

Abstract
Objective
To characterize modifications of high-density lipoprotein (HDL) in autoimmune gld (generalized lymphoproliferative disorder) mice that may be relevant to premature atherosclerosis in systemic lupus erythematosus and assess their relationship to specific aspects of autoimmune disease.
Methods
HDL-cholesterol (HDL-C), apolipoprotein-A1 (ApoA1), paraoxonase-1 (PON1) activity, hepatic gene expression and HDL biogenesis were measured in ageing female gld and wild-type (WT) congenic mice. Autoantibodies, lymphoid organs and cytokines were analyzed by enzyme-linked immunosorbent assay, flow cytometry and multiplex assay respectively.
Results
Plasma HDL-C, HDL-ApoA1 and HDL-associated PON1 activity were reduced in ageing gld mice in association with the development of autoimmunity independently of changes in hepatic ApoA1 and PON1 expression or HDL biogenesis. Hepatic induction of the acute phase reactant, serum amyloid A-1, resulted in its incorporation onto HDL in gld mice. Deletion of the lipid-sensitive receptor, G2A, in gld mice (G2A-/-gld) attenuated reductions in HDL-C and PON1 activity without altering hepatic ApoA1 and PON1 expression, HDL biogenesis or levels of acute phase pro-inflammatory cytokines. Plasma anti-ApoA1 autoantibodies were elevated in ageing gld mice commensurate with detectable increases in ApoA1 immune complexes. Autoantibodies were lower in ageing G2A-/-gld mice compared to gld mice and anti-ApoA1 autoantibody levels were significantly related to HDL-C concentration (r=-0.645, p<0.00004) and PON1 activity (r=-0.555, p<0.0007) amongst autoimmune gld and G2A-/-gld mice.
Conclusion
Autoantibodies against ApoA1 contribute to reducing HDL-C and PON1 activity in autoimmune gld mice independently of hepatic HDL biogenesis, suggesting that functional impairment and premature clearance of HDL immune complexes may be principal mechanisms involved.
Reverse cholesterol transport (RCT) and paraoxonase-1 (PON1)-mediated anti-oxidant activity are considered major functional properties of high-density lipoprotein (HDL) responsible for its atheroprotective effects (1,2). RCT maintains normal cholesterol homeostasis in peripheral tissues by promoting the efflux of excess cholesterol onto HDL for transport to the liver and subsequent excretion into bile and feces (3). In addition to apolipoprotein-A1 (ApoA1), the main lipid-binding protein component of HDL, HDL-associated PON1 contributes to the protective anti-inflammatory effects of HDL by reducing the generation of pro-inflammatory oxidized phospholipids during the oxidative modification of low-density lipoprotein (LDL) in the vascular wall and eliminating bioactive lipids generated under conditions of oxidative stress (2).
The acute phase response (APR) is a protective systemic inflammatory reaction to infection orchestrated largely through the modulation of hepatic synthesis of specific plasma proteins by systemic increases in pro-inflammatory cytokines (4). The APR is accompanied by transient down-modulation of plasma HDL-cholesterol (HDL-C) levels, PON1 activity and HDL-associated anti-inflammatory properties (5,6). Similar reductions of HDL-C and PON1 activity have been reported in rodent models of infection and inflammation (7). Hepatic synthesis of the positive acute phase protein, serum amyloid A-1 (SAA1), is increased during the APR (4,5) and subsequently incorporated onto HDL (8). Although SAA1 incorporation onto HDL in vitro has been proposed to displace ApoA1, the major protein constituent of HDL mediating its cholesterol transport function and essential for maintaining optimal PON1 activity on HDL (9,10), it remains unclear whether SAA1 can displace ApoA1 from HDL in vivo (11). Systemic inflammation may also modulate HDL levels via transcriptional changes in the hepatic expression of major proteins involved in HDL metabolism such as ATP binding cassette (ABC) transporters and the HDL-C scavenger receptor class B type 1 (SRB1) (12,13). Studies in mice demonstrating down-modulatory effects of HDL and Liver X Receptor-dependent cholesterol efflux on lymphocyte proliferation and activation (14,15) support a scenario in which these inhibitory effects of the APR on HDL may serve to facilitate subsequent immune responses by transiently overriding the anti-inflammatory influence of HDL and attenuating RCT to optimize cholesterol pools in immune cells required for their proliferative expansion. However, if prolonged due to a failure to eliminate the inflammatory stimulus, or in the context of chronic inflammatory disease, they can result in the sustained impairment of HDL which may provoke an increased risk for atherosclerosis (16).
