

Abstract
Background
X-box binding protein 1 (XBP1), a transcription factor involved in unfolded protein response, is also an estrogen-regulated gene and strongly correlates with estrogen receptor alpha (ERα) expression in breast cancers. We investigated the functional role of XBP1 in estrogen responsive breast and endometrial cancer cells as its functions are not fully understood.
Materials and methods
ERα positive breast (MCF7) and endometrial (ECC1) cancer cells were used to study XBP1 gene regulation by 17-β-estradiol (E2) and to investigate the role of XBP1 in E2-mediated growth using short interfering RNA. Quantitative real-time PCR and Western blot were used to assess RNA and protein levels. Recruitment of ERα and other cofactors at the promoter and enhancer region of the XBP1 gene was investigated by chromatin immunoprecipitation. Estrogen responsive element (ERE)-mediated transcriptional activity was evaluated by a luciferase reporter assay.
Results
E2 induced the transcription of XBP1 in both MCF7 and ECC1 cells. E2-dependent recruitment of ERα, steroid receptor coactivator (SRC)-1 and SRC-3, and RNA polymerase II were observed at the promoter and/or enhancer region of the XBP1 gene. Depletion of XBP1 markedly inhibited the E2-induced growth in MCF7 and ECC1 cells. However, ERE-mediated transcription was not altered in XBP1-overexpressing or XBP1-depleted MCF7 cells.
Conclusion
Our results confirm E2-induced transcription of XBP1 and demonstrate the crucial role of XBP1 in E2-induced growth of ERα positive breast and endometrial cancer cells without modulating the classical ERE-mediated transcription by ER. This knowledge creates new opportunities for therapeutic interventions.
Keywords: breast cancer, estrogen, estrogen receptor, X-box binding protein 1 (XBP1)
Introduction
Estrogen is the principal growth mediator of the estrogen receptor (ER) positive breast and endometrial cancers (1). Estrogen acts by binding to ERα or β and the resulting complex can activate transcription of estrogen responsive genes. Examples of estrogen responsive genes include the transcription factors which crucially regulate estrogen-dependent growth. X-box binding protein 1 (XBP1) is a transcription factor, identified as basic region leucine zipper belonging to the ATF/CREB family, strongly coexpressed in ERα positive luminal epithelial breast cancers (2, 3). Several DNA microarray studies have also found XBP1 as an estrogen-regulated gene in ER positive breast cancer cell lines as well as in breast cancers (4–9). In addition, recruitment of ERα on the XBP1 promoter as well as enhancer regions has been confirmed using chromatin immunoprecipitation (ChIP) followed by tiled microarray on human chromosomes 21 and 22 (10).
XBP1 is an important component of unfolded protein response (UPR) where it activates a distinct set of genes and regulates endoplasmic reticulum stress-mediated apoptosis (11). Studies have found that XBP1 is essential for survival of mouse embryonic fibroblasts and is also required for tumor growth of human fibrosarcoma cells under hypoxic conditions, as XBP1-deficient cells show impaired survival (12). Consistent with these findings, XBP1 knockout mice are found to be embryonic lethal as embryonic livers at 13.5 day from XBP−/− mouse exhibited increased apoptosis compared with wild type embryos (13). Further studies show that embryonic lethality of the XBP−/− can be rescued by selectively expressing XBP1 in the hepatocytes. However, these animals died in early postnatal period with pancreatic insufficiency (14). Likewise, XBP1 is essential for UPR and differentiation of plasma cells (15). In multiple myeloma, a plasma cell malignancy, XBP1 deficiency can induce apoptosis in response to endoplasmic reticulum stress (16). A recent study observed apoptosis in XBP1-depleted intestinal epithelial cells from mouse and a concurrent increase in their susceptibility to developing inflammatory bowel disease (17).
Interestingly, XBP1 is reported (18) to interact with ERα in a ligand-independent manner and can also induce transcription from estrogen responsive element (ERE) containing luciferase reporter gene even in the absence of estrogen. Further studies found large-scale chromatin unfolding associated with XBP1-mediated increase in ERα transcriptional activity (19). Although these findings strongly suggest an interaction of XBP1 with ERα and its involvement in the ERα-mediated transcriptional process, the precise underlying mechanisms are unknown in ER positive breast and endometrial cancers.
Overexpression of XBP1 in ER positive breast cancer cells not only induces estrogen-independent growth of ER positive breast cancer cells but also confers resistance to the anti-estrogen tamoxifen (20). However, no data are available to explain the relevance of endogenous level of XBP1 and also how estrogen mediated upregulation of XBP1 could have a functional role in estrogen-induced growth of ERα positive breast and endometrial cancer cells.
We report the estrogen regulation of endogenous XBP1 and show that coactivators steroid receptor coactivator (SRC)-1 and SRC-3 along with ERα are recruited at the promoter and/or enhancer elements of the XBP1 gene. By depleting XBP1 levels using siRNA, we also show that XBP1 is required to mediate the estrogen-induced growth of MCF7 breast and ECC1 endometrial cancer cells.
