

Abstract
Current understanding of specific defense mechanisms in the context of neutropenic infections is limited. It has previously been reported that invasive aspergillosis, a prototypic opportunistic infection in neutropenic hosts, is associated with marked accumulation of inflammatory dendritic cells (DCs) in the lungs. Given recent data indicating that neutrophils can modulate immune responses independent of their direct microbial killing, we hypothesized that neutropenia impacts the host response to Aspergillus by determining the migration and phenotype of lung DCs. Inflammatory DCs, but not other DC subsets, were found to accumulate in the lungs of neutropenic hosts challenged with killed or live-attenuated Aspergillus as compared to non-neutropenic hosts, indicating that the accumulation was independent of neutrophil microbicidal activity. The mechanism of this accumulation in neutropenic hosts was found to be augmented influx of DCs, or their precursors, from the blood to the lungs. This effect was attributable to greatly elevated lung TNF expression in neutropenic as compared to non-neutropenic animals. This resulted in greater lung expression of the chemokine ligands CCL2 and CCL20 which, in turn, mediated enhanced recruitment of TNF-producing inflammatory DCs resulting in a positive-feedback cycle. Finally, in the context of neutropenic invasive aspergillosis, depletion of DCs resulted in impaired fungal clearance, indicating that this mechanism is protective for the host. These observations identify a novel defense mechanism in invasive aspergillosis that is the result of alterations in DC traffic and phenotype and is specific to neutropenic hosts.
Keywords: Cells-neutrophils, Infections-fungal, Molecules-chemokines, molecules-cytokines, processes-inflammation, pneumonia
Introduction
Neutropenia, defined as reduced concentration of circulating neutrophils in the blood, is a common clinical problem that complicates cytotoxic chemotherapy, transplantation and hematologic malignancies. Neutropenic hosts are dramatically more susceptible to many infections, but our understanding of specific host defense mechanisms in the context of neutropenic infections is limited (1). Invasive aspergillosis is a severe infection caused by common environmental molds of the Aspergillus species and is a prototypic opportunistic infection of neutropenic hosts. Aspergillus conidia are ubiquitous in air and, when inhaled, can bypass the physical barriers of the respiratory tract and reach beyond the ciliated epithelium, where they become swollen and metabolically active. In normal hosts, swollen conidia are eliminated at this stage but in immunocompromised patients, they germinate to form hyphae that penetrate the lung epithelium and cause invasive pneumonia. The severity and duration of neutropenia, as well as qualitative defects in neutrophil function, are the best characterized clinical risk factors for the development of this infection [reviewed in (2, 3)]. Thus, host responses to Aspergillus represent a clinically relevant setting to assess mechanisms of host defense in neutropenic hosts.
Dendritic cells (DC) are antigen-presenting cells critical to shaping T cell responses in many contexts, including in response to A. fumigatus. DCs recognize Aspergillus cells via pattern-recognition receptors (4–7) and transport them from the lungs to secondary lymphoid tissues to initiate acquired immunity (8–10). The internalization of Aspergillus conidia and hyphae by DCs initiates qualitatively different CD4+ T cell responses: conidia-activated DCs lead to the priming of Th1 response whereas hyphal phagocytosis by lung DCs result in the generation of IL-4 producing CD4 T cells (8, 11). Inflammatory (CD11bhi CD11c+) DCs are a subset of DCs that accelerate their traffic from the blood to tissues to lymphatic organs during inflammatory responses (12) and have been shown to expand in the lung following a respiratory challenge of Aspergillus (10, 13, 14). We previously reported an unexpectedly large accumulation of these cells in the lungs of neutropenic mice with invasive aspergillosis that was, in part, dependent on the interaction of the chemokine ligand-receptor pair, CCL20–CCR6 (13).
Since such large numbers of inflammatory DCs are not observed in other models of lung inflammation and given evidence of cross-talk between neutrophils and DCs (15), we posited that the absence of neutrophils from infected tissues alters the local inflammatory environment independent of neutrophil-mediated microbial killing and, as a result, modulates the behavior of lung DCs. We therefore tested the hypothesis that neutropenia impacts the host response to Aspergillus by determining the migration and phenotype of lung DCs.
Materials and Methods
Animals and in vivo procedures
Wildtype mice, transgenic mice bearing the simian diphtheria toxin receptor gene under the control of the mouse CD11c promoter (CD11c-DTR mice) (16) or mice heterozygous for targeted replacement of the endogenous CX3CR1 with an EGFP reporter gene (CX3CR1GFP/+ mice) (17), all on C57Bl/6 background, were purchased from Jackson Laboratories (Bar Harbor, Maine). Animals were bred and maintained under pathogen-free conditions. Age- and gender-matched 6- to 8-week old animals were used in all experiments. All animal experiments were approved by the Animal Care and Use Committee of University of Virginia.
Neutrophil depletion was achieved with a single i.p. injection of 80μg of a monoclonal Ab (Gr1, anti-Ly6G/C, clone RB6–8C5) 1 day before an intratracheal challenge with Aspergillus fumigatus, as described (18). This resulted in peripheral blood neutropenia (absolute circulating neutrophil count less than 50 cells/μL) on days 1 and 3 after injection in both infected and uninfected mice, with a return of peripheral counts to pretreatment levels (>1000 cells/μL) by day 5 (19, 20). Administration of the mAb did not influence the number of non-neutrophil peripheral blood leukocytes, nor lung and spleen lymphocytes or DC subsets [(13) and Supplement Figure 3A–B]. In some experiments, neutrophil depletion was achieved by i.p. administration of 200 μg of anti-Ly6G (clone 1A8, BioXcell, West Lebanon, NH); this resulted in peripheral blood neutropenia for 3–4 days, as described (21). Non-neutropenic mice received the equivalent concentration of isotype control mAbs (clones LTF2 and 2A3, BioXcell). For depletion of DCs, mice heterozygous for the CD11c-DTR transgene were injected with diphtheria toxin (Sigma-Aldrich, St Louis, MO), as described (16). In in vivo TNF neutralization experiments, animals received i.p. administration of 300 μg of anti-TNF or isotype control mAbs (clones XT3.11 and HRPN, respectively; BioXcell) 1 day before Aspergillus challenge. For the day 3 time point, an additional i.p. administration of anti-TNF or control Ab was given 48 h later for a total of 2 doses.
