixture was continuously stirred at room temperature for 2 hours under argon. 5 mL of methanol were added into the above mixture and the resulting precipitated polymer stabilized QDs were collected by centrifugation at 8000 rpm. The polymer stabilized QDs were redispersed in toluene and precipitated again by addition of methanol in order to remove the dissociated TOPO and large excess of polymers.
Hydrolysis of the acrylate groups on the surface of QDs into carboxylic acid groups was carried out in a toluene-H2O-NaOH heterogeneous system. The above polymer coated QDs were dispersed in 1 mL of toluene and mixed with 1 mL of 1% NaOH aqueous solution. After the heterogeneous mixture was vigorously stirred for 30 min under argon, the QDs were partitioned into the aqueous phase, which turned brown due to the formation of water-soluble QDs. The water-soluble QDs were separated from toluene using a separatory funnel and kept at pH 8 at 4 °C.
2.8 Characterization
1H NMR spectra were recorded on a 400 MHz Bruker instrument. UV-vis spectra were recorded on a Cary 5000 spectrometer using a 1.0 cm quartz cuvette. Emission spectra were measured on a Hitachi F-7000 fluorescent spectrometer using a 1.0 cm quartz cuvette. The molecular weights of polymers were determined using a PL-GPC 50 gel permeation chromatograph (Polymer Laboratories) using THF as eluent and toluene as the internal standard (flow rate of 1.0 mL/min and differential refractive index (RI) detector). The apparent molecular weights and polydispersity were determined with a calibration based on polystyrene standards. Fourier transformed infrared spectra (FTIR) were collected on polymer/KBr pellets from 4000 to 650 cm-1, using a Magna-IR 550 Spectrometer Series II (Nicolet). The polymer was mixed with 70 mg of spectroscopic grade KBr at a 1% (w/w) and ground into fine powder. The pellets (7 mm diameter) were pressed with a ThermobSpectra-Tech's Qwik Handi-Press for 5 min. Transmission electron micrographs (TEM) were taken by a JEOL 3010 HREM operating at 100 kV. Sample for TEM was prepared by depositing a drop of suspension of QDs onto carbon-coated Cu grids followed with drying at room temperature.
3. Results and Discussion
3.1 Preparation of the multidentate triblock copolymer by successive atom transfer radical polymerization (ATRP)
The multidentate triblock copolymer 6 (figure 1) was designed for surface coating of QDs. The first block containing multiple thiols was designed as the multidentate ligand for surface coating of QDs. Recent studies show that, in comparison to low molecular weight monothiol, bidentate thiol can effectively improve the stability and resistance of QDs to chemical degradation [40,41]. Consequently, it is expected that multidentate thiols may lead to greater stability of QDs. Through the chelating effect, the multidentate mercapto groups will bind strongly to the surface of QDs, resulting in greater stability of QDs [40]. In addition, because this block is a mercapto group based stabilizer, it will reduce the possibility of the exchange reaction between TOPO on QDs and biomolecules such as proteins containing thiol groups. The second block is a cross-linked hydrophobic block which will form a densely compacted hydrophobic shell around QDs. This hydrophobic shell will prohibit the diffusion of other water-soluble molecules or ions within living systems into QDs core, so as to protect QDs from hydrolytic and enzymatic degradation. The third block consists of multiple t-butyl acrylates which can be hydrolyzed into carboxylic acid groups. This polycarboxylic block will improve the water solubility and biocompatibility of QDs, and provide reaction sites for subsequent bioconjugation for targeted delivery [30].
The triblock copolymer was prepared by successive atom transfer radical polymerization (ATRP) (figure 1). The first block, the multimercapto block, was generated from an initiator, disulfide 4, which can be cleaved into thiols for QDs coating. The reason to use disulfide 4, instead of thiol 1 as the initiator is because an unwanted chain transfer radical polymerization from thiol will occur if the initiator contains a mercapto group [50]. The disulfide initiator 4 was synthesized from 2,2′-dihydroxyethyl disulfide 2 and 2-bromoisobutyryl bromide 3. The bromoisobutyryl group within the resulting disulfide initiator decomposed in the presence of CuBr/bispyridine to generate free radicals to initiate the successive ATRP with ST, DVB, tBA to generate the triblock copolymer. Finally, reductive cleavage of the disulfide bonds in the first block generated the multi-dentate merapto groups, which are the binding groups to stabilize QDs.