Materials and methods
Cell culture and reagents
Cell culture media were purchased from Invitrogen Inc. (Grand Island, NY, USA) and fetal calf serum (FCS) was obtained from HyClone Laboratories (Logan, UT, USA). The ER positive breast cancer cells MCF7:WS8 used in this study were derived from MCF7 cells obtained from the American Type Culture Collection as reported previously (21). The ER positive endometrial cancer cells ECC1 cells were originally from Dr. Myles Brown's lab. MCF7 cells were routinely maintained in RPMI media and ECC1 cells were maintained in Dulbecco's Modified Eagle Medium media supplemented with 10% FCS, 6 ng/mL bovine insulin and penicillin and streptomycin. Three to four days prior to harvesting, cells were switched to phenol red-free media containing 10% charcoal dextran treated FCS. Media was changed every other day. The cells were treated with indicated reagents for the specified time and were subsequently harvested for total RNA isolation or protein lysate. Cycloheximide (CHX) and 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) were purchased from Sigma chemicals (St. Louis, MO, USA) and fulvestrant (FUL) was from Tocris Pharmaceuticals (Ellisville, MI, USA). Cells were pretreated for 30 min with CHX, DRB or FUL before treating with 17β-estradiol (E2) or vehicle for specified times as mentioned in Figure legends. All experiments were repeated at least three times, in triplicate to confirm the results.
Total RNA isolation and real-time polymerase chain reaction (PCR)
Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and an RNeasy kit (Qiagen Inc.) according to the manufacturer's instructions. Real-time PCR was performed by reverse transcribing 1 μg of total RNA in a total volume of 20 μL using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA) as per manufacturer's instructions and subsequently diluted to 200 μL with sterile water. The real-time PCR was performed in a 20 μL reaction which included 1 × SYBR green PCR master mix (Applied Biosystems), 100 nM each of forward and reverse primers and 2 μL of diluted cDNA using an ABI Prism 7900 HT Sequence Detection System (Applied Biosystems) for 40 cycles (95°C for 15 s, 60°C for 1 min) following an initial 10 min incubation at 95°C. The fold change in expression of transcripts was calculated using the ΔΔCt method, with the ribosomal protein 36B4 mRNA as the internal control (22). The primer sequences used were the same as previously reported for XBP1 (10) and 36B4 (23).
Growth assay
Cells were grown in 10 cm plates in charcoal-stripped media for 4 days before plating for the growth assay. In total, 12,000 cells were plated in each well of 24-well plates and allowed to attach for at least 16 h before treating them with vehicle or 1 nM E2. One untreated plate was collected at the start day of treatment and this served as the baseline for the comparison of the growth of cells. Cells were then collected on the second, fourth and sixth days of treatment and frozen in −30°C. The growth was assessed by measuring the DNA content in each well using a fluorescent DNA quantitation kit (Bio-Rad, Hercules, CA, USA) according to the manufacturer's instructions. Calf thymus DNA was used to plot the standard curve for the DNA assay with each set of quantitation. The experiments were repeated three times in quadruplicates to confirm the data.
Chromatin immunoprecipitation (ChIP) assay
ChIP was performed as described by Shang et al. (24) with minor modifications. Cells were grown in 15 cm plates in phenol red-free RPMI media containing 10% charcoal stripped fetal bovine serum for 3 days before treating with vehicle or 1 nM estradiol for 45 min. Cells were then washed with phosphate buffered saline (PBS) followed by crosslinking with 1% formaldehyde at room temperature for 15 min and then stopped it using 125 mM glycine. Cells were then rinsed with PBS and collected in PBS containing protease inhibitors (Roche Diagnostics, Indianapolis, IN, USA) and 10 mM dithiothreitol (DTT), followed by centrifuging at 2000×g for 5 min at 4°C. Subsequently, cells were resuspended in nuclei isolation buffer (50 mM Tris Cl, 60 mM KCl, 0.5% NP40, protease inhibitors and 10 mM DTT) followed by centrifugation to isolate the nuclei. Isolated nuclei were resuspended in sodium dodecyl sulfate (SDS) lysis buffer (50 mM Tris Cl, 1% SDS, 10 mM EDTA, pH 8.1 with protease inhibitors) and sonicated (Microson ultrasonic dismembrator) three times at setting `10' followed by centrifugation at 14,000×g for 20 min at 4°C. Fixed chromatin supernatant was diluted using ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl with 1 × protease inhibitors) followed by immunoclearing using normal rabbit serum and 60 μL of protein A agarose (Upstate Cell Signaling Solutions, Temecula, CA, USA). Immunoprecipitation was performed overnight with antibodies against ERα, SRC-1, SRC-3 and phospho-2-serine-RNA polymerase II (p-RNA polII). The immunocomplexes were precipitated using 60 μL of protein A agarose and incubated for an additional 2 h followed by centrifugation at 700×g for 5 min. The beads bound to immunocomplexes were sequentially washed 10 min each using buffer I (20 mM Tris Cl, 2 mM EDTA, 0.1% SDS, 1% Triton X-100 and 150 mM NaCl), buffer II (20 mM Tris Cl, 2 mM EDTA, 0.1% SDS, 1% Triton X-100 and 250 mM NaCl), buffer III (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl, pH 8.1). Precipitates were then washed twice with TE buffer and extracted twice with freshly made 1% SDS and 0.1 M NaHCO3. Pooled elutes were decrosslinked using 200 μM of NaCl and heated at 65°C overnight.