In experiments designed to track the movement of cells the lungs, we used previously described protocols with minor modifications (22, 23): circulating blood monocytes were labeled with latex beads by i.v. administration of 100μl of a 1:10 dilution of 0.5μm yellow-green latex microspheres (Polysciences, Warrington, PA) 24 hrs after administration of Aspergillus hyphae. In adoptive transfer experiments, 4–5×106 DC (prepared as described below) were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen Life Technologies) and administrated via the lateral tail vein in 100μl saline on day 2 following Aspergillus challenge, as described (13).
Preparation and administration of A. fumigatus
We used a previously characterized mouse model of neutropenic invasive aspergillosis (13, 18, 24–27) with some modifications. In experiments where animals were challenged with killed fungal elements, A. fumigatus (strain 13073, American Type Culture Collection) conidia were collected in 0.1% Tween in PBS from 7- to 14- day old cultures on Sabouraud’s dextrose agar plates, filtered through sterile gauze and counted under a hemacytometer. In some experiments, resting conidia were then grown in RPMI-1640 in a shaking 37°C incubator for 5 hours to obtain swollen conidia or overnight to obtain short hyphae; the resulting fungal forms were then killed by resuspension in 70% ethanol in sterile water for 48 hours. Viability of the resulting suspension was determined to be <1:2.6×107 cfu by serial dilution and culture. Fungal forms were administered intratracheally in inocula ranging from 2–5×106 for swollen conidia and 6 to 9×105 for hyphae in 30μl per mouse. In other experiments, mice were infected with mutant temperature-sensitive strain of Aspergillus that has been shown to have attenuated virulence due to impaired growth at 37°C but not at room temperature (28). The mutant organism was grown on minimal media agar plates with phleomycin at room temperature for 5 days before harvesting of conidia.
Histology
We obtained tissue blocks of surgical lung biopsy samples from 16 patients with the histological diagnosis of invasive mold infection who had Aspergillus fumigatus isolated from their respiratory samples during that hospital admission and obtained peripheral blood absolute neutrophil counts from the day of surgery. Histologically normal lungs were used as controls. The use of anonymous human samples for this study was reviewed by the University of Virginia Institutional Review Board for Human Subject Research and classified as exempt.
Representative 4μm paraffin-embedded sections were deparaffinized in xylene and rehydrated through graded ethanol to water, subjected to heat-induced target retrieval (for S100 antigen only, Dako Target Retrieval solution, Dako North America, Carpinteria, CA), had endogenous peroxidase activity quenched (Dual Endogenous Enzyme Block, Dako), were labeled with Ab against S100 (Dako, code Z0311) or CD1a (Dako, code M3571) followed by incubation with the labeled polymer and 3-3′ diaminobenzidine (DAB+) substrate chromogen. Slides were counterstained with hematoxylin and bluing reagent (Thermo Scientific Anatomical Pathology, Pittsburgh, PA), dehydrated through graded alcohol to xylene and coverslipped.
Identification of leukocyte subsets
At designated time points, animals were euthanized by CO2 asphyxiation, the pulmonary vasculature was perfused, and whole lungs were removed and leukocyte-enriched lung single cell suspensions were prepared as previously described (13, 19, 24, 25, 27). Peripheral blood was collected from the right ventricle into heparinized tubes. The following antibodies were used to label cells for flow cytometry (from BD Biosciences, San Jose, CA, eBiosciences, San Diego, CA, Miltenyi, Auburn, CA, or R&D Systems, Minneapolis, MN): anti-B220-pacific blue (clone RA3–6B2), anti-CD3e-pacific blue (clone 500-A2), anti-CD11b-allophycocyanin-Cy7 (clone M1/70), anti-CD11c-PE-Cy7 (clone HL3), CD40-PE (clone 3/23), anti-CD45-peridinin chlorophyll protein (clone 30-F11), CD80-FITC (clone 16-10A1), anti-CD86-PE (clone GL1), anti-CD103-biotin (clone 2E7), anti-CD115-PE (clone AFS98), anti-F4/80-PE (clone A31), anti-I-A/I-E-FITC, -biotin, allophycocyanin and -pacific blue (clone M5/114.15.2), anti-Ly-6C-FITC (clone AL-21), anti-Ly-6G-PE and -FITC (clone 1A8), anti-Ly-6G/C-PE and -pacific blue (clone RB6-8C5), anti-PDCA1-PE (clone JF05-IC2.4.1), and anti-TNF-PE (clone MP6-XT22). To determine the lung cells capable of producing TNF, lung suspensions were incubated with brefeldin A (10ng/mL), PMA (10ng/mL) and ionomycin (100ng/mL) in RPMI-1640 with 5% FBS for 5 hours and intracellular staining was detected using a commercial kit (Cytofix/Cytoperm, BD Biosciences). Samples were analyzed on a FACS Canto II instrument using Diva software (BD Biosciences). The absolute number of each leukocyte subset was determined as the product of the percentage of the cell type and the total number of cells in the sample, as determined under a hemocytometer or on an automated cell counter (Countess, Invitrogen, Carlsbad, CA).
Cytokine and chitin assays
Aspergillus fumigatus grows as multicellular branching hyphae and does not form distinct reproductive structures in infected tissues. We therefore used a previously characterized assay for chitin, a carbohydrate component of hyphal wall that is absent from mammalian tissues and conidia, to quantify the burden of hyphae in infected lungs, as detailed previously (29). Organ chitin content in animal models of invasive aspergillosis has been shown by several groups to correlate with histopathological evidence of fungal invasion and mortality from the infection (14, 27, 30–32). TNF, CCL2 and CCL20 protein levels in filtered supernatant of lung homogenates were determined either using commercial ELISA kits (Duoset ELISA Development, R&D Systems) or multiplex bead array kits (Milliplex Map, Millipore, Billerica, MA), according to the manufacturer’s instructions.
Culture and adoptive transfer of dendritic cells
Immature bone marrow-derived conventional DCs were prepared as described previously (13). In brief, bone-marrow cells were cultured in 20ng/mL mGM-CSF for 5 days and positively selected by immunomagnetic selection of CD11c+ cells (Miltenyi, Auburn, CA) resulting in >95% purity. Cells were then labeled with the vital fluorochrome carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen Life Technologies) according to manufacturers’ instructions and uniform staining was verified by epifluorescent microscopy. The resulting cells were >95% viable by trypan blue exclusion, and consistent with immature phenotype, had low expression (MFI) of CD86, CD40, CD80, and MHC class II molecules by flow cytometry. Cells were transferred to animals via lateral tail vein of 4–5 × 106 cells in 100 μl PBS.