Premature atherosclerosis remains a major cause of morbidity and mortality in patients with systemic lupus erythematosus (SLE) and other rheumatic autoimmune diseases (17). The prevalence of reduced HDL-C levels and PON1 activity in SLE patients suggests that autoimmune-mediated HDL dysfunction may be a significant factor contributing to their predisposition to premature atherosclerosis (18-20). Furthermore, elevated levels of ApoA1-binding autoantibodies have been reported in SLE patients (18,19) which may lead to premature clearance and/or functional impairment of HDL immune complexes. Although hypercholesterolemic models of atherosclerosis such as apolipoprotein-E deficient (ApoE-/-) and LDL receptor deficient (LDLR-/-) C57BL/6J mice on SLE-prone genetic backgrounds recapitulate the synergistic relationship between SLE and atherosclerosis observed in humans (21,22), characterization of autoimmune-mediated effects on HDL in these mice is hampered by the significant disruptions in normal lipoprotein metabolism and inflammatory processes caused by deficiency of ApoE or the LDL receptor (23,24). We therefore examined modifications of HDL in normolipidemic SLE-prone Fas ligand mutant C57BL/6J gld (generalized lymphoproliferative disorder) mice to determine whether modulatory effects that may be mechanistically relevant to premature atherosclerosis in SLE are recapitulated in this model and to assess their relationship to specific aspects of autoimmune disease.
Materials and Methods
Animals
Congenic G2A+/+ (WT), G2A-/-, G2A+/+gld (gld) and G2A-/-gld mice were derived by breeding N10 C57BL/6J G2A-/-LDLR-/- (25) with C57BL/6J gld mice (B6Smn.C3-Tnfsf6gld/J; Jackson Laboratories) and subsequently intercrossing compound heterozygotes. Female WT, G2A-/-, gld and G2A-/-gld mice (all LDLR+/+) were maintained on a standard rodent chow diet (Diet #5015; Harlan Teklad).
Measurement of lipoprotein profiles, PON1 activity and FPLC
Plasma lipids were measured as previously described (25). Plasma (200μl pooled from 3 mice) was fractionated using a Superose-6 column (Amersham Biosciences) on a Biologic DuoFlow FPLC system (Bio-Rad) as previously described (26). PON1 activity was measured using 1mM Paraoxon (Supelco) as substrate (1 unit of PON1 activity=1 nmole of 4-nitrophenol/minute).
Hepatocyte isolation and culture
Hepatocytes were isolated and cultured for analysis of ApoA1 and SAA1 secretion as previously described (26).
SDS-polyacrylamide gel electrophoresis and immunoblotting
FPLC fractions or hepatic protein lysates prepared as described previously (26) were separated on 10-20% gradient SDS acrylamide/bis-tris gels (BioRad) and transferred to Immobilon-P membranes (Millipore). Membranes were blocked with PBS containing 0.05% Tween-20 and 5% non-fat milk, incubated with primary antibodies (rabbit anti-ApoA1; Biodesign International, rabbit anti-SAA1; R&D Systems, rabbit anti-SR-B1; Novus Biologicals) followed by horseradish peroxidase (HRP)-conjugated secondary antibodies. Immunoreactive proteins were visualized using enhanced chemiluminescence and quantified densitometrically using Image J (NIH).
Quantitative real-time PCR
Quantitative real-time PCR analysis of hepatic gene expression was performed as previously described (26). Data were analyzed using the relative expression method: Relative expression = 2-(SΔCT-CΔCT) where ΔCT is the average difference in cycle threshold (CT) between the gene of interest and the housekeeping gene, cyclophilin (CYC), in G2A-/-, gld or G2A-/-gld (S) and WT (C) cells. Forward (F) and reverse (R) primers were as follows: CYC F:TGGAGAGCACCAAGACAGACA, CYC R:TGCCGGAGTCGACAATGAT. ApoA1 F:TTCGCTAATGTGTATGTG, ApoA1 R:TCTTTCTCCAGGTTATCC. SAA1 F:GGACTGCCTGACAAATACTGAG, SAA1 R:GAGCATCTTCAGTGTTCCTAGG. PON1 F:CACTCTTGCATCTGAAAACCATC, PON1 R:AGACCCAAGTACATTTCCCAG. ABCA1 F:TACCCACCCTACGAACAAC, ABCA1 R:TGAGAACAGGCGAGACAC. SRB1 F:GCAAATTTGGCCTGTTTGTT, SRB1 R:AGGATTCGGGTGTCATGAAG.