The DNA fragments were purified using a Qiaquick PCR purification kit (Qiagen Inc.). Then, 1–2 μL of eluted DNA was used for real-time PCR analysis. The primer sequences used are as follows: XBP1 promoter: 5′TCTGGAAAGCTCTCGGTTTG3′ (forward); 5′AATCCCTGGCCAAAGGTACT3′ (reverse); XBP1 enhancer: 5′ATACTTGGCAGCCTGTGACC3′ (forward); 5′GTGCCACAAAGCAGGAAAAA3′ (reverse). The data are expressed as percent input of 1/20th part of starting chromatin material and are representative of three separate experiments with similar results.
Western immunoblotting
MCF7 and ECC1 cells were grown in phenol red-free media containing charcoal stripped serum. After treatments with the compounds for the indicated time periods, cells were rinsed with cold PBS and then lysed by RIPA buffer (Sigma Chemicals, St. Louis, MO, USA) supplemented with protease inhibitors (Roche Diagnostics, Indianapolis, IN, USA) and phosphatase inhibitor cocktails I and II (Calbiochem, La Jolla, CA, USA). Cell lysates were collected, sonicated (3 × for 10 s, on ice) and centrifuged at 14,000 rpm for 20 min at 48°C. Cell supernatants were aliquoted and stored at −80°C. Protein concentration was determined using a BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Proteins (20–40 μg) were separated by electrophoresis using 10% polyacrylamide gels containing sodium dodecyl sulfate (SDS-PAGE) and transferred onto PVDF transfer membranes (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA). Primary antibodies used for Western blotting were raised against XBP1 (Santa Cruz, CA, USA) and b-actin (Sigma-Aldrich Corp., St. Louis, MO, USA). The bands were visualized using an ECL Western blotting detection system (GE Healthcare Bio-Sciences Corp) as per manufacturer's instructions. The experiments were repeated at least three times with similar results.
Short interfering RNA (siRNA) experiments
For siRNA experiments, MCF7 or ECC1 cells were grown in 10 cm plates, in antibiotic-free media. Cells were transfected with 100 nM XBP1 siRNA (on target plus, cat #009552; Dharmacon, Inc.) or control siRNA (on target plus, non-targeting pool, cat #D-001810; Dharmacon, Inc.) using 20 μL of Dharmafect transfection reagent as per manufacturer's instruction, for 48 h. Cells were allowed to recover in complete medium (without antibiotics) for 16 h, followed by reseeding in 24-well plates for growth assay or in 6-well plates for RNA and protein isolation. Cells were then treated with vehicle or E2 for indicated times. RNA and protein was extracted by methods as mentioned earlier. The experiments were repeated at least three times, in triplicate to confirm the results.
Luciferase reporter assays
Plasmid pERE(5X)TA-ffLuc (25) containing five tandem copies of the consensus palindromic ERE and firefly luciferase was transfected to assess the ERE-mediated transcriptional activity in the MCF7 cells. Plasmid pTA-srluc (25) expressing renilla luciferase reporter gene was cotransfected as an internal control. Then, 300 ng of pERE(5×)TA-ffLuc and 50 ng of pTA-srluc was cotransfected in each well containing 105 cells treated with control or XBP1 siRNA. Cells were further treated with vehicle or E2 for 48 h before harvesting for the assay.
In another set of experiments, cells were cotransfected with either 20 ng or 500 ng of XBP1 expressing plasmid along with 300 ng of pERE(5×)TA-ffLuc and 50 ng of pTA-srluc. The entire assay was performed using dual luciferase assay kits (Promega). All data are represented in terms of ratio of firefly/renilla RLU values. The experiments were repeated three times, in quadruplicates to confirm the results.
Statistics
Statistical significance of our data were assessed using the Student t-test. A p-value <0.05 was considered as statistically significant.
Results
Estrogen upregulates XBP1 in MCF7 and ECC1 cells and is a primary responsive gene
We first studied the dose-response of E2 on XBP1 mRNA regulation in MCF7 breast cancer cells after 8 h of E2 treatment using quantitative real-time PCR. Our data show that XBP1 mRNA was induced by E2 in a dose-dependent manner (Figure 1A). Low dose of E2 (10−14 M) was not able to induce any upregulation of XBP1 levels, whereas 10−12 M of E2 achieved the peak induction. Higher doses of E2 treatment induced XBP1 levels similar to 10−12 M of E2. We then studied the regulation of the XBP1 at various time points after 1 nM (10−9 M) E2 treatment and found that it was upregulated as early as 2 h after estrogen treatment and maintained an elevated level even after 48 h of estrogen treatment in breast cancer (MCF7) as well as in endometrial cancer (ECC1) cells (Figures 1B and 2A). This upregulation was completely abrogated in the presence of FUL, a complete anti-estrogen, indicating an ERα-mediated mechanism (Figures 1C and 2B). Pretreatment with CHX, an inhibitor of protein synthesis, in the presence of E2 did not alter the E2-mediated upregulation suggesting that de novo protein synthesis is not required for the estrogen-mediated upregulation of XBP1 (Figures 1C and 2B). Conversely, pretreatment with DRB, a transcriptional inhibitor, completely blocked the upregulation of XBP1 demonstrating involvement of transcriptional machinery in upregulation of XBP1 by estrogen (Figures 1C and 2B).