The second block, a cross-linked hydrophobic block, was polymerized from ST and DVB. There are three reasons to select these two monomers. First, both ST and DVB are hydrophobic monomers, which can protect the core of QDs by preventing the diffusion of biological molecules and ions in physiological milieu (usually hydrophilic) into the polymeric shell. Second, the polymer block from these monomers tend to form a densely compacted structure through the strong π-π interaction between the benzene moieties within ST and DVB. Third, the densely compacted hydrophobic polymeric shell will be further strengthened as a result of cross-linking of the ethylene groups of DVB during polymerization. Recent studies indicate that such cross-linking of polymer can protect QDs core. The study of poly(maleic anhydride-alt-tetradecene) indicated that the stability was increased by cross-linking of the polymers [35]. Another report used lysines as linkers to generate such a cross-linked shell [51,52]. In comparison to the amide groups from lysines, the block copolymerized from ST and DVB within our triblock copolymer will have greater hydrophobicity, thus it is expected that this polymer has greater capability to prevent the diffusion of biological molecules and ions into the core.
The third block containing multiple carboxylic acids makes QDs water-soluble and biocompatible, while furnishing reaction sites for pegylation or conjugation with tumor-targeting ligands. Direct polymerization of acrylic acids is not suitable for ATRP, because the acid groups can coordinate with the copper ions and poison the catalysts. In addition, bispyridine, the complex ligand for CuBr catalyst, can be protonated by carboxylic acid, resulting in decomplexation of copper bipyridyl complex catalyst. An alternative approach to prepare polymethacrylic acids by ATRP is to polymerize the protected monomers such as t-butyl methacrylate, and benzyl methacrylate followed by hydrolysis [53]. In the present approach, the carboxylic block was generated from tBA, which were subsequently hydrolyzed to carboxylic acids.
The polymerization of the monomers ST or DVB, individually and in combination, was examined in a number of solvents, such as acetone, toluene and diphenyl ether. The progress of polymerization was primarily monitored by the precipitation of the polymer from reaction mixture with methanol. Polymerization was not observed even after being refluxed in acetone overnight, probably due to the lower boiling point of acetone. Similar result was found from the solvent of toluene when the temperature is below 60 °C (experiment 3 in table 1). However, polymerization of ST and DVB occurred at temperatures above 65° C in toluene or diphenyl ether as solvents. Temperature plays a critical role in the progress of ATRP and control of the molecular weights of polymers because high temperature leads to a faster polymerization. Linear polyST homopolymer as well as its block copolymer with poly(methyl acrylate) containing mercapto groups have been reported in the literature [48,49]. In ref [48], polymerization of ST initiated by a similar disulfide initiator at 90 °C led to a polymer with molecular weight around 1.5×104 g/mol. Our first experiment carried out at 90°C immediately resulted in a gelated structure within only 20 mins at the molar ratio of 1:25:12.5 (initiator:ST:DVB) (experiment 1 in table 1). This suggests that the polymerization rate is too fast at such a high temperature to control the degree of cross-linking.
Therefore, in order to prepare polymers with relatively low and controllable molecular weights, copolymerization of ST and DVB was carried out at a reaction temperature below 90 °C. However, if the temperature is below 60 °C, no obvious polymerization occurred even though the reaction mixture was stirred overnight. Eventually, the temperature between 65–75 °C was selected for copolymerization of ST and DVB for controllable molecular weight.
The feeding ratio of monomer is central to controlling molecular weight and the degree of cross-linking. Polymerization from single monomer and co-monomers at variable feeding ratio is shown in table 1. The result of polymerization and the degree of cross-linking can be primarily observed by eye on the experimental phenomenon and indicated by the solubility of the resulting polymer within variable solvents, e.g. THF or dichloromethane. The homopolymer of polyST generated after 2.5 hours at 65 - 70 °C in diphenyl ether was completely soluble in both THF and dichloromethane (experiment 7 in table 1). However, the homopolymer from DVB alone (polyDVB) was completely insoluble in either THF or dichloromethane (experiment 8 in table 1). The insolubility and swelling property of polyDVB in THF and dichloromethane implies its highly cross-linked structure.
The combination of ST and DVB will lead to copolymers with cross-linked structures, but with a relatively low degree of cross-linking. In addition, the degree of cross-linking and the molecular weights of the copolymers, which can be indicated by its solubility, can be controlled by the molar ratio of ST and DVB (DVB/ST). As expected, the solubility of poly(ST-co-DVB) copolymer is intermediate between that of polyST and poly DVB. If the molar ratio of DVB/ST was increased, then the solubility of copolymer decreased, indicating the increase in degree of cross-linking. When the feeding ratio of ST and DVB is 2:1, 2.0 mg of poly(ST-co-DVB) is completely soluble in 200 μL of dichloromethane and THF completely (experiment 2 in table 1). When the molar ratio increases to 1:1, the solubility in THF decreases sharply, but the polymer can still be dissolved in dichloromethane (0.8 mg insoluble residual from the original 2.0 mg of copolymer, experiment 9 in table 1). If the molar ratio increases to 1:2 (ST/DVB), 2 mg of copolymer is hardly soluble in THF and dichloromethane, which is similar to polyDVB (experiment 10 in table 1).