Statistical analysis
Data were analyzed on a Macintosh Powerbook G4 computer using Prism statistical package (v.4.0a, Graphpad Software, San Diego, CA). Values between 2 groups over multiple times were compared with 2-way ANOVA, comparisons between 2 groups at a single time were performed with unpaired two-tailed Mann-Whitney (non-parametric) test, and comparisons between multiple groups at a single time were compared using the Kruskal-Wallis test with Bonferroni post-test. Probability values were considered statistically significant if they were less than 0.05.
Results
Absence of neutrophils results in accumulation of DCs in the lungs in response to Aspergillus
Given prior observation of marked accumulation of inflammatory DCs in the lungs of neutropenic mice with invasive aspergillosis (13), we sought to determine whether the absence of neutrophils is the direct cause of increased number of lung DCs. We reasoned that, in the context of a neutropenic host, Aspergillus conidia germinate into hyphae resulting in invasive infection whereas, in immunocompetent hosts, they are killed at the conidial stage before forming hyphae (33) -- as a result, the microbial antigens encountered by neutropenic and immunocompetent hosts after challenge with viable Aspergillus conidia are not comparable and any differences in host response may be related to differences in the antigenic stimulus. We therefore challenged the animals with killed fungal elements in order to retain a comparable microbial stimulus that is independent of the presence of neutrophils. Increase in the number of lung DCs was observed in both neutropenic and non-neutropenic hosts challenged with killed Aspergillus elements, but was greatly enhanced in neutropenic hosts, resulting in a 4-fold increase in the number of inflammatory DCs in the lungs in neutropenic mice as compared to the non-neutropenic mice 3 days after challenge with hyphae (Figure 1A–B). Moreover, the effect was observed when animals were challenged with killed hyphae as well as swollen conidia (Supplemental Figure 1A). In contrast, depletion of neutrophils did not influence the number of airway mucosal DC or lung plasmacytoid DC after challenge with Aspergillus (Figure 1C–D).
Figure 1.

Effect of neutrophil depletion on the number of lung DCs in response to Aspergillus. Number of lung neutrophil (A), inflammatory DCs (B), mucosal DCs (C) and plasmacytoid DCs (D) are shown at various times after challenge with killed Aspergillus hyphae in animals with Ab-mediated neutrophil depletion and mice treated with isotype control Ab. Data shown represent mean ± SEM; n = 20 mice per group per time point pooled from 4 independent experiments for panels A–B and n = 8 mice per group per time point pooled from 2 independent experiments for panels C–D. Time 0 represents unchallenged animals; *, p < 0.05 comparing trend between the two groups over time.
We performed additional studies to ensure that the observed effect was not an epiphenomenon related to the specific experimental conditions. We found a similar increase in lung inflammatory DCs after challenge with killed hyphae when neutropenia was induced using an alternative mAb (Supplemental Figure 1B). Similar results were found when animals were challenged with live hyphae from an attenuated strain of Aspergillus that is growth-impaired at body temperatures (28) (Supplemental Figure 1C). Lastly we found that, similar to observations in the animal model, neutropenia was associated with a substantial increase in the number of DCs in the airways and alveolar spaces of patients with invasive aspergillosis compared to the number of DCs found in normal lungs and in non-neutropenic patients with invasive aspergillosis (Supplementary Figure 2).
Neutropenia results in increased influx and differentiation of inflammatory DCs from the blood to the lungs following challenge with Aspergillus
We next test the hypothesis that the accumulation DCs in the lung is the result of enhanced influx or local differentiation. We tested this hypothesis by tracing the fate of peripheral blood monocytes labeled in the circulation with intravenously delivered fluorescent latex beads, as previously described (22, 23). We first confirmed that neutrophil depletion did influence blood monocyte subsets in this system (Supplemental Figure 3A–B) and that, irrespective of neutrophil depletion, bead-labeled peripheral blood leukocytes after i.v. delivery of beads consisted of monocytes (Supplemental Figure 3C), similar to prior reports (22, 23). We found that few bead-positive cells arrived in the lungs of unchallenged mice after i.v. administration of labeled beads but that, within hours after i.v. delivery of beads, >2-fold more bead-positive cells arrived in the lungs of neutropenic mice challenged with killed Aspergillus hyphae as compared to non-neutropenic mice (Figure 2). In addition, the bead-positive cells arriving in the lungs of neutropenic mice consisted mostly of CD11bhi CD11c+ inflammatory DCs as early as 1hr after administration of beads, with smaller numbers of CD11bhi CD11c− inflammatory monocytic cells; in contrast, bead-positive cells arriving in the lungs of non-neutropenic mice consisted almost entirely of inflammatory monocytic cells (Figure 2C–D).
Figure 2.

Effect of neutrophil depletion on DC influx to the lung in response to Aspergillus. Mice with Ab-mediated neutrophil depletion and mice treated with isotype control Ab were challenged with killed Aspergillus hyphae 24 hours before i.v. administration of FITC-labeled latex beads. (A) Representative flow cytometry plots and gating strategy of whole lung single cell suspensions gated on CD45+ cells, stained for CD11b and CD11c. The bottom flow cytometry plots show the same populations gated on FITC+ beads. (B–D) Bead-associated cell subsets in the lungs were identified based on surface expression of CD11b and CD11c. Day 0 represents unchallenged mice. Data shown represent mean ± SEM; n = 5 mice per group per time point. *, p < 0.05 comparing trend between neutropenic and non-neutropenic challenged mice. (E) Bone marrow-derived DCs were labeled with CFSE and transferred intravenously into mice with Ab-mediated neutrophil depletion and mice treated with isotype control Ab on day 2 after challenge with killed Apergillus hyphae. Labeled cells were identified and enumerated in the lung after 4 hours by flow cytometry. n=8 mice per group per time point, pooled results of 2 experiments. *, p = 0.038 compared to the non-neutropenic group.
The rapidity of appearance of bead-positive DCs in the lungs of neutropenic mice suggested the possibility of selection of small numbers of circulating DCs from the blood in addition to in situ differentiation from recruited DC precursors in the lungs. To assess whether the recruitment of circulating cells was influenced by neutropenia, we intravenously delivered cultured and fluorochrome-labeled immature conventional DCs and tracked their arrival in the lungs after intrapulmonary challenge with killed Aspergillus hyphae. We found ~ 2-fold higher numbers of transferred DCs in the lungs of neutropenic as compared to non-neutropenic animals (Figure 2E). Taken together, these results suggest that in the context of host response to Aspergillus, neutropenia results in enhanced influx of DCs, DC precursors, or both, to the lungs.