Figure 1.

E2-mediated upregulation of XBP1 in MCF7 cells. MCF7 cells were treated with different concentrations of E2 for 8 h and expression of XBP1 was measured using quantitative real-time PCR and compared with vehicle-treated cells (A). MCF7 cells treated with E2 (1 nM) for 2, 4, 8, 16, 24 or 48 h and expression of XBP1 was measured using quantitative real-time PCR and compared with vehicle-treated cells (B). MCF7 cells were treated with CHX (10 μg/mL), DRB (75 μM) or FUL (1 mM) in absence or presence of E2 (1 nM) for 24 h and expression of XBP1 was assessed using real-time PCR (C). *p<0.05 compared with respective vehicle-treated group.
Figure 2.

E2-mediated upregulation of XBP1 in ECC1 cells. ECC1 cells treated with E2 (1 nM) for 2, 4, 8, 16, 24 or 48 h and expression of XBP1 was measured using quantitative real-time PCR and compared with vehicle-treated cells (A). ECC1 cells were treated with CHX (10 μg/mL), DRB (75 mM) or FUL (1 mM) in absence or presence of E2 (1 nM) for 24 h and expression of XBP1 was assessed using real-time PCR (B). SKBR-3 and MDA-MB-231 cells were treated with E2 (1 nM) or vehicle (0.1% ethanol) for 24 h and expression of XBP1 was measured using quantitative real-time PCR and compared with vehicle-treated cells (C). Total RNA from MCF7 and MCF:5C was isolated and expression of XBP1 was measured using quantitative real-time PCR relative to MCF7 cells (D). MCF7:5C cells were treated with vehicle (0.1% ethanol) or E2 (1 nM) for 48 h and expression of XBP1 was measured using quantitative real-time PCR and compared with vehicle-treated cells (E). *p<0.05 compared with vehicle-treated group (B). **p<0.05 compared with MCF7 cells (D) or vehicle-treated group (E).
We also studied the regulation of XBP1 in ER negative breast cancer cells SKBR3 and MDA MB 231 cells. As expected, XBP1 was not regulated by E2 in these cells (Figure 2C). Furthermore, we investigated the levels of XBP1 in long-term estrogen-deprived MCF7 cells, known as MCF7:5C cells (26) which are estrogen-deprived resistant cells. Paradoxically, low levels of E2 induce apoptosis in these cells (27). Basal levels of XBP1 mRNA were found to be around 23-fold higher in MCF7:5C cells compared with MCF7 cells (Figure 2D). Interestingly, E2 treatment for 48 h drastically downregulated the XBP1 levels in MCF7:5C cells (Figure 2E), which coincides with estrogen-induced apoptosis in these cells.
Recruitment of ERα and other factors at the promoter and enhancer regions of the XBP1 gene
To further confirm the direct involvement of ERα in transcriptional induction of the XBP1 gene, we performed ChIP assay to assess the recruitment of ERa, SRC-1, SRC-3 and serine-2-phosphorylated RNA polymerase II at ~0.3 kb (promoter) and ~13 kb (enhancer) upstream of the transcription start site of the XBP1 gene (Figure 3A) in the MCF7 cells treated with vehicle or 1 nM E2 for 45 min. We found higher occupancy of ERα and SRC-3 at the enhancer region but not at the promoter region. The occupancy of these factors at the enhancer region was further stimulated after 45 min of E2 treatment compared with vehicle treatment (Figure 3B and C). In contrast, serine-5-phosphorylated RNA polymerase II was found to be recruited 4-fold more at the promoter region than at the enhancer region after 45 min of E2 treatment (Figure 3D). Occupancy of SRC-1 was stimulated after E2 treatment in both the promoter and enhancer regions of the XBP1 gene (Figure 3E). These results indicate that the enhancer region of XBP1 is involved in the regulation of estrogen-induced transcriptional stimulation of the XBP1 gene.
Figure 3.

Recruitment of ERα, phospho-serine-2-RNA polII, SRC-1 and SRC-3, at the proximal promoter and distal enhancer region of the XBP1 gene assessed by chromatin immunoprecipitation (ChIP) assay. MCF7 cells were treated with vehicle or E2 (1 nM) for 45 min and ChIP assay was performed as mentioned in the materials and methods section. Schematic representation of the promoter and enhancer regions of the XBP1 gene (A). The extent of recruitment of the factors indicated is shown for promoter and enhancer region of the XBP1 gene. The data are expressed as percent input of 1/20th part of starting chromatin material in each case after subtracting non-specific binding. The data shown are representative of three separate experiments with similar results.