Quantitatively, GPC measurements on the prepared polymers are in agreement with their solubility characteristics in THF or dichloromethane. When the same amount (2.0 mg) of the polymers was used for GPC analysis, the GPC profiles of polyST and polyDVB were quite different (figure 2). PolyST shows a longer elution time (i.e., larger elution volume) indicating a lower molecular weight (1730 g/mol with a polydispersity of 1.60, line A in figure 2). The GPC profile of polyDVB consists of an almost flat elution pattern with very weak intensity, except for a small peak around elution volume of 17 mL (line B in figure 2). The small peak may be due to the residual monomer, rather than small molecular weight polymeric species. The weak intensity indicates that hardly any soluble polymer can be detected. This supports the insolubility of polyDVB in THF, indicating a gelated structure with high degree of cross-linking.
Figure 2.

GPC (gel permeation chromatography) profiles of homopolymers or copolymer from monomers: styrene (ST) and divinylbenzene (DVB). All polymerizations were carried out for 2.5 hours at 65°C. THF was used as the eluting solvent at a flow rate of 1.0 mL/min and toluene was used as an internal standard with refractive index detection. The instrument was calibrated using standard polystyrene samples.
Line C in figure 2 shows a GPC profile of the copolymer (poly(ST-co-DVB)) from the mixed monomers in a molar ratio of 2/1 (ST/DVB). It shows a relatively low elution volume and an intermediate molecular weight of 3470 g/mol. The molecular weight is greater than that of polyST but is certainly lower than that of polyDVB, the highly cross-linked gelated structure. By adjusting the molar ratio of monomer ST and DVB, the degree of cross-linking and molecular weight of polymer can be tuned.
Unlike the feeding ratio of monomers, the effect of monomer concentration at a fixed molar ratio of ST/DVB to the molecular weight was not observed. In order to examine the concentration effect, an experiment using a fixed molar ratio of disulfide initiator, ST and DVB (initiator/ST/DVB = 1:25:12.5) was conducted using different volumes of solvent. When the solvent volume was taken as 2 mL and 4 mL for the same amount of monomers (experiments 2 and 5 in table 1), the resulting polymers possessed similar molecular weights shown by GPC (data not shown). However, the yield of polymer from the low concentration is less than that from high concentration at the same time of polymerization. This implies that the polymer molecular weight was probably independent of the concentration at the tested range. However, if the solvent is as little as 0.5 mL, monomer concentration is so high as a bulk polymerization that only gelated product with a very high molecular weight was formed (experiment 6 in table 1).
Time dependent polymerization showed a gradual increase in the amount of polymer, but did not demonstrate an obvious molecular weight change during a 4 hour period. The time dependent polymerization was examined at a constant molar ratio of the initiator to monomers (initiator/ST/DVB = 1/25/12.5) between 65–75 °C. In order to monitor the progress of polymerization, a sample (0.1 mL) was taken out every 30 minute during a polymerization period of 4 hours. Then, 1.0 mL of methanol was added to each sample to precipitate the polymer for GPC measurements. In the first 30 min, hardly any polymer could be precipitated from methanol. With increasing reaction time, more polymers were precipitated (i.e. 0.5 mg, 1.8 mg, 4.2 mg from the samples polymerized for 1 h, 1.5 h and 2.5 h, respectively). This indicates the continuous polymerization over time. The gradual increase in the amount of polymer indicates that the polymerization process can be well controlled by the polymerization time within such a temperature range. However, GPC measurements showed no obvious effect of reaction time on molecular weight of polymer. The molecular weights of poly(ST-co-DVB) sampled at different times are close to each other. For example, the molecular weights of poly(ST-co-DVB)s from polymerization reactions after 1 h and 2.5 h are approximately 3470 g/mol (lines A and B in figure 3). This is in agreement with the chain reaction mechanism of addition polymerization of unsaturated monomers which leads to formation of products with invariable molecular weight [54]. Primary activation of a monomer is followed by the addition of other monomers in rapid succession until the growing chain is eventually deactivated under our experimental conditions.
Figure 3.

GPC profiles of copolymers showing the effect of polymerization time and monomers on molecular weights. Polymerization was conducted at 65 °C. Styrene (ST) and divinylbenzene (DVB) were copolymerized from the beginning, while t-butyl acrylate (tBA) was added at 2.5 h of polymerization to form the block copolymer. GPC condition is the same as that in Figure 2.