Measurement of autoantibody levels
96-well Maxisorp plates (Nunc) were coated with double-stranded DNA (dsDNA) (Sigma; 0.24μg/well) histone (Sigma; 2.4μg/well) or mouse ApoA1 (Biodesign International; 0.1μg/well) in carbonate buffer (150mM Na2CO3, 350mM NaHCO3, pH9.7) overnight at 4°C. Plates were washed with PBS 0.5% Tween (PBST) using an automated plate-washer (BioTek) and blocked with PBST containing 3% non-fat milk (blocking buffer). Plates were subsequently incubated with diluted plasma (1:100 in blocking buffer) for 2 hours, washed, and incubated with a 1:10,000 dilution of HRP-conjugated anti-mouse IgG1 (BD Biosciences), anti-mouse IgG2b, anti-mouse IgG2c antibody or anti-mouse IgG antibody (Southern Biotech). Plates were washed, developed with TMB substrate (Pierce) and measured spectrophotometrically at 450nm. For measurement of plasma ApoA1 (HDL)-IgG immune complexes, maxisorp plates were coated overnight at 4°C with 5μg/ml of goat anti-mouse IgG (Southern Biotech). Plates were blocked with blocking buffer and subsequently incubated with diluted plasma (1:50 in blocking buffer) for 2 hours, washed and incubated with a 1:10,000 dilution of rabbit anti-mouse ApoA1 antibody (Biodesign International). Plates were incubated with a 1:10,000 dilution of HRP-conjugated anti-rabbit IgG (BioRad), washed, developed with TMB substrate and measured spectrophotometrically at 450nm.
Plasma cytokines and SAA1
Plasma concentrations of interferon-γ, IL-6, TNFα and IL-1β were measured using a custom Bioplex mouse cytokine array (BioRad) following the manufacturer's protocol. Total plasma SAA1 levels were measured using a mouse SAA1 ELISA kit from Biosource following the manufacturers protocol.
Flow cytometric analysis
CD4+ and CD8+ T lymphocytes, B220+ B lymphocytes and effector/memory CD4+ T lymphocytes (CD4+CD44highCD62Llow) were analyzed in single cell suspensions prepared from collagenase-digested lymph nodes (iliac, axillary and superficial cervical) of 28 week-old WT, G2A-/-, gld and G2A-/-gld mice by flow cytometry using a BD FACS Calibur. All antibodies (CD4PerCP, CD8APC, B220PerCP, CD44FITC, CD62LPE) were from BD Biosciences. Data analysis was performed using WinMDI software.
LPC measurement
LPC was measured in plasma from 28 week-old WT, G2A-/-, gld and G2A-/-gld mice by electrospray ionization tandem mass spectrometry (ESI-MS/MS) as previously described (27) except that lipid samples were delivered into the ESI source without prior thin-layer chromatographic separation.
Statistics
Statistical analysis of plasma lipoprotein levels, PON1 activity, relative gene expression, cytokine concentrations and autoantibody levels was performed by ANOVA. Pair-wise statistical comparisons between mice were performed by unpaired Student's t test. Correlation analysis was performed by Pearson Product Moment test. Statistical tests were performed using SigmaPlot 11 software (Systat Software).
Results
Plasma HDL-C levels and HDL-associated PON1 activity are reduced in ageing gld mice
Female C57BL/6J gld mice developed statistically significant reductions in HDL-C after 12 weeks of age (47% reduction at 20 weeks and 56% reduction at 28 weeks of age compared to age-matched female congenic WT mice) (Figure 1A). Despite these reductions in HDL-C, the proportion of esterified cholesterol (EC) was unaffected in gld mice (EC:TC ratio in Table 1), reflecting the absence of a significant effect of autoimmunity on HDL-associated lecithin acyl-cholesterol transferase (LCAT) activity. Plasma PON1 activity was also reduced in ageing gld mice, albeit to a lesser extent compared to HDL-C levels (average 24% reduction at 20 weeks and 26% reduction at 28 weeks of age compared to age-matched WT mice) (Figure 1A). Reductions in plasma HDL-C and HDL-associated PON1 activity were confirmed in 28 week-old gld mice by measurement of cholesterol and PON1 activity on lipoprotein fractions separated by fast performance liquid chromatography (FPLC) (Figure 1C).
Figure 1.

Reduction of HDL-cholesterol (HDL-C) levels and PON1 activity associated with the development of autoimmunity in gld mice. (A) Plasma HDL-C concentrations (mg/dL) and PON1 activity (nmoles/min/ml) in ageing WT (n=14) and gld mice (n=17) (age indicated in weeks: wk). Means with standard deviation and statistically significant differences shown: *p<0.05. (B) Reductions in HDL-C and PON1 activity are attenuated in G2A-/-gld mice. Plasma HDL-C concentrations (mg/dL) and PON1 activity (nmoles/min/ml) in ageing G2A-/- (n=10) and G2A-/-gld mice (n=14) (age indicated in weeks: wk). Means with standard deviation and statistically significant differences shown: *p<0.05. (C) Cholesterol levels (μg/fraction) and PON1 activity (nmoles/min/fraction) in FPLC separated plasma lipoproteins from 28 week-old WT, gld, G2A-/- and G2A-/-gld mice. Data shown is representative of 3 independent experiments.