XBP1 depletion inhibits estrogen-mediated growth
To investigate the functional importance of XBP1, we evaluated the effect through loss-of-function using a pool of siRNA against XBP1 in MCF7 and ECC1 cells. The extent of XBP1 knockdown was confirmed by real-time PCR and Western blotting (Figure 4A and B; Figure 4D and E, respectively). A growth assay was performed after XBP1 knockdown by siRNA and total DNA content was used as a measure to determine the cell growth over a 6-day period. A parallel identical growth assay was performed using pool of non-targeting control siRNA for comparison. XBP1 knockdown attenuated the E2-induced growth of MCF7 and ECC1 cells by 49% and 30%, respectively, compared with cells treated with control siRNA (Figure 4C and F). These data indicated that the level of XBP1 expression is critical for inducing estradiol-mediated growth of breast and endometrial cancer cells.
Figure 4.

Short interfering RNA (siRNA)-mediated knockdown of XBP1 inhibits growth of MCF7 and ECC1 cells and its effect on estrogen-mediated growth. MCF7 and ECC1 cells, transfected with XBP1 siRNA or control siRNA, were treated with E2 (1 nM) or vehicle for 24 h and the extent of knockdown was assessed using quantitative real-time PCR compared with control siRNA, vehicle-treated cells (A and D) and Western blotting (B and E). Subsequently, cells were reseeded and the growth of the cells was monitored over a 6-day period. Total DNA content was measured as a marker of growth and the fold change in DNA content was calculated compared with the number of cells at the time of the start of the treatment (baseline) (C and F). *p<0.05 compared with control siRNA group and **p<0.005 using the unpaired Student t-test. The Western blots were scanned and quantified. Levels of XBP1 normalized for β-actin, relative to control siRNA-vehicle treated cells, are indicated below each band.
We further investigated if the levels of ERα were altered in the XBP1-depleted MCF7 cells compared with control siRNA-treated MCF7 cells. No differences were detected in levels of ERa in XBP1-depleted cells compared with control siRNA-treated cells in presence of vehicle or E2 for 24 h (Figure 5). This rules out the possibility that growth inhibition of XBP1-depleted cells was due to altered ERα levels.
Figure 5.
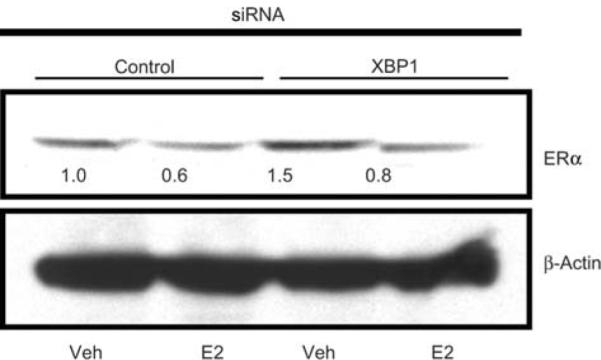
ERα levels in MCF7 cells treated with control or XBP1 siRNA. MCF7 cells were transfected with control or XBP1 siRNA and subsequently treated with vehicle or E2 for 24 h. ERa levels were assessed by Western blotting. Levels of β-actin are shown as loading control. The Western blots were scanned and quantified. Levels of ERα protein normalized for β-actin, relative to control siRNA-vehicle treated cells, are indicated below each band.
XBP1 overexpression or XBP1 depletion does not affect ERE-mediated transcriptional activity
To understand the underlying mechanism by which XBP1 can influence estrogen-mediated growth, we examined the effect of XBP1 overexpression or depletion on the transcriptional activity of ER from a classical ERE. We performed an ERE-luciferase reporter assay in the MCF7 cells transiently transfected with XBP1 expression plasmid or XBP1 siRNA. No differences were observed (Figure 6A and B) in transcriptional activity (as measured by luciferase activity) of the ERE-luciferase reporter in the cells either overexpressing XBP1 or the cells depleted of XBP1 compared with their respective controls. This result suggests that levels of XBP1 in the cell might not affect the classical transcriptional activity of ERa mediated through the direct binding of ERE.
Figure 6.

ERE-mediated luciferase activity in XBP1 overexpressing or XBP1 depleted cells. MCF7 cells were transfected with empty vector (none), 20 ng or 500 ng of XBP1-expressing plasmid and ERE-mediated luciferase activity was assessed in absence or presence of 1 nM E2 (A). MCF7 cells transfected with control or XBP1 siRNA were used to assess ERE-mediated luciferase activity in presence or absence of 1 nM E2 (B). Renilla luciferase activity was used as internal control and all values are represented as a ratio of ERE-luciferase and renilla luciferase activity. The values are average of at least four replicates ±SD. *p<0.05 compared with respective vehicle-treated group.