Absence of neutrophils results in increased local production of inflammatory cytokines in response to Aspergillus
Since neutropenia appeared to cause a much greater influx of DCs and/or their precursors to the lungs after challenge with Aspergillus, we next sought to determine the mechanism for this effect. TNF is a key proximal cytokine in innate defenses against many infections (34) and has been shown to be critical to host defense against Aspergillus both in humans (35) and in animal models, where it induces the expression of multiple chemokines in the lungs (36, 37). We noted a rapid and marked induction of TNF in the lungs of neutropenic but not in non-neutropenic mice that were challenged with killed Aspergillus hyphae (Figure 3) supporting the hypothesis that TNF may be a proximal signal responsible for recruitment of DCs or their precursors to the lungs in neutropenic hosts. To address this possibility, we also measured lung levels of CCL2 and CCL20, chemokines known to be strongly induced by TNF and previously shown to be critical to recruitment of DCs and monocytes (10, 13, 36), and noted a similar marked induction of CCL2 and CCL20 in the lungs of neutropenic mice in response to Aspergillus hyphae as compared to non-neutropenic animals (Figure 3A–C). Similar inductions were observed when neutrophil depletion was achieved with an alternative mAb (Figure 3D).
Figure 3.

Protein levels of TNF, CCL2, and CCL20 in whole lung homogenates in mice challenged with Aspergillus. (A–C) Cytokine levels in animals with Ab-mediated neutrophil depletion with RB6-8C5 or isotype control mAb were measured at various times following challenge with killed hyphae. Day 0 represents unchallenged mice. Data represent mean ± SEM of pooled data from 2 experiments; n = 6–8 mice per group per time point. *, p < 0.05 comparing trend between the two groups over time. (D) Lung cytokine levels in mice with with Ab-mediated neutrophil depletion with 1A8 or isotype control mAb on day 1 after challenge with with killed hyphae. Data represent mean ± SEM; n = 10 mice per group. *, p < 0.05 compared to non-neutropenic group.
We next sought to establish the cellular sources of the elevated lung TNF in neutropenic mice. As expected, we found ~2-fold greater number of TNF-producing cells in the lungs of neutropenic, as compared to non-neutropenic, mice challenged with Aspergillus hyphae (Figure 4). Interestingly, the TNF-producing cells consisted of approximately equal numbers of CD11bhi CD11c+ inflammatory DCs and CD11bhi CD11c− inflammatory monocytic cells early after challenge with Aspergillus in both groups, but mostly of CD11bhi CD11c+ inflammatory DCs in neutropenic mice late after challenge (Figure 4B). Resident lung macrophages (identified as CD11blo/− CD11c+ cells) were found to be a minor population of TNF-producing cells in both groups (Figure 4B).
Figure 4.

Cellular source of lung TNF in mice challenged with Aspergillus. (A) Representative flow cytometry plots and gating strategy of whole lung single cell suspensions in animals with Ab-mediated neutrophil depletion and mice treated with isotype control Ab after challenge with killed hyphae. Panels were initially gated on CD45+ cells. (B) TNF-expressing cell subsets in the lungs were quantified based on surface expression of CD11b and CD11c. Day 0 represents unchallenged mice. Data represents mean ± SEM; n = 6 mice per group per time point. *, p < 0.05 comparing trend between the two groups over time.
Our data show that the absence of neutrophils results in increased expression of lung TNF, CCL2 and CCL20 in response to Aspergillus and that, in this setting, TNF is also a product of recruited DCs. We therefore hypothesized a causal series of events: that in the lungs of neutropenic hosts infected with Aspergillus, DC-derived TNF results in induction of CCL2 and CCL20 recruiting DCs to the lungs, which, in turn, act as a source of TNF, thereby setting up a positive feed-back loop. To test this hypothesis, we examined the effect of Ab-mediated neutralization of TNF on lung expression of CCL2 and CCL20, and accumulation of inflammatory DCs in the lungs after challenge with killed Aspergillus hyphae. Immunoneutralization of TNF in neutropenic mice led to a marked reduction in both lung CCL2 and CCL20 levels, indicating that TNF is necessary for optimal expression of CCL2 and CCL20 in this setting (Figure 5A–B). In addition, TNF neutralization resulted in reduction of the number of lung inflammatory DCs to levels observed in non-neutropenic animals (Figure 5C). Together with previous data supporting the role of CCL20 and its receptor, CCR6, in recruitment of inflammatory DCs to the lungs (13), these data provide evidence for an amplification loop in the lungs of neutropenic hosts challenged with Aspergillus, that results in accumulation of large numbers of TNF-producing inflammatory DCs.
Figure 5.
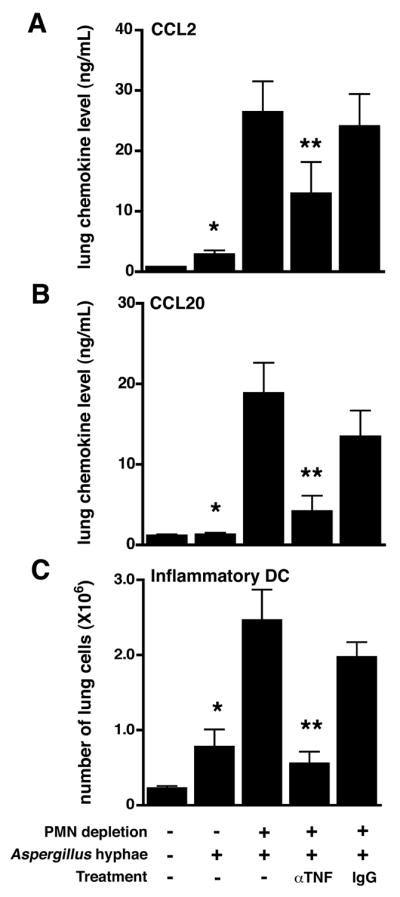
Effect of TNF neutralization on lung chemokine levels and lung inflammatory DC number in response to Aspergillus. Mice with Ab-mediated neutrophil depletion and mice treated with isotype control Ab were challenged with killed Aspergillus hyphae. (A–B) Protein levels of CCL2 and CCL20 in whole lung homogenates were measured 1 day after hyphae challenge. (C) Number of lung inflammatory DC in whole lung single cell suspensions on day 3 after hyphal challenge. *, p < 0.05 compared with unchallenged animals; **, p < 0.05 compared with animals receiving control IgG and challenged mice not receiving any treatment. Data shown represent mean ± SEM; n = 5–6 for each group at each time point, representative data from 2 experiments.