Once the cross-linked polymer of poly (ST-co-DVB) was formed, poly (t-butyl acrylate) (polytBA) block can successively grow to generate the triblock copolymer. The successive growth of polytBA is evidenced by GPC measurement. Although there is no obvious increase of molecular weight of poly(ST-co-DVB) with polymerization time, the molecular weight increased immediately to 4921 g/mol after addition of tBA (lines C and D in figure 3). The GPC elution peak around 3470 g/mol (poly(ST-co-DVB)) disappeared when tBA was added. The control experiment without addition of tBA still kept the molecular weight of polymer around 3470 g/mol of poly(ST-co-DVB). This confirms that polytBA block was growing from the existing poly(ST-co-DVB) macroinitiator to form the final block copolymer: poly (ST-co-DVB-b-tBA).
Fourier transformed infrared spectroscopy (FTIR) confirmed the formation of the block copolymer. Figure 4 is the FTIR spectrum of the prepared polymer before and after addition of tBA. In both spectra, the strong peaks at 3025 and 2927 cm-1 are attributed the stretch of CH2. The peaks at 1600, 1493 and 1454 cm-1 from both spectra are the typical characteristics of benzene within styrene and DVB. This is in agreement with the previous conclusion that ST has been cross-linked with DVB (figure 2). Significantly, in addition to the major peaks from ST and DVB, the spectrum of polymer after addition of tBA shows additional two strong peaks at 1728 cm-1 (C=O) and 1151 cm-1 (C-O from ester). Because the residual tBA monomers have been completely removed by reprecipitation with methanol/dichloromethane, the co-existence of t-butylacrylate and styrene in the FTIR confirms the formation of the triblock copolymer, poly(ST-co-DVB-b-tBA).
Figure 4.
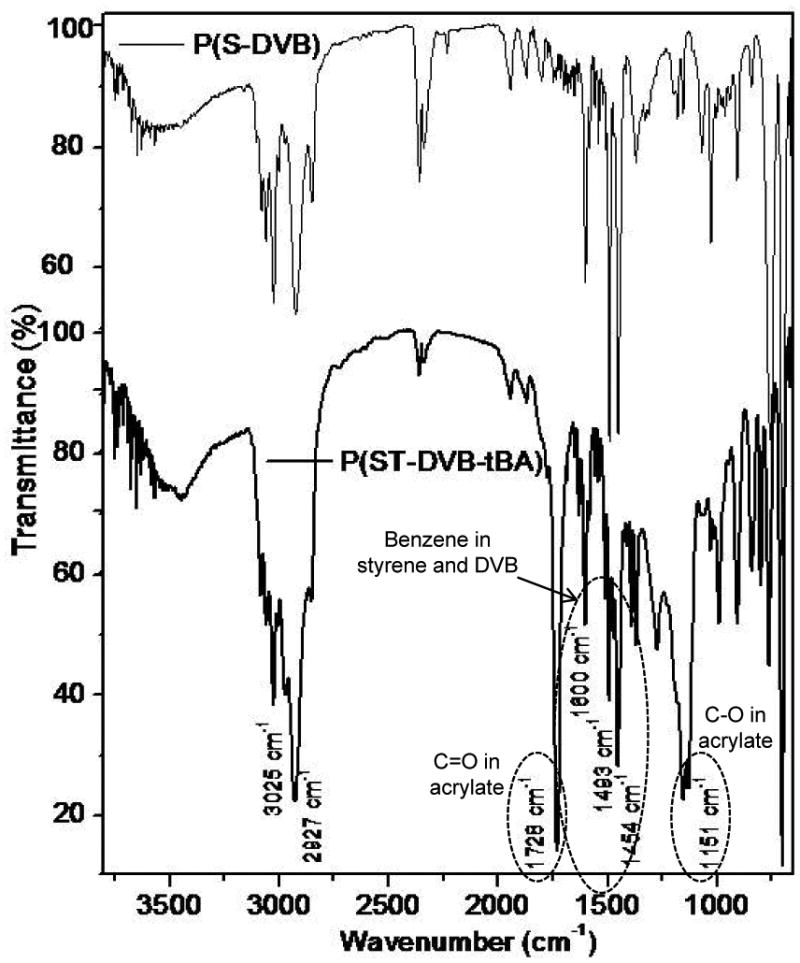
FTIR spectra of polymer from styrene and divinylbenezene before (spectrum of P(ST-co-DVB)) and after (spectrum of P(ST-co-DVB-b-tBA)) addition of t-butyl acrylate. The co-existing of the vibration peak of benzene (1600, 1493 and 1454 cm-1) and ester (1728 and 1151 cm-1) indicates the formation of the block copolymer, P(ST-co-DVB-b-tBA).
Conclusively, polymerization of ST and DVB can be initiated by the newly prepared disulfide initiator 4. The temperature and molar ratio of monomers are critical to control the cross linking degree and molecular weight of copolymer. tBA block can grow from the poly(ST-co-DVB) macroinitiator to generate the block copolymer poly(ST-co-DVB-b-tBA). Disulfide in the first block will be cleaved into mercapto groups for surface coating of QDs. The intermediate ST/DVB block will form a cross-linked hydrophobic shell to protect QDs. The tBA within the third block will be hydrolyzed into carboxylic acid so as to make QDs water-soluble and offer reaction site for further conjugation with targeting delivery ligand.