Discussion
Estrogen is the prime growth regulator of ER positive breast and endometrial cancer cells. To better understand the induction of estrogen-mediated growth in breast cancer, some studies (6, 28) have explored the estrogen-induced transcriptional network using DNA microarrays to identify downstream pathways. In these studies, many of the estrogen-regulated genes are identified as transcription factors which could be collectively responsible for the phenotypic manifestations of estrogen-induced growth of ER positive cancer cells. However, in the majority of cases, the precise role of the downstream events in growth is not understood. One solution which can be used to dissect the complexities of clinical tissues is to interrogate estrogen responsive cell lines. One such recent study (29) has noted that the genes are similarly regulated by estrogen in breast cancer cells in vitro and human breast tumors. This provides a unique opportunity to study the underlying mechanism by manipulating specific genes in the cells, which can influence the progression of ER positive cancers and also provide potential targets for therapeutic intervention. In this context, some recent reports have identified the important roles played by the estrogen-regulated genes such as FOXA1 (10, 30, 31), GREB1 (32) and GATA-3 (30, 33) in regulating estrogen-induced growth in breast cancers.
In the present study, we evaluated the phenotypic effects of the E2-regulated gene XBP1, which has been consistently shown to be highly coexpressed with ERα in breast cancer patients and is also known to be upregulated by estrogen in the ER positive breast cancer cells in vitro (4–9, 29). Although it is well established that XBP1 plays a key role in UPR and endoplasmic reticulum stress by acting as a transcription factor for the genes involved in UPR, its role in E2-dependent ER positive cancers is not fully understood.
Our results confirm that XBP1 is an E2-regulated gene which is in agreement with previous studies (6, 10). The E2-induction of XBP1 is mediated by ERα and does not need de novo protein synthesis for the upregulation, as CHX did not alter the regulation. Treatment with DRB, an inhibitor of transcription, completely blocked the upregulation of XBP1 by estrogen, indicating transcriptional regulation. The ChIP data further confirmed direct binding of ERα, SRC-1, SRC-3 and serine-2-phosphorylated RNA polymerase II at the promoter and/or enhancer region of the XBP1 gene. Interestingly, recruitment of ERα and SRC-3 (AIB1) was higher at the enhancer region of the XBP1 gene and very minimal at the promoter region. ERa recruitment was induced dramatically at the enhancer region after 45 min of E2 treatment. Recruitment of ERα was also accompanied by SRC-1 and SRC-3 at the enhancer region. However, as expected, recruitment of serine-2-phosphorylated RNA polymerase II was higher at the promoter region than the enhancer region. These data strongly suggest that the enhancer region of the XBP1 gene, which is approximately 13 kb upstream of the transcription start site, is involved in the transcriptional regulation of XBP1 by ER. Indeed, recent studies (34–36) have indicated that distal enhancers of E2-induced genes GREB1 and carbonic anhydrase 12 are involved in the transcriptional regulation by ER. It has been shown that the distal enhancer can interact with the proximal promoter region of these estrogen-regulated genes by intrachromosomal looping.
This study reports for the first time that the XBP1 level is critical for E2-induced growth of ER positive breast and endometrial cancer cells, as evidenced by marked inhibition of E2-induced growth of XBP1-deficient MCF7 and ECC1 cells. This specifically demonstrates that the endogenous level of XBP1 and its upregulation by estrogen is intimately involved in the growth regulation of estrogen responsive breast and endometrial cancer cells. A recent study demonstrated that overexpression of XBP1 in ER positive breast cancer cells can lead to anti-estrogen resistance, by regulating genes associated with apoptosis and cell cycle progression (20).
To further understand the mechanism by which XBP1 can influence E2-induced growth, we hypothesized that levels of XBP1 could affect the ERE-mediated transcriptional activity of ER. To test this we depleted or overexpressed XBP1 and performed an ERE-luciferase reporter assay. Our data show that the level of XBP1 in the MCF7 cells does not affect the transcriptional activity of ER mediated through classical ERE binding. This indicates that XBP1 can influence the growth of the cells by either regulating a subset of genes directly under the control of XBP1 or can also modulate the E2 regulation of the genes which are not exclusively regulated by classical ERE-mediated transcription. These data are, however, in contrast to a previous study (19) where XBP1 overexpression activated ER transcriptional activity in a ligand-independent manner. The differences in the results could be attributed to the exogenous overexpression of ERa in the previous study, whereas in the present study we relied on the intrinsic activity of ER in the MCF7 cells. Further investigations are required to address the associated mechanism of action.
In summary, our results demonstrate that XBP1 expression is estrogen regulated at the transcriptional level and the enhancer region of the XBP1 gene can play a critical role in regulating E2-mediated transcriptional activation. Our findings show that expression level of XBP1 is critical in achieving optimal E2-induced growth in breast and endometrial cancer cells without influencing the classical ERE-mediated transcriptional activity. Our findings also provide an explanation for the strong correlation observed between ERa and XBP1 expression in breast cancer patients. Taken together, we suggest that this novel mechanism for the regulation of cancer cell growth via XBP1 can be exploited as a novel drug target in future studies of anti-hormonal resistance in ER positive cancer cells.
Acknowledgements
This research was supported by the Department of Defense Breast Program under award number BC050277 Center of Excellence; Fox Chase Cancer Center Core Grant NIH P30 CA006927; the Weg Fund of Fox Chase Cancer Center; the Genuardi fund and the Hollenbach Family Fund. The views and opinions of the author(s) do not reflect those of the US Army or the Department of Defense.