Accumulation of inflammatory DCs in the lungs of neutropenic mice is beneficial during the early phase of invasive aspergillosis
Lastly, we sought to determine the contribution of the accumulated lung DCs to host defense in mice with neutropenic invasive aspergillosis. We used transgenic mice expressing the simian diphtheria toxin receptor driven by the mouse CD11c promoter (16) to achieve conditional depletion of DCs in neutropenic mice with invasive aspergillosis. We confirmed that diphtheria toxin administration in neutrophil-depleted transgenic mice challenged with live Aspergillus conidia resulted in depletion of lung conventional DCs, but did not affect other lung DCs or macrophage subsets (Figure 6A–B). Using this system, ablation of lung inflammatory DCs in neutropenic invasive aspergillosis resulted in a >7-fold increase in lung fungal burden on day 3 of infection as compared to neutropenic wildtype mice (Figure 6C), thus providing evidence for a protective role for the inflammatory DCs that accumulate in the lung in neutropenic hosts with invasive aspergillosis.
Figure 6.
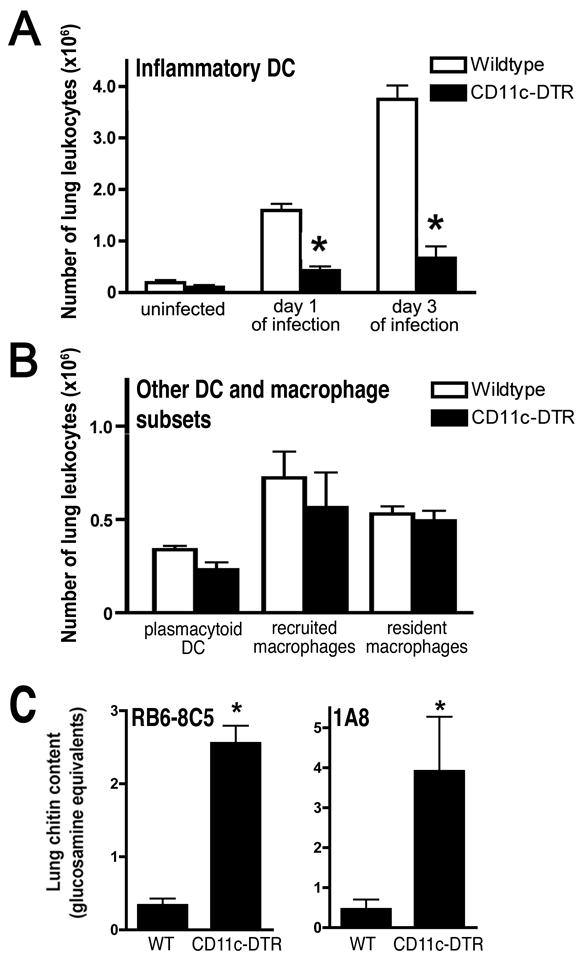
Effect of DC depletion in neutropenic mice with invasive aspergillosis. Mice with Ab-mediated neutrophil depletion were inoculated intratracheally with live Aspergillus conidia. (A) Lung conventional DC in CD11c-DTR and wildtype mice treated with diphtheria toxin on the day before the measurements were obtained. (B) Other lung DC and macrophage subsets in CD11c-DTR and wildtype mice treated with diphtheria toxin, measured on day 1 of infection. DC subsets as defined in Figure 1; macrophage subsets as defined in Figure 2 and (13). (C) Lung chitin content in neutropenic wildtype and CD11c-DTR mice treated with diphtheria toxin, measured on day 3 of infection. Neutropenia was induced with mAbs indicated on the graph; data represents mean ± SEM; n = 4–10 for each group; *, p < 0.05 compared with respective neutropenic wildtype mice.
Discussion
In the present manuscript, we demonstrate that neutropenia fundamentally alters the inflammatory environment of the lung in response to the opportunist mold, Aspergillus, resulting in dramatic changes in the traffic and phenotype of lung inflammatory DCs. As a mechanism for the observed accumulation of inflammatory DCs in the lungs, we found greatly accelerated recruitment of DCs to the lungs in neutropenic mice, which contrasts with prior reports of neutrophils mediating the recruitment of DCs in other experimental systems (38–40). This enhanced recruitment was dependent on a positive-feedback cycle involving lung DC-derived TNF, driving the local production of the chemokine ligands CCL2 and CCL20, and resulting in further recruitment of TNF-producing DCs to the lungs. This observation provides an explanation for prior observations of very high expression of these mediators in the lungs of neutropenic mice with invasive aspergillosis (25, 36). In addition, this paradoxical finding -- enhanced inflammatory cytokine milieu in the absence of neutrophils, which are the cellular hallmarks of acute inflammation -- is consistent with the recently described role of neutrophils in dampening immune responses (41).
An interesting finding of the current work pertains to the specific role of the accumulated lung DCs in defense against neutropenic invasive aspergillosis. In prior studies, interruption of the CCL20–CCR6 axis resulted in reduced lung DC numbers and worsened outcome of infection (13); given the well-documented role of DCs in initiating T cell-mediated protective immunity, this observation may have been attributable to failure to mount acquired immunity. Absence of CCR7, which resulted in failure of efflux of lung DCs, however, was found to result in an unexpected improvement of the outcome of the infection (14), suggesting one of two possibilities: either that DC migration to mediastinal lymph nodes was detrimental [for example by initiating T regulatory responses (42)] or that retaining activated DCs in the lung was beneficial. As an additional consideration, observations in CCR6- and CCR7-deficient mice could conceivably be attributable to lack of expression of these receptors on cells other than DCs. In the context of this literature, we found that the depletion of conventional DCs in neutropenic hosts resulted in substantial worsening of pathogen clearance at a very early phase of the infection, suggesting that the accumulation of inflammatory DCs in the lungs of neutropenic hosts, rather than their maturation and efflux to mediastinal lymph nodes, is the key protective mechanism.