Table 1.
Plasma lipid profiles and PON1 activity in 28 week-old control and lupus-prone gld mice (n≥10). Means with standard deviations shown. Statistically significant differences between control and gld mice of the same G2A genotype († p<0.05) and betwen gld and G2A-/-gld mice (*p<0.05). TC: Total cholesterol. EC: Esterified cholesterol. LDL-C: LDL-cholesterol. HDL-C: HDL-cholesterol. PON1 activity in nmoles/min/ml plasma.
| TC (mg/dl) | EC (mg/dl) | EC:TC ratio | LDL-C (mg/dl) | HDL-C (mg/dl) | PON1 activity | |
|---|---|---|---|---|---|---|
| WT | 92.2 ± 9.9 | 70.1 ± 8.8 | 0.75 ± 0.02 | 20.5 ± 4.6 | 72.4 ± 7.7 | 381.6 ± 55.5 |
| gld | 56.7 † ± 17.5 | 42.4 † ± 13 | 0.74 ± 0.08 | 20.8 ± 4.7 | 32.0 † ± 13.24 | 283.9 † ± 106.5 |
| G2A-/- | 96.5 ± 12.4 | 68.5 ± 4.9 | 0.76 ± 0.02 | 18.6 ± 4.7 | 75.5 ± 6.9 | 345 ± 74.4 |
| G2A-/-gld | 75.2 * † ± 17.6 | 60.6 * ± 13.3 | 0.72 ± 0.15 | 20.3 ± 7.0 | 47.7 * † ± 13 | 391.3 ± 146.4 |
Attenuated reductions in HDL-C and PON1 activity in G2A-deficient gld mice
G2A is lipid-sensitive receptor (28) whose deletion in mice on a mixed BALBc/129Sv genetic background resulted in the development of a spontaneous late-onset systemic autoimmune syndrome reminiscent of SLE (29). Although a similar predisposition to autoimmunity in G2A-/- mice on a pure genetic background and the consequences of G2A deficiency in an SLE-prone mouse model have not been reported, we reasoned that G2A deficiency might promote the development of autoimmunity in gld mice and thereby exacerbate the reductions in HDL-C and PON1 activity. We therefore generated G2A-deficient C57BL/6J gld mice (G2A-/-gld). HDL-C levels and PON1 activity were comparable in WT and G2A-/- mice at all ages examined. Surprisingly, reductions in plasma HDL-C levels in ageing G2A-/-gld mice were significantly attenuated compared to those in gld mice (average 30% reduction at 20 weeks and 37% reduction at 28 weeks of age compared to age-matched G2A-/- mice) (Figure 1B). HDL-C levels were reduced by 32% in G2A-/-gld mice between 12 and 28 weeks of age compared to 48% in gld mice. Furthermore, the moderate reductions in PON1 activity in ageing gld mice were essentially ameliorated in the absence of G2A (Figure 1B-C).
Interestingly, reductions in HDL-C levels in gld mice were associated with significantly lower plasma concentrations of major species of lysophosphatidylcholine (LPC), a lipid product of PON1-mediated oxidized phosphatidylcholine (PC) hydrolysis (Supplementary figure 1A) (30). Furthermore, similar LPC reductions were not observed in age-matched G2A-/-gld mice in which PON1 activity remained comparable to that in their WT and G2A-/- counterparts (Supplementary figure 1A). Finally, the concentrations of all LPC species measured correlated significantly with HDL-C levels and PON1 activity in gld and G2A-/-gld mice (Supplementary figure 1B).