References
- 1.Jensen EV, Jordan VC. The estrogen receptor: a model for molecular medicine. Clin Cancer Res. 2003;9:1980–9. [PubMed] [Google Scholar]
- 2.van't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 3.Finlin BS, Gau CL, Murphy GA, Shao H, Kimel T, Seitz RS, Chiu YF, Botstein D, Brown PO, Der CJ, Tamanoi F, Andres DA, Perou CM. RERG is a novel ras-related, estrogen-regulated and growth-inhibitory gene in breast cancer. J Biol Chem. 2001;276:42259–67. doi: 10.1074/jbc.M105888200. [DOI] [PubMed] [Google Scholar]
- 4.Wang DY, Fulthorpe R, Liss SN, Edwards EA. Identification of estrogen-responsive genes by complementary deoxyribonucleic acid microarray and characterization of a novel early estrogen-induced gene: EEIG1. Mol Endocrinol. 2004;18:402–11. doi: 10.1210/me.2003-0202. [DOI] [PubMed] [Google Scholar]
- 5.Tozlu S, Girault I, Vacher S, Vendrell J, Andrieu C, Spyratos F, Cohen P, Lidereau R, Bieche I. Identification of novel genes that co-cluster with estrogen receptor alpha in breast tumor biopsy specimens, using a large-scale real-time reverse transcription-PCR approach. Endocr Relat Cancer. 2006;13:1109–20. doi: 10.1677/erc.1.01120. [DOI] [PubMed] [Google Scholar]
- 6.Cunliffe HE, Ringner M, Bilke S, Walker RL, Cheung JM, Chen Y, Meltzer PS. The gene expression response of breast cancer to growth regulators: patterns and correlation with tumor expression profiles. Cancer Res. 2003;63:7158–66. [PubMed] [Google Scholar]
- 7.Bertucci F, Nasser V, Granjeaud S, Eisinger F, Adelaide J, Tagett R, Loriod B, Giaconia A, Benziane A, Devilard E, Jacquemier J, Viens P, Nguyen C, Birnbaum D, Houlgatte R. Gene expression profiles of poor-prognosis primary breast cancer correlate with survival. Hum Mol Genet. 2002;11:863–72. doi: 10.1093/hmg/11.8.863. [DOI] [PubMed] [Google Scholar]
- 8.Bertucci F, Houlgatte R, Benziane A, Granjeaud S, Adelaide J, Tagett R, Loriod B, Jacquemier J, Viens P, Jordan B, Birnbaum D, Nguyen C. Gene expression profiling of primary breast carcinomas using arrays of candidate genes. Hum Mol Genet. 2000;9:2981–91. doi: 10.1093/hmg/9.20.2981. [DOI] [PubMed] [Google Scholar]
- 9.Fujimoto T, Onda M, Nagai H, Nagahata T, Ogawa K, Emi M. Upregulation and overexpression of human X-box binding protein 1 (hXBP-1) gene in primary breast cancers. Breast Cancer. 2003;10:301–6. doi: 10.1007/BF02967649. [DOI] [PubMed] [Google Scholar]
- 10.Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, Fox EA, Silver PA, Brown M. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 11.Koong AC, Chauhan V, Romero-Ramirez L. Targeting XBP-1 as a novel anti-cancer strategy. Cancer Biol Ther. 2006;5:756–9. doi: 10.4161/cbt.5.7.2973. [DOI] [PubMed] [Google Scholar]
- 12.Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, Mori K, Glimcher LH, Denko NC, Giaccia AJ, Le QT, Koong AC. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004;64:5943–7. doi: 10.1158/0008-5472.CAN-04-1606. [DOI] [PubMed] [Google Scholar]
- 13.Reimold AM, Etkin A, Clauss I, Perkins A, Friend DS, Zhang J, Horton HF, Scott A, Orkin SH, Byrne MC, Grusby MJ, Glimcher LH. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000;14:152–7. [PMC free article] [PubMed] [Google Scholar]
- 14.Lee AH, Chu GC, Iwakoshi NN, Glimcher LH. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005;24:4368–80. doi: 10.1038/sj.emboj.7600903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwakoshi NN, Lee AH, Glimcher LH. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol Rev. 2003;194:29–38. doi: 10.1034/j.1600-065x.2003.00057.x. [DOI] [PubMed] [Google Scholar]
- 16.Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH. Protea-some inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci USA. 2003;100:9946–51. doi: 10.1073/pnas.1334037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, Nieuwenhuis EE, Higgins DE, Schreiber S, Glimcher LH, Blumberg RS. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–56. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding L, Yan J, Zhu J, Zhong H, Lu Q, Wang Z, Huang C, Ye Q. Ligand-independent activation of estrogen receptor alpha by XBP-1. Nucleic Acids Res. 2003;31:5266–74. doi: 10.1093/nar/gkg731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang Y, Yan J, Ding L, Liu Y, Zhu J, Huang C, Zhao H, Lu Q, Zhang X, Yang X, Ye Q. XBP-1 increases ERa transcriptional activity through regulation of large-scale chromatin unfolding. Biochem Biophys Res Commun. 2004;323:269–74. doi: 10.1016/j.bbrc.2004.08.100. [DOI] [PubMed] [Google Scholar]