The phenotype of lung DCs observed in neutropenic mice with invasive aspergillosis as inflammatory TNF-producing cells that are important in antimicrobial defense is reminiscent of TNF- and iNOS-producing DCs (Tip-DCs). Tip-DCs were first described as monocyte-derived inflammatory DCs in the spleens of mice early after intra-peritoneal Listeria infection (43) and have subsequently been described in several bacterial and protozoal infections [reviewed in (12)]. Tip-DCs contribute to host defense in some (43, 44) but not other (45, 46) models of infection. The present work relates to this literature in several ways: first, the inflammatory DCs that accumulate in the lungs of mice with neutropenic invasive aspergillosis differ from the description of Tip-DCs in that they do not express iNOS -- specifically, the iNOS producing cells in this system consisted of inflammatory monocytes/macrophages (data not shown). Interestingly, In the context of the lung, cells with a Tip-DC-like phenotype were identified in the lungs in experimental influenza pneumonia and were found to contribute to lung injury but not to viral clearance (47). Our findings also contrast with prior reports in that the absolute number of accumulated inflammatory DCs in lungs of neutropenic mice with aspergillosis is an order of magnitude greater than in other models.
In humans, neutropenia is typically the result of treatment with cytotoxic drugs that influence both the number and function of multiple, often incompletely defined, lineages of cells in addition to neutrophils. In experimental models that seek to examine the consequences of neutropenia, the means of depleting neutrophils is an important methodological consideration. There are currently no genetic approaches to achieve neutropenia in experimental models, and inherent to Ab-mediated approaches are concerns over specificity of neutrophil depletion and potential off target immune complex-mediated effects (48). Specifically, RB6-8C5, the most commonly used mAb, reacts with 2 cell surface markers: Ly-6G, an antigen expressed by neutrophils and inflammatory monocytes (49) and, with 10- to 100-fold lower affinity, Ly-6C, an antigen expressed by multiple leukocyte subsets [(50) and our unpublished observations]. While administration of large quantities of RB6–8C5 depletes Ly-6C-expressing cells (including subsets of DCs and monocytes), the administration of titrated doses results in neutropenia without detectable effect on the number of other leukocyte subsets in naive animals and the administration of isotype control Ab does not appear to influence leukocyte numbers [current manuscript and (13, 19, 20)]. Finally, depletion of neutrophils using an alternative mAb against Ly-6G (clone 1A8), previously shown to deplete neutrophils without affecting monocytes (21), resulted in comparable accumulation of lung DCs to that seen with RB6–8C5. Finally, treatment with the alkylating agent cyclophosphamide, which results in depletion of multiple cell lines including neutrophils, was recently reported to result in expansion of CD11b+ CD11c+ DCs (51).
In summary, we report that neutropenia causes enhanced lung inflammatory response to the common environmental mold, Aspergillus, and is associated with augmented influx of TNF-producing inflammatory DCs to the lungs, resulting in an accumulation of these cells in the lungs to the benefit of the host. These findings have several ramifications for future investigations: First, while we have demonstrated influx and local differentiation as major factors contributing to lung dendritic cell accumulation, neutrophils may also have a role in DC efflux from the lungs to draining lymph nodes. Indeed, in vitro studies with human neutrophils and DCs (52–54) have shown neutrophils to be involved in mediating maturation of DC (which may, in turn, result in their efflux). Second, given recent evidence for the existence of phenotypically and functionally distinct sub-populations of neutrophils (55, 56) and an immunoregulatory role for neutrophils (41), determining the mechanism of neutrophil-mediated down regulation of lung inflammatory responses is of interest. Finally, we provide data in support of the paradigm that inflammatory DCs, like other myeloid cells, can act as effector cells in the early phase of infection, suggesting a function that is independent of their antigen presentation properties.
Supplementary Material
Acknowledgments
Source of funding: Supported by NIH grant HL73848 (Mehrad)
Non-standard abbreviations
- DC
dendritic cell
Footnotes
Conflict of interest: The authors have no conflicts to declare.
References
- 1.Viscoli C, Varnier O, Machetti M. Infections in patients with febrile neutropenia: epidemiology, microbiology, and risk stratification. Clin Infect Dis. 2005;40(Suppl 4):S240–245. doi: 10.1086/427329. [DOI] [PubMed] [Google Scholar]
- 2.Hohl TM, Feldmesser M. Aspergillus fumigatus: principles of pathogenesis and host defense. Eukaryot Cell. 2007;6:1953–1963. doi: 10.1128/EC.00274-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park SJ, Mehrad B. Innate immunity to Aspergillus species. Clin Microbiol Rev. 2009;22:535–551. doi: 10.1128/CMR.00014-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serrano-Gomez D, Dominguez-Soto A, Ancochea J, Jimenez-Heffernan JA, Leal JA, Corbi AL. Dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin mediates binding and internalization of Aspergillus fumigatus conidia by dendritic cells and macrophages. J Immunol. 2004;173:5635–5643. doi: 10.4049/jimmunol.173.9.5635. [DOI] [PubMed] [Google Scholar]
- 5.Hohl TM, Van Epps HL, Rivera A, Morgan LA, Chen PL, Feldmesser M, Pamer EG. Aspergillus fumigatus triggers inflammatory responses by stage-specific beta-glucan display. PLoS Pathog. 2005;1:e30. doi: 10.1371/journal.ppat.0010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steele C, Rapaka RR, Metz A, Pop SM, Williams DL, Gordon S, Kolls JK, Brown GD. The beta-glucan receptor dectin-1 recognizes specific morphologies of Aspergillus fumigatus. PLoS Pathog. 2005;1:e42. doi: 10.1371/journal.ppat.0010042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bellocchio S, Montagnoli C, Bozza S, Gaziano R, Rossi G, Mambula SS, Vecchi A, Mantovani A, Levitz SM, Romani L. The contribution of the Toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. J Immunol. 2004;172:3059–3069. doi: 10.4049/jimmunol.172.5.3059. [DOI] [PubMed] [Google Scholar]