3.2 Surface coating of QDs with multidentate block copolymers
TOPO stabilized CdSe-ZnS QDs (TOPO/QDs) were synthesized for ligand exchange with multimercapto copolymers. The TOPO/QDs were synthesized using cadmium oxide and selenium as reactants and dimethyl zinc and hexamethyldisilathiane as ZnS coating precursors according to the method described in the literature [30]. Fractionation by centrifugation was followed by size selective precipitation from toluene or hexane solution by addition of methanol, yielding QDs with size distribution within a narrow range. Transmission electron microscopy (TEM) measurement indicates that these QDs are nearly spherical particles at an average diameter of 4.5 nm together with some rodlike structures at about 6.8 nm length.
The optical properties of QDs were confirmed by fluorescent emission and UV-vis absorption spectra (figure 5). The broad absorption around 500 nm in UV-vis spectrum is a typical characteristic of QDs. The maximum absorption around 500 nm in UV-vis spectrum and maximum fluorescent emission at 617 nm in fluorescent spectrum are in agreement with earlier studies [41].
Figure 5.

Absorption spectra and photoluminescence spectra of TOPO/QDs in toluene.
The above TOPO/QDs are soluble in non-polar solvent such as toluene or chloroform but are insoluble in methanol or water (sample A in figure 6). For biological application, additional surface coating is necessary to improve the water solubility and biocompatibility of QDs.
Figure 6.
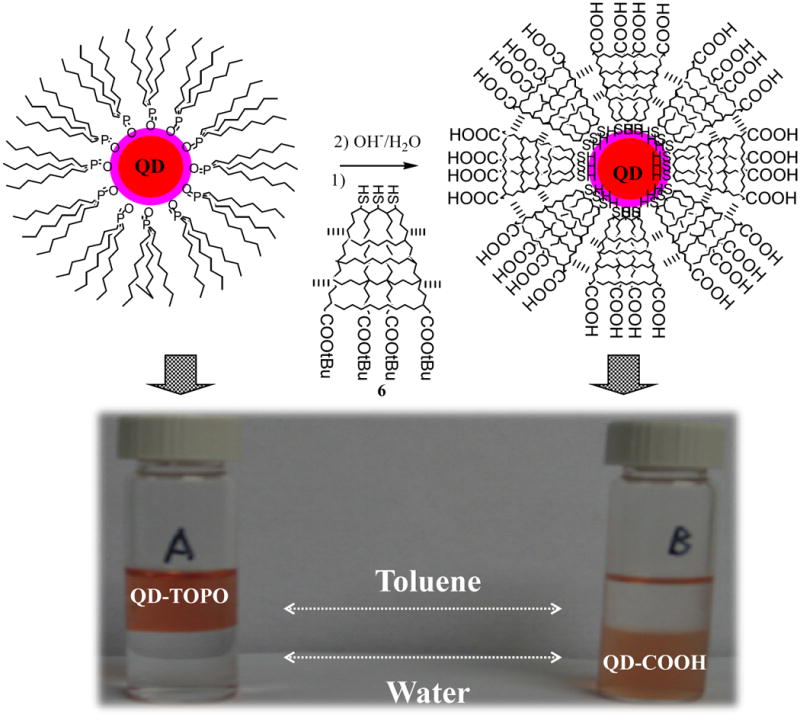
Scheme of ligand exchange reaction between multidentate block copolymers and TOPO/QDs and transferring QDs from organic phase to aqueous phase. (A) A control experiment of TOPO/QDs before ligand exchange reaction and (B) The toluene-water system after ligand exchange reaction, showing QDs in the aqueous phase.
The new triblock copolymer containing multidentate mercapto groups can effectively coat QDs and make them water-soluble. Water-soluble QDs were prepared by the ligand surface exchange reaction between polymer and TOPO/QDs in toluene followed by hydrolysis of t-butyl acrylates into carboxylic acids [40].
Disulfides bind poorly with QDs, whereas thiols bind well with QDs. Therefore, the multiple disulfides within polymers need to be cleaved into thiols for strong binding to QDs. The disulfide bond can be reductively cleaved into thiols either by free thiol (e.g., dithiothreitol or 2-mercaptoethanol) or by phosphines (e.g., Bu3P) [55]. In this research, the disulfides were cleaved using 2-mercaptoethanol in DMF according to the published method [48]. Reduction was carried out under argon atmosphere in order to avoid air oxidation of the thiol groups within polymers and 2-mercaptoethanol.