Reciprocal alterations in HDL ApoA1 and SAA1 content in gld mice
ApoA1 immunoreactivity of FPLC-separated HDL fractions from gld mice was reduced (Figure 2A) and the attenuated reduction in plasma HDL-C levels in G2A-/-gld mice compared to their gld counterparts was associated with higher ApoA1 levels in the former group of mice (Figure 2A). SAA1 incorporation was observed in FPLC separated HDL fractions from gld mice compared to those from WT mice (Figure 2A). Interestingly, the higher ApoA1 content of HDL fractions from G2A-/-gld mice compared to gld mice was associated with a lower SAA1 content (Figure 2A). Although a trend towards decreased hepatic SAA1 gene expression was observed in G2A-/-gld mice, this was not statistically significant (Figure 2B). Similarly to hepatic SAA1 mRNA levels (Figure 2B), SAA1 protein levels in HDL fractions from gld mice were highly variable, although consistently lower in G2A-/-gld mice (Supplementary figure 2A). Total plasma concentrations of SAA1 were similarly highly variable and reduced in G2A-/-gld mice compared to their age-matched gld counterparts, although not to a statistically significant extent (Supplementary figure 2A). Finally, in accordance with the lack of a statistically significant reduction in hepatic SAA1 gene expression in G2A-/-gld mice compared to their gld counterparts, systemic levels of acute phase pro-inflammatory cytokines mediating hepatic SAA1 induction were highly variable and not significantly different in gld and G2A-/-gld mice (Supplementary figure 2B).
Figure 2.
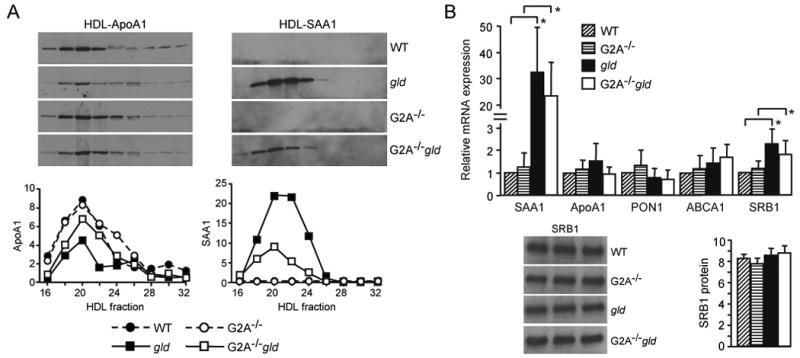
(A) HDL ApoA1 reduction and incorporation of SAA1 onto HDL in autoimmune gld mice. Immunoblot of the indicated FPLC-separated HDL fractions from 28 week-old WT, gld, G2A-/- and G2A-/-gld mice (as in Figure 1) showing reduced levels of ApoA1 (left panel) and incorporation of SAA1 onto HDL (right panel) in gld mice. Densitometric quantification of ApoA1 and SAA1 immunoreactivity in HDL fractions is shown below. Data shown is representative of 3 independent experiments. (B) Quantitative RT-PCR analysis of the expression of the indicated genes in livers from 28 week-old WT, gld, G2A-/- and G2A-/-gld mice. Data is the average from 6 mice in each group (means with standard deviation shown). *p<0.05. Inset in (B): Immunoblot analysis of liver SRB1 expression in 3 WT, 3 G2A-/-, 3 gld and 3 G2A-/-gld mice with densitometric quantification of SRB1 immunoreactivity shown alongside (means with standard deviation).
Hepatic HDL biogenesis is unaffected by autoimmunity in gld mice
We investigated whether decreased hepatic HDL biogenesis contributed to the reductions in HDL-C levels in gld mice. No statistically significant differences in hepatic ApoA1, PON1 or ABCA1 expression were observed between WT, gld, G2A-/- and G2A-/-gld mice by real-time PCR (Figure 2B). Furthermore, hepatocytes from WT, gld, G2A-/- and G2A-/-gld mice secreted comparable amounts of ApoA1 in culture (Figure 3). Thus, alterations in hepatic HDL biogenesis do not contribute to the reduced plasma HDL-C levels in gld mice or the attenuated reductions in HDL-C levels in G2A-/-gld mice compared to their gld counterparts. Finally, although hepatic SRB1 mRNA levels were increased approximately 2-fold in gld and G2A-/-gld mice compared to their age-matched WT and G2A-/- counterparts, significant increases in SRB1 protein levels were not detected (Figure 2B).
Figure 3.