- 8.Bozza S, Gaziano R, Spreca A, Bacci A, Montagnoli C, di Francesco P, Romani L. Dendritic cells transport conidia and hyphae of Aspergillus fumigatus from the airways to the draining lymph nodes and initiate disparate Th responses to the fungus. J Immunol. 2002;168:1362–1371. doi: 10.4049/jimmunol.168.3.1362. [DOI] [PubMed] [Google Scholar]
- 9.Bozza S, Perruccio K, Montagnoli C, Gaziano R, Bellocchio S, Burchielli E, Nkwanyuo G, Pitzurra L, Velardi A, Romani L. A dendritic cell vaccine against invasive aspergillosis in allogeneic hematopoietic transplantation. Blood. 2003;102:3807–3814. doi: 10.1182/blood-2003-03-0748. [DOI] [PubMed] [Google Scholar]
- 10.Hohl TM, Rivera A, Lipuma L, Gallegos A, Shi C, Mack M, Pamer EG. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe. 2009;6:470–481. doi: 10.1016/j.chom.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bozza S, Gaziano R, Lipford GB, Montagnoli C, Bacci A, Di Francesco P, Kurup VP, Wagner H, Romani L. Vaccination of mice against invasive aspergillosis with recombinant Aspergillus proteins and CpG oligodeoxynucleotides as adjuvants. Microbes Infect. 2002;4:1281–1290. doi: 10.1016/s1286-4579(02)00007-2. [DOI] [PubMed] [Google Scholar]
- 12.Dominguez PM, Ardavin C. Differentiation and function of mouse monocyte-derived dendritic cells in steady state and inflammation. Immunol Rev. 234:90–104. doi: 10.1111/j.0105-2896.2009.00876.x. [DOI] [PubMed] [Google Scholar]
- 13.Phadke AP, Akangire G, Park SJ, Lira SA, Mehrad B. The role of CC chemokine receptor 6 in host defense in a model of invasive pulmonary aspergillosis. Am J Respir Crit Care Med. 2007;175:1165–1172. doi: 10.1164/rccm.200602-256OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartigan AJ, Westwick J, Jarai G, Hogaboam CM. CCR7 deficiency on dendritic cells enhances fungal clearance in a murine model of pulmonary invasive aspergillosis. J Immunol. 2009;183:5171–5179. doi: 10.4049/jimmunol.0901027. [DOI] [PubMed] [Google Scholar]
- 15.Yang D, de la Rosa G, Tewary P, Oppenheim JJ. Alarmins link neutrophils and dendritic cells. Trends Immunol. 2009;30:531–537. doi: 10.1016/j.it.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mehrad B, Strieter RM, Moore TA, Tsai WC, Lira SA, Standiford TJ. CXC chemokine receptor-2 ligands are necessary components of neutrophil-mediated host defense in invasive pulmonary aspergillosis. J Immunol. 1999;163:6086–6094. [PubMed] [Google Scholar]
- 19.Park SJ, Wiekowski MT, Lira SA, Mehrad B. Neutrophils regulate airway responses in a model of fungal allergic airways disease. J Immunol. 2006;176:2538–2545. doi: 10.4049/jimmunol.176.4.2538. [DOI] [PubMed] [Google Scholar]
- 20.Mehrad B, Park SJ, Akangire G, Standiford TJ, Wu T, Zhu J, Mohan C. The lupus-susceptibility locus, Sle3, mediates enhanced resistance to bacterial infections. J Immunol. 2006;176:3233–3239. doi: 10.4049/jimmunol.176.5.3233. [DOI] [PubMed] [Google Scholar]
- 21.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 22.Jakubzick C, Tacke F, Ginhoux F, Wagers AJ, van Rooijen N, Mack M, Merad M, Randolph GJ. Blood monocyte subsets differentially give rise to CD103+ and CD103- pulmonary dendritic cell populations. J Immunol. 2008;180:3019–3027. doi: 10.4049/jimmunol.180.5.3019. [DOI] [PubMed] [Google Scholar]
- 23.Tacke F, Ginhoux F, Jakubzick C, van Rooijen N, Merad M, Randolph GJ. Immature monocytes acquire antigens from other cells in the bone marrow and present them to T cells after maturing in the periphery. J Exp Med. 2006;203:583–597. doi: 10.1084/jem.20052119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mehrad B, Wiekowski M, Morrison BE, Chen SC, Coronel EC, Manfra DJ, Lira SA. Transient lung-specific expression of the chemokine KC improves outcome in invasive aspergillosis. Am J Respir Crit Care Med. 2002;166:1263–1268. doi: 10.1164/rccm.200204-367OC. [DOI] [PubMed] [Google Scholar]
- 25.Morrison BE, Park SJ, Mooney JM, Mehrad B. Chemokine-mediated recruitment of NK cells is a critical host defense mechanism in invasive aspergillosis. J Clin Invest. 2003;112:1862–1870. doi: 10.1172/JCI18125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mehrad B, Moore TA, Standiford TJ. Macrophage inflammatory protein-1 alpha is a critical mediator of host defense against invasive pulmonary aspergillosis in neutropenic hosts. J Immunol. 2000;165:962–968. doi: 10.4049/jimmunol.165.2.962. [DOI] [PubMed] [Google Scholar]
- 27.Park SJ, Hughes MA, Burdick M, Strieter RM, Mehrad B. Early NK cell-derived IFN-{gamma} is essential to host defense in neutropenic invasive aspergillosis. J Immunol. 2009;182:4306–4312. doi: 10.4049/jimmunol.0803462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhabhra R, Miley MD, Mylonakis E, Boettner D, Fortwendel J, Panepinto JC, Postow M, Rhodes JC, Askew DS. Disruption of the Aspergillus fumigatus gene encoding nucleolar protein CgrA impairs thermotolerant growth and reduces virulence. Infect Immun. 2004;72:4731–4740. doi: 10.1128/IAI.72.8.4731-4740.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lehmann PF, White LO. Chitin assay used to demonstrate renal localization and cortisone-enhanced growth of Aspergillus fumigatus mycelium in mice. Infect Immun. 1975;12:987–992. doi: 10.1128/iai.12.5.987-992.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.D’Angelo C, De Luca A, Zelante T, Bonifazi P, Moretti S, Giovannini G, Iannitti RG, Zagarella S, Bozza S, Campo S, Salvatori G, Romani L. Exogenous pentraxin 3 restores antifungal resistance and restrains inflammation in murine chronic granulomatous disease. J Immunol. 2009;183:4609–4618. doi: 10.4049/jimmunol.0900345. [DOI] [PubMed] [Google Scholar]