It is well-known that TOPO on QDs can be easily replaced by mercapto groups [11,38,39]. Through the multidentate mercapto groups, the new triblock copolymer will replace TOPO and link with QDs. A large excess of polymers were used for ligand exchange reaction to avoid cross-linking between QDs. Argon atmosphere is critical to prevent oxidation of the multiple mercapto groups during ligand exchange reaction. Because the hydrophobicity of tBA, the resulted polymer coated QDs are soluble in chloroform or toluene. Following hydrolysis, the QDs were completely water soluble implying the formation of carboxylic acids on the surface of QDs.
Hydrolysis of tBA into carboxylic acid was conducted in a heterogeneous mixture of diluted NaOH (1%) aqueous solution and polymer coated QDs in toluene in an argon atmosphere. The progress of hydrolysis was monitored by the solubility of the resulting QDs in water. After hydrolysis for about half an hour, the QDs were completely transferred into aqueous phase, indicating by the brown colour of aqueous phase (sample B in figure 6). The formation of water-soluble QDs confirms the generation of carboxylic acid from tBA.
Therefore, the new triblock copolymer can be successfully used as surface coating material to prepare water-soluble QDs. The TOPO ligand on the surface of TOPO/QDs can be replaced with the new multimercapto groups. By hydrolyzing the carboxylate groups, QDs can be transferred into aqueous phase. In addition to improve the water solubility, the carboxylic acids on QDs can also be used as functional groups to conjugate with biomolecules (e.g. protein or antibodies) for targeted imaging. A drawback of such aqueous QDs suspensions is their low stability in water, shown by the aggregation after 1 hour at room temperature. Possible reasons might be the easy oxidation of mercapto groups or/and the over-cross-linking of the multi-dentate polymer with too high molecular weight. Further research will be focused on improvement of the chemical stability of such aqueous QDs through disulfide stabilizing groups and/or molecular weight control of polymer. Conjugation of QDs with biomolecules for cellular labeling and in vivo imaging studies in live animals will also be explored.
4. Conclusions
A new kind of polymer was successfully synthesized through successive atom transfer radical polymerization from disulfide initiator and three different monomers: styrene, divinylbenzene and t-butyl acrylate. This polymer – designed for surface coating of QD nanocrystals – is composed of three different blocks: polythiol, poly(styrene-co-divinylbenzene) and polycarboxylic acid. The multiple mercapto groups of the polythiol block act as multidentate ligands to stabilize QDs. The polycarboxylic acid block generated from poly (t-butyl acrylate) not only makes QDs water-soluble but also offers reaction sites for further bioconjugation of QDs for targeted molecular imaging. The intermediate poly(styrene-co-divinylbenzene) block is a hydrophobic block formed from cross-linked poly(styrene-co-divinylbenzene). The cross-linking degree of this block can be controlled through reaction time, monomer molar ratio and reaction temperature. This block will form a densely compacted hydrophobic shell during coating of QDs and will effectively prevent the diffusion of other molecules into the QDs core, so as to better protect the QD cores against decomposition.
By using this triblock copolymer, QDs can be transferred from toluene to water efficiently. Because the multidentate mercapto groups instead of TOPO are the functional groups to bind with QDs, such QDs will be less likely to be displaced by the mercapto or disulfide groups within proteins or other biomolecules. An existing problem of the prepared QDs is the insufficient stability, which might be due to the oxidation the mercapto groups. With the improvement of the stability of QDs in the future study, this research will provide an alternative coating material to produce QDs which could be more suitable for in vivo use under complex physiological conditions than the current TOPO stabilized QDs.