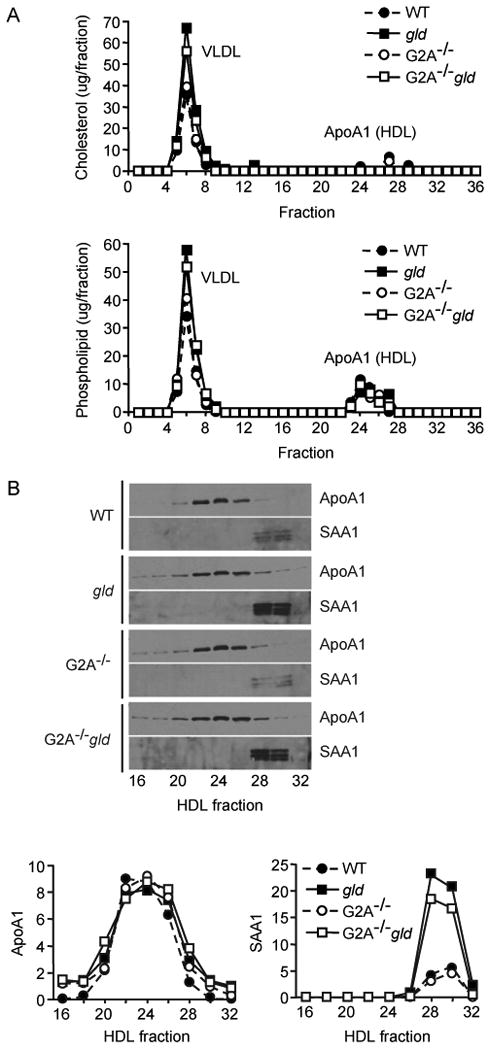
Hepatocyte ApoA1-HDL secretion is unaffected by autoimmunity in gld mice. (A) FPLC-separated lipoproteins secreted into culture medium over 12 hours by freshly isolated hepatocytes from 28 week-old WT, gld, G2A-/- and G2A-/-gld mice. (B) Immunoblot of HDL fractions showing comparable levels of ApoA1 in HDL fractions and induction of hepatocyte SAA1 secretion in gld and G2A-/-gld mice. Densitometric quantification of ApoA1 and SAA1 immunoreactivity in the indicated HDL fractions shown below. Data shown is representative of 3 independent experiments.
Hepatic SAA1 secretion was markedly increased in gld mice (Figure 3B), consistent with the increase in hepatic SAA1 mRNA levels (Figure 2B) and SAA1 content of plasma HDL fractions (Figure 2A). Furthermore, only minor reductions were observed in the amount of SAA1 secreted by hepatocytes from G2A-/-gld mice compared to those from gld mice (Figure 3B), suggesting that differences in autoimmune-mediated effects on processes other than hepatic SAA1 production may be responsible for the lower levels of SAA1 on circulating HDL in G2A-/-gld mice.
HDL-C levels and PON1 activity are inversely related to anti-ApoA1 autoantibody levels in gld and G2A-/-gld mice
Autoantibodies recognizing ApoA1 are increased in SLE patients and have been suggested to contribute to reducing HDL-C and PON1 activity by eliciting the formation of HDL immune complexes (18,19,31). We therefore measured the relative levels of plasma autoantibodies in ageing WT, gld, G2A-/- and G2A-/-gld mice. Surprisingly, while anti-dsDNA and anti-histone autoantibody levels were comparable in ageing WT and G2A-/- mice (Figure 4), levels of most anti-dsDNA and anti-histone IgG autoantibody isotypes were moderately reduced in ageing G2A-/-gld mice compared to their age-matched gld counterparts (Figure 4). Although a trend towards reduced expansion of lymph node CD4+ T lymphocytes, CD8+ T lymphocytes and B220+ B lymphocytes was observed in G2A-/-gld mice compared to their gld counterparts, this was not statistically significant (Supplementary figure 3A). However, the number of effector/memory CD4+CD44highCD62Llow T cells was significantly lower in lymph nodes from G2A-/-gld mice (Supplementary figure 3B). In addition, the increase in average spleen weight was significantly attenuated in G2A-/-gld mice compared to their gld counterparts (Supplementary figure 3C) and this was similarly associated with reduced numbers of each major lymphocyte subset (CD4+, CD8+ and B220+; data not shown).
Figure 4.

Relative levels of plasma anti-dsDNA and anti-histone IgG autoantibodies in ageing WT, G2A-/-, gld, and G2A-/-gld mice. Means with standard deviation shown. (wk: age in weeks).
Plasma anti-ApoA1 autoantibodies were elevated in ageing gld mice compared to age-matched WT mice (Figure 5A). Despite the significantly lower levels of plasma HDL (ApoA1) in gld mice compared to age-matched WT mice (Figure 2A), significant increases in ApoA1-IgG immune complexes (IC) could be detected by measuring ApoA1 immunoreactivity in plasma IgG fractions captured using an anti-mouse IgG antibody (Figure 5A). Anti-ApoA1 autoantibody and ApoA1-IgG IC levels were significantly lower in G2A-/-gld mice compared to their gld counterparts (Figure 5A). As a statistically significant reduction in HDL-C was first apparent at 20 weeks of age in gld and G2A-/-gld mice (Figure 1), we performed statistical correlation analyses at this time-point to determine the relationship of HDL-C concentrations to autoantibody levels. A significant inverse relationship was found between HDL-C concentration and the levels of most autoantibodies (Supplementary table 1). Similar findings were obtained with an identical analysis of combined data from 20 and 28 week-old gld and G2A-/-gld mice (Supplementary table 2), indicating that the extent of autoimmunity was a significant determinant of the degree to which HDL-C levels were reduced in autoimmune gld and G2A-/-gld mice. Inverse correlations between HDL-C and anti-ApoA1 autoantibody levels, and between PON1 activity and anti-ApoA1 autoantibody levels were highly significant amongst gld and G2A-/-gld mice (Figure 5B and Supplementary tables 1-2), supporting a causal relationship between anti-ApoA1 autoantibodies and HDL reduction in SLE-prone gld mice.