- 31.Jaillon S, Peri G, Delneste Y, Fremaux I, Doni A, Moalli F, Garlanda C, Romani L, Gascan H, Bellocchio S, Bozza S, Cassatella MA, Jeannin P, Mantovani A. The humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J Exp Med. 2007;204:793–804. doi: 10.1084/jem.20061301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stephens-Romero SD, Mednick AJ, Feldmesser M. The pathogenesis of fatal outcome in murine pulmonary aspergillosis depends on the neutrophil depletion strategy. Infect Immun. 2005;73:114–125. doi: 10.1128/IAI.73.1.114-125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blease K, Mehrad B, Lukacs NW, Kunkel SL, Standiford TJ, Hogaboam CM. Antifungal and airway remodeling roles for murine monocyte chemoattractant protein-1/CCL2 during pulmonary exposure to Asperigillus fumigatus conidia. J Immunol. 2001;166:1832–1842. doi: 10.4049/jimmunol.166.3.1832. [DOI] [PubMed] [Google Scholar]
- 34.Strieter RM, Belperio JA, Keane MP. Cytokines in innate host defense in the lung. J Clin Invest. 2002;109:699–705. doi: 10.1172/JCI15277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsiodras S, Samonis G, Boumpas DT, Kontoyiannis DP. Fungal infections complicating tumor necrosis factor alpha blockade therapy. Mayo Clin Proc. 2008;83:181–194. [PubMed] [Google Scholar]
- 36.Mehrad B, Strieter RM, Standiford TJ. Role of TNF-alpha in pulmonary host defense in murine invasive aspergillosis. J Immunol. 1999;162:1633–1640. [PubMed] [Google Scholar]
- 37.Phadke AP, Mehrad B. Cytokines in host defense against Aspergillus: recent advances. Med Mycol. 2005;43(Suppl 1):S173–176. doi: 10.1080/13693780500052099. [DOI] [PubMed] [Google Scholar]
- 38.de la Rosa G, Yang D, Tewary P, Varadhachary A, Oppenheim JJ. Lactoferrin acts as an alarmin to promote the recruitment and activation of APCs and antigen-specific immune responses. J Immunol. 2008;180:6868–6876. doi: 10.4049/jimmunol.180.10.6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chertov O, Ueda H, Xu LL, Tani K, Murphy WJ, Wang JM, Howard OM, Sayers TJ, Oppenheim JJ. Identification of human neutrophil-derived cathepsin G and azurocidin/CAP37 as chemoattractants for mononuclear cells and neutrophils. J Exp Med. 1997;186:739–747. doi: 10.1084/jem.186.5.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bennouna S, Bliss SK, Curiel TJ, Denkers EY. Cross-talk in the innate immune system: neutrophils instruct recruitment and activation of dendritic cells during microbial infection. J Immunol. 2003;171:6052–6058. doi: 10.4049/jimmunol.171.11.6052. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X, Majlessi L, Deriaud E, Leclerc C, Lo-Man R. Coactivation of Syk kinase and MyD88 adaptor protein pathways by bacteria promotes regulatory properties of neutrophils. Immunity. 2009;31:761–771. doi: 10.1016/j.immuni.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 42.Montagnoli C, Fallarino F, Gaziano R, Bozza S, Bellocchio S, Zelante T, Kurup WP, Pitzurra L, Puccetti P, Romani L. Immunity and tolerance to Aspergillus involve functionally distinct regulatory T cells and tryptophan catabolism. J Immunol. 2006;176:1712–1723. doi: 10.4049/jimmunol.176.3.1712. [DOI] [PubMed] [Google Scholar]
- 43.Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 44.Copin R, De Baetselier P, Carlier Y, Letesson JJ, Muraille E. MyD88-dependent activation of B220-CD11b+LY-6C+ dendritic cells during Brucella melitensis infection. J Immunol. 2007;178:5182–5191. doi: 10.4049/jimmunol.178.8.5182. [DOI] [PubMed] [Google Scholar]
- 45.Engel D, Dobrindt U, Tittel A, Peters P, Maurer J, Gutgemann I, Kaissling B, Kuziel W, Jung S, Kurts C. Tumor necrosis factor alpha- and inducible nitric oxide synthase-producing dendritic cells are rapidly recruited to the bladder in urinary tract infection but are dispensable for bacterial clearance. Infect Immun. 2006;74:6100–6107. doi: 10.1128/IAI.00881-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guilliams M, Movahedi K, Bosschaerts T, VandenDriessche T, Chuah MK, Herin M, Acosta-Sanchez A, Ma L, Moser M, Van Ginderachter JA, Brys L, De Baetselier P, Beschin A. IL-10 dampens TNF/inducible nitric oxide synthase-producing dendritic cell-mediated pathogenicity during parasitic infection. J Immunol. 2009;182:1107–1118. doi: 10.4049/jimmunol.182.2.1107. [DOI] [PubMed] [Google Scholar]
- 47.Aldridge JR, Jr, Moseley CE, Boltz DA, Negovetich NJ, Reynolds C, Franks J, Brown SA, Doherty PC, Webster RG, Thomas PG. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc Natl Acad Sci U S A. 2009;106:5306–5311. doi: 10.1073/pnas.0900655106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cassatella MA, Locati M, Mantovani A. Never underestimate the power of a neutrophil. Immunity. 2009;31:698–700. doi: 10.1016/j.immuni.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 49.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 50.Fleming TJ, Fleming ML, Malek TR. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6–8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J Immunol. 1993;151:2399–2408. [PubMed] [Google Scholar]
- 51.Salem ML, Al-Khami AA, El-Naggar SA, Diaz-Montero CM, Chen Y, Cole DJ. Cyclophosphamide induces dynamic alterations in the host microenvironments resulting in a Flt3 ligand-dependent expansion of dendritic cells. J Immunol. 184:1737–1747. doi: 10.4049/jimmunol.0902309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Gisbergen KP, Sanchez-Hernandez M, Geijtenbeek TB, van Kooyk Y. Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN. J Exp Med. 2005;201:1281–1292. doi: 10.1084/jem.20041276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Gisbergen KP, I, Ludwig S, Geijtenbeek TB, van Kooyk Y. Interactions of DC-SIGN with Mac-1 and CEACAM1 regulate contact between dendritic cells and neutrophils. FEBS Lett. 2005;579:6159–6168. doi: 10.1016/j.febslet.2005.09.089. [DOI] [PubMed] [Google Scholar]
- 54.Megiovanni AM, Sanchez F, Robledo-Sarmiento M, Morel C, Gluckman JC, Boudaly S. Polymorphonuclear neutrophils deliver activation signals and antigenic molecules to dendritic cells: a new link between leukocytes upstream of T lymphocytes. J Leukoc Biol. 2006;79:977–988. doi: 10.1189/jlb.0905526. [DOI] [PubMed] [Google Scholar]
- 55.Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsuda Y, Takahashi H, Kobayashi M, Hanafusa T, Herndon DN, Suzuki F. Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin-resistant Staphylococcus aureus. Immunity. 2004;21:215–226. doi: 10.1016/j.immuni.2004.07.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.