Acknowledgments
This work was supported by DoD USAMRMC W81XWH-05-1-0291, NIH/NCRR/RCMI G12 RR003048 and NIH 5U54CA091409-06. Mr. Haiyong He and Dr. Xingjie Liang are acknowledged for TEM measurements. The authors also thank Prof. Zaver Bhujwalla and Prof. Oladapo Bakare for helpful discussions and support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pomper MG, Lee JS. Current Pharm Design. 2005;11:3247. doi: 10.2174/138161205774424681. [DOI] [PubMed] [Google Scholar]
- 2.Alivisatos AP, Gu W, Larabell C. Annu Rev Biomed Eng. 2005;7:55. doi: 10.1146/annurev.bioeng.7.060804.100432. [DOI] [PubMed] [Google Scholar]
- 3.Smith AM, Gao X, Nie S. Photochem Photobio. 2004;80:377. doi: 10.1562/0031-8655(2004)080<0377:QDNFIV>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 4.Dabbousi BO, Rodriguez-Viejo J, Mikulec FV, Heine JR, Mattoussi H, Ober R, Jensen KF, Bawendi MG. J Phys Chem B. 1997;101:9463. [Google Scholar]
- 5.Leatherdale CA, Woo WK, Mikulec FV, Bawendi MG. J Phys Chem B. 2002;106:7619. [Google Scholar]
- 6.Murphy CJ. Anal Chem. 2002;74:520A. doi: 10.1021/ac022124v. [DOI] [PubMed] [Google Scholar]
- 7.Parak WJ, Gerion D, Pellegrino T, Zanchet D, Micheel C, Williams SC, Boudreau R, Le Gros MA, Larabell CA, Alivisatos AP. Nanotechnology. 2003;14:R15. [Google Scholar]
- 8.Niemeyer CM. Angew Chem Int Ed. 2001;40:4128. doi: 10.1002/1521-3773(20011119)40:22<4128::AID-ANIE4128>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 9.Alivisatos AP. Nature Biotechnol. 2004;22:47. doi: 10.1038/nbt927. [DOI] [PubMed] [Google Scholar]
- 10.Bruchez M, Moronne M, Gin P, Weiss S, Alivisatos AP. Science. 1998;281:2013. doi: 10.1126/science.281.5385.2013. [DOI] [PubMed] [Google Scholar]
- 11.Chan WCW, Nie S. Science. 1998;281:2016. doi: 10.1126/science.281.5385.2016. [DOI] [PubMed] [Google Scholar]
- 12.Dubertret B, Skourides P, Norris DJ, Noireaux V, Brivanlou AH, Libchaber A. Science. 2002;298:1759. doi: 10.1126/science.1077194. [DOI] [PubMed] [Google Scholar]
- 13.Larson DR, Zipfel WR, Williams RM, Clark SW, Bruchez MP, Wise FW, Webb WW. Science. 2003;300:1434. doi: 10.1126/science.1083780. [DOI] [PubMed] [Google Scholar]
- 14.Ishii D, Kinbara K, Ishida Y, Ishii N, Okochi M, Yohda M, Aida T. Nature. 2003;423:628. doi: 10.1038/nature01663. [DOI] [PubMed] [Google Scholar]
- 15.Jaiswal JK, Mattoussi H, Mauro JM, Simon SM. Nat Biotechnol. 2003;21:47. doi: 10.1038/nbt767. [DOI] [PubMed] [Google Scholar]
- 16.Medintz IL, Clapp AR, Mattoussi H, Goldman ER, Fisher B, Mauro JM. Nat Mater. 2003;2:630. doi: 10.1038/nmat961. [DOI] [PubMed] [Google Scholar]
- 17.Dahan M, Levi S, Luccardini C, Rostaing P, Riveau B, Triller A. Science. 2003;302:442. doi: 10.1126/science.1088525. [DOI] [PubMed] [Google Scholar]
- 18.Rosenthal SJ, Tomlinson I, Adkins EM, Schroeter S, Adams S, Swafford L, McBride J, Wang Y, DeFelice LJ, Blakely RD. J Am Chem Soc. 2002;124:4586. doi: 10.1021/ja003486s. [DOI] [PubMed] [Google Scholar]
- 19.Mahtab R, Harden HH, Murphy CJ. J Am Chem Soc. 2000;122:14. [Google Scholar]
- 20.Sun B, Xie W, Yi G, Chen D, Zhou Y, Cheng J. J Immunological Methods. 2001;249:85. doi: 10.1016/s0022-1759(00)00331-8. [DOI] [PubMed] [Google Scholar]
- 21.Pathak S, Choi SK, Arnheim N, Thompson ME. J Am Chem Soc. 2001;123:4103. doi: 10.1021/ja0058334. [DOI] [PubMed] [Google Scholar]
- 22.Klarreich E. Nature. 2001;413:450. doi: 10.1038/35097256. [DOI] [PubMed] [Google Scholar]
- 23.Mitchell P. Nat Biotechnol. 2001;19:1013. doi: 10.1038/nbt1101-1013. [DOI] [PubMed] [Google Scholar]