- 20.Gomez BP, Riggins RB, Shajahan AN, Klimach U, Wang A, Crawford AC, Zhu Y, Zwart A, Wang M, Clarke R. Human X-box binding protein-1 confers both estrogen independence and antiestrogen resistance in breast cancer cell lines. FASEB J. 2007;21:4013–27. doi: 10.1096/fj.06-7990com. [DOI] [PubMed] [Google Scholar]
- 21.Pink JJ, Jordan VC. Models of estrogen receptor regulation by estrogens and antiestrogens in breast cancer cell lines. Cancer Res. 1996;56:2321–30. [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003;144:4562–74. doi: 10.1210/en.2003-0567. [DOI] [PubMed] [Google Scholar]
- 24.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–52. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 25.Ariazi EA, Kraus RJ, Farrell ML, Jordan VC, Mertz JE. Estrogen-related receptor a1 transcriptional activities are regulated in part via the ErbB2/HER2 signaling pathway. Mol Cancer Res. 2007;5:71–85. doi: 10.1158/1541-7786.MCR-06-0227. [DOI] [PubMed] [Google Scholar]
- 26.Lewis JS, Osipo C, Meeke K, Jordan VC. Estrogen-induced apoptosis in a breast cancer model resistant to long-term estrogen withdrawal. J Steroid Biochem Mol Biol. 2005;94:131–41. doi: 10.1016/j.jsbmb.2004.12.032. [DOI] [PubMed] [Google Scholar]
- 27.Lewis JS, Meeke K, Osipo C, Ross EA, Kidawi N, Li T, Bell E, Chandel NS, Jordan VC. Intrinsic mechanism of estradiol-induced apoptosis in breast cancer cells resistant to estrogen deprivation. J Natl Cancer Inst. 2005;97:1746–59. doi: 10.1093/jnci/dji400. [DOI] [PubMed] [Google Scholar]
- 28.DeNardo DG, Kim HT, Hilsenbeck S, Cuba V, Tsimelzon A, Brown PH. Global gene expression analysis of estrogen receptor transcription factor cross talk in breast cancer: identification of estrogen-induced/activator protein-1-dependent genes. Mol Endocrinol. 2005;19:362–78. doi: 10.1210/me.2004-0267. [DOI] [PubMed] [Google Scholar]
- 29.Creighton CJ, Cordero KE, Larios JM, Miller RS, Johnson MD, Chinnaiyan AM, Lippman ME, Rae JM. Genes regulated by estrogen in breast tumor cells in vitro are similarly regulated in vivo in tumor xenografts and human breast tumors. Genome Biol. 2006;7:R28. doi: 10.1186/gb-2006-7-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lacroix M, Leclercq G. About GATA3, HNF3A, and XBP1, three genes co-expressed with the oestrogen receptor-alpha gene (ESR1) in breast cancer. Mol Cell Endocrinol. 2004;219:1–7. doi: 10.1016/j.mce.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 31.Laganiere J, Deblois G, Lefebvre C, Bataille AR, Robert F, Giguere V. From the cover: location analysis of estrogen receptor alpha target promoters reveals that FOXA1 defines a domain of the estrogen response. Proc Natl Acad Sci USA. 2005;102:11651–6. doi: 10.1073/pnas.0505575102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rae JM, Johnson MD, Scheys JO, Cordero KE, Larios JM, Lippman ME. GREB 1 is a critical regulator of hormone dependent breast cancer growth. Breast Cancer Res Treat. 2005;92:141–9. doi: 10.1007/s10549-005-1483-4. [DOI] [PubMed] [Google Scholar]
- 33.Eeckhoute J, Keeton EK, Lupien M, Krum SA, Carroll JS, Brown M. Positive cross-regulatory loop ties GATA-3 to estrogen receptor alpha expression in breast cancer. Cancer Res. 2007;67:6477–83. doi: 10.1158/0008-5472.CAN-07-0746. [DOI] [PubMed] [Google Scholar]
- 34.Barnett DH, Sheng S, Charn TH, Waheed A, Sly WS, Lin CY, Liu ET, Katzenellenbogen BS. Estrogen receptor regulation of carbonic anhydrase XII through a distal enhancer in breast cancer. Cancer Res. 2008;68:3505–15. doi: 10.1158/0008-5472.CAN-07-6151. [DOI] [PubMed] [Google Scholar]
- 35.Deschenes J, Bourdeau V, White JH, Mader S. Regulation of GREB1 transcription by estrogen receptor alpha through a multipartite enhancer spread over 20 kb of upstream flanking sequences. J Biol Chem. 2007;282:17335–9. doi: 10.1074/jbc.C700030200. [DOI] [PubMed] [Google Scholar]
- 36.Sun J, Nawaz Z, Slingerland JM. Long-range activation of GREB1 by estrogen receptor via three distal consensus estrogen-responsive elements in breast cancer cells. Mol Endocrinol. 2007;21:2651–62. doi: 10.1210/me.2007-0082. [DOI] [PubMed] [Google Scholar]