Figure 5.

Plasma HDL-C concentration and PON1 activity inversely correlate with anti-ApoA1 autoantibody levels in autoimmune gld and G2A-/-gld mice. (A) Anti-ApoA1 autoantibody levels in ageing WT, G2A-/-, gld and G2A-/-gld mice (n=8). Plasma ApoA1-IgG immune complex (IC) levels in WT, G2A-/-, gld and G2A-/-gld mice shown alongside (20 and 28 week-old mice combined, *p<0.05). Means with standard deviation shown. (B) Statistically significant inverse correlation between anti-ApoA1 autoantibody levels and HDL-C, and between anti-ApoA1 autoantibody levels and PON1 activity in gld and G2A-/-gld mice (20 and 28 week-old mice combined). Correlation coefficient (r) and statistical significance (p) are indicated in each plot.
Discussion
The development of autoimmunity in C57BL/6J gld mice was associated with reductions in HDL-C levels and PON1 activity. HDL-C levels and PON1 activity were inversely related to anti-ApoA1 autoantibody levels amongst gld and G2A-/-gld mice, corroborating observations in SLE patients with respect to the potential involvement of anti-ApoA1 autoantibodies in mediating reductions in HDL-C and PON1 activity that may contribute to their increased risk for atherosclerosis (18,19,31). However, given the association of most autoantibodies in gld mice with HDL-C levels (Supplemental tables 1 and 2), further studies are required to provide definitive proof that anti-ApoA1 autoantibodies directly contribute to HDL reduction in these mice.
Similar reductions in HDL-C were not reported in a previous study of normolipidemic SLE-prone mouse strains on different genetic backgrounds and higher plasma HDL-C levels were reported in autoimmune MRL/lpr and NZB/BINJ mice compared to WT mice of different genetic backgrounds (32,33). A possible explanation for this apparent discrepancy between the current data in congenic C57BL/6J WT and C57BL/6J gld mice and those in the afore-mentioned studies may be the heterogeneity of genetic backgrounds between the SLE-prone and control WT strains examined in the latter (32,33). Indeed, a quantitative trait locus analysis using MRL/lpr × BALB/cJ second generation (F2) intercross mice revealed a significant locus for plasma HDL-C levels resulting from the mutant Fas gene carried in the parental MRL/lpr strain (33). Furthermore, HDL-C levels inversely correlated with autoantibody levels in these F2 mice, leading the authors of the study to infer an autoimmune-mediated reduction in HDL-C levels (33). It is also important to consider the inherent differences in immune responses between C57BL/6J mice and other mouse strains. In this respect, C57BL/6J and BALB/cJ mice typically exhibit TH1 and TH2 biased immunity respectively, producing distinct patterns of cytokine and humoral responses that may influence autoimmunity. Indeed, the penetrance of autoimmune disease in SLE-prone mice is strongly influenced by genetic background (34). In this respect, our finding that G2A deficiency reduced anti-dsDNA and anti-histone autoantibody levels in C57BL/6J gld mice, while one previous study reported the spontaneous development of a late-onset SLE-like autoimmune syndrome in G2A-/- mice on a mixed BALBc/129Sv genetic background (29) is not necessarily conflicting. Indeed, a similar predisposing effect of G2A deficiency to spontaneous autoimmunity in comparative studies of G2A-/- mice with their congenic WT counterparts has not been reported and was not detected in the current study. Furthermore, we have monitored ageing WT and G2A-/- C57BL/6J congenic mice up to 1 year of age and found no evidence for an increased susceptibility of the latter to spontaneous autoimmunity (Parks, BW and Kabarowski JH, unpublished data). It may also be noteworthy that G2A deficiency in C57BL/6J mice did not promote autoimmunity in the context of experimental autoimmune encephalomyelitis (35). The spontaneous development of a late-onset autoimmune syndrome in BALBc/129Sv G2A-/- mice may therefore have been conferred by differences in uncharacterized genetic loci.