- 24.Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS, Weiss S. Science. 2005;307:538. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim S, Lim YT, Soltesz EG, De Grand AM, Lee J, Nakayama A, Parker JA, Mihaljevic T, Laurence RG, Cohn LH, Bawendi MG, Frangioni JV. Nat Biotechnol. 2004;22:93. doi: 10.1038/nbt920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaiswal JK, Simon SM. Trends Cell Biol. 2004;14:497. doi: 10.1016/j.tcb.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 27.Medintz IL, Uyeda HT, Goldman ER, Mattoussi H. Nat Mater. 2005;4:435. doi: 10.1038/nmat1390. [DOI] [PubMed] [Google Scholar]
- 28.Wu X, Liu H, Haley KN, Treadway JA, Larson JP, Ge N, Peale F, Bruchez MP. Nat Biotech. 2003;21:41. doi: 10.1038/nbt764. [DOI] [PubMed] [Google Scholar]
- 29.Cai W, Shin DW, Chen K, Gheysens O, Cao O, Wang SX, Gambhir SS, Chen X. Nano Lett. 2006;6:669. doi: 10.1021/nl052405t. [DOI] [PubMed] [Google Scholar]
- 30.Gao X, Cui Y, Levenson R, Chung L, Nie S. Nat Biotech. 2004;22:969. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- 31.Derfus AM, Chan WCW, Bhatia SN. Nano Lett. 2004;4:11. doi: 10.1021/nl0347334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murray CB, Norris DJ, Bawendi MG. J Am Chem Soc. 1993;115:8706. [Google Scholar]
- 33.Gerion D, Pinaud F, Williams SC, Parak WJ, Zanchet D, Weiss S, Alivisatos AP. J Phys Chem B. 2001;105:8861. [Google Scholar]
- 34.Rogach AL, Nagesha D, Ostrander JW, Giersig M, Kotov NA. Chem Mater. 2000;12:2676. [Google Scholar]
- 35.Pellegrino T, Manna L, Kudera S, Liedl T, Koktysh D, Rogach AL, Keller S, Rädler J, Natile G, Parak WJ. Nano Lett. 2004;4:703. [Google Scholar]
- 36.Fan H, Leve EW, Scullin C, Gabaldon J, Tallant D, Bunge S, Boyle T, Wilson MC, Brinker C. J Nano Lett. 2005;5:645. doi: 10.1021/nl050017l. [DOI] [PubMed] [Google Scholar]
- 37.Berg JM, Tymoczko JL, Stryer L. Biochemistry. 6. W H Freeman & Company; 2007. p. 25. [Google Scholar]
- 38.Wuister SF, Swart I, Driel van F, Hickey SG, de Mello Donega C. Nano Lett. 2003;3:503. [Google Scholar]
- 39.Aldana J, Wang YA, Peng X. J Am Chem Soc. 2001;123:8844. doi: 10.1021/ja016424q. [DOI] [PubMed] [Google Scholar]
- 40.Mattoussi H, Mauro JM, Goldman ER, Anderson GP, Sundar VC, Mikulec FV, Bawendi MG. J Am Chem Soc. 2000;122:12142. [Google Scholar]
- 41.Uyeda HT, Medintz IL, Jaiswal JK, Simon SM, Mattoussi H. J Am Chem Soc. 2005;127:3870. doi: 10.1021/ja044031w. [DOI] [PubMed] [Google Scholar]
- 42.Duan H, Nie S. J Am Chem Soc. 2007;129:3333. doi: 10.1021/ja068158s. [DOI] [PubMed] [Google Scholar]
- 43.Nann T. Chem Commun. 2005:1735. doi: 10.1039/b414807j. [DOI] [PubMed] [Google Scholar]
- 44.Smith AM, Duan HW, Rhyner MN, Ruan G, Nie S. Phys Chem Chem Phys. 2006;8:3895. doi: 10.1039/b606572b. [DOI] [PubMed] [Google Scholar]
- 45.Nikolic MS, Krack M, Aleksandrovic V, Kornowsko A, Forster S, Weller H. Angew Chem Int Ed. 2006;45:6577. doi: 10.1002/anie.200602209. [DOI] [PubMed] [Google Scholar]
- 46.Wisher AC, Bronstein I, Chechik V. Chem Commun. 2006:1637. doi: 10.1039/b518115a. [DOI] [PubMed] [Google Scholar]
- 47.Wang M, Felorzabihi N, Guerin G, Haley JC, Scholes GD, Winnik MA. Macromolecules. 2007;40:6377. [Google Scholar]
- 48.Tsarevsky NV, Matyjaszewski K. Macromolecules. 2002;35:9009. [Google Scholar]
- 49.Tsarevsky NV, Matyjaszewski K. Macromolecules. 2005;38:3087. [Google Scholar]
- 50.Matyjaszewski K, Davis TP. Handbook of Radical Polymerization. John Wiley & Sons, Inc; Hoboken: 2002. [Google Scholar]
- 51.Jiang W, Mardyani S, Fischer H, Chan WCW. Chem Mater. 2006;18:872. [Google Scholar]
- 52.Yong KT, Qian J, Roy I, Lee HH, Bergey EJ, Tramposch KM, He S, Swihart MT, Maitra A, Prasad PN. Nano Lett. 2007;7:761. doi: 10.1021/nl063031m. [DOI] [PubMed] [Google Scholar]
- 53.Davis KA, Charleux B, Matyjaszewski K. J Polym Sci, Part A: Polym Chem. 2000;38:2274. [Google Scholar]
- 54.Flory PJ. Principles of Polymer Chemistry. Cornell University Press; 1953. p. 51. [Google Scholar]
- 55.Jocelyn PC. Methods Enzymol. 1987;143:246. doi: 10.1016/0076-6879(87)43048-6. [DOI] [PubMed] [Google Scholar]