Although the lower autoantibody levels in G2A-/-gld mice compared to their age-matched gld counterparts is suggestive of a suppression of autoimmunity, additional studies, including assessment of activation markers on B lymphocytes and other immunoregulatory cell-types, will be required to support this conclusion and to establish the cellular and molecular basis for the effects of G2A deficiency. In this respect, it may be noteworthy that anti-histone (but not anti-dsDNA) autoantibody IgG2c:IgG1 ratios were significantly lower in G2A-/-gld mice at 20 and 28 weeks of age compared to their age-matched gld counterparts (1.1±0.82 vs 0.43±0.29 and 1.2±0.77 vs 0.44±0.29 for 20 and 28 week-old gld vs G2A-/-gld mice respectively), suggesting a TH2 bias in anti-histone autoantibody responses in G2A-/-gld mice. Despite the current absence of a mechanistic explanation for these effects of G2A deficiency in gld mice, the cumulative data pointed to a possible causal relationship between anti-ApoA1 autoantibodies and HDL-C reduction in gld and G2A-/-gld mice. Several mechanisms have been proposed to explain how anti-ApoA1 autoantibodies may lead to quantitative and qualitative changes in HDL, including premature clearance of HDL immune complexes. Indeed, reductions in HDL cholesterol, ApoA1 and PON1 activity in gld mice are consistent with a reduction in the number of circulating HDL particles. Furthermore, the reduction in HDL levels in gld mice was not due to decreased hepatic HDL biogenesis (Figure 3), supporting increased HDL clearance as a mechanism involved. However, we cannot exclude the possibility that a lower stoichiometry of ApoA1 on HDL particles may also contribute to the reduced ApoA1 immunoreactivity observed in HDL fractions from gld mice (Figure 2A). Indeed, reductions in HDL ApoA1 were associated with increases in HDL SAA1 content in gld mice (Figure 2A). Considering that SAA1 was reported to displace ApoA1 from HDL particles in vitro (9), it is possible that ApoA1 displacement by SAA1 may contribute to the reduced ApoA1 immunoreactivity of HDL fractions from gld mice. However, there is considerable debate as to whether SAA1 can displace ApoA1 from HDL in vivo. Notably, hepatic SAA1 overexpression alone in the absence of an APR or chronic inflammation failed to reduce HDL ApoA1 content or HDL-C levels in mice (11), although it did attenuate RCT in a rodent macrophage-to-feces model (36). This suggests that other modulatory processes associated with inflammation may be required in concert with hepatic SAA1 induction to affect ApoA1 stoichiometry on HDL. A corollary to this is that the amount of SAA1 incorporated onto HDL in the context of SLE may be significantly influenced by factors other than its level of hepatic production. Considering the significant relationship between anti-ApoA1 autoantibodies and HDL-C levels (Figure 5B and Supplementary table 1), it is perhaps worth considering that the binding of these autoantibodies to ApoA1 on HDL particles may also facilitate SAA1 incorporation by disrupting apolipoprotein/lipid interactions and/or promoting ApoA1 dissociation.
Although not directly addressed in the current study, oxidative modification leading to compositional changes in phospholipids is a major feature of dysfunctional HDL in certain inflammatory states and is normally counteracted by the action of PON1 (37). The maintenance of higher levels of HDL-associated PON1 activity in G2A-/-gld mice compared to their gld counterparts may therefore be significant in terms of minimizing the extent of oxidative phospholipid modification on HDL particles and preserving their functional properties. Indeed, the lower plasma HDL-C and PON1 activity in gld mice compared to WT, G2A-/- and G2A-/-gld mice was associated with reduced plasma concentrations of certain species of LPC, a product of PON1-mediated oxidized PC hydrolysis (Supplementary figure 1A) (30). Importantly, the concentrations of all LPC species measured correlated with HDL-C levels and PON1 activity amongst gld and G2A-/-gld mice (Supplementary figure 1B). However, it is important to note that in addition to PON1, the hydrolytic activity of platelet activating factor-acetylhydrolase (PAF-AH), which is present largely on HDL in mice, is also a major source of LPC (38). Decreased activity of this enzyme may therefore contribute to the reduced plasma LPC levels in gld mice. However, there are conflicting reports of both decreased and increased PAF-AH activity in SLE patients (39,40) as well as in rodent models of the APR (41,42). Nevertheless, while LPC has been implicated as an autoantibody-binding antigenic epitope in SLE (43), to our knowledge, this is the first evidence for an association between plasma LPC concentration and autoimmune-mediated HDL-C/PON1 reduction in SLE, suggesting that LPC species may be useful biomarkers for HDL dysfunction in SLE.
Despite statistically significant reductions in HDL-C levels in G2A-/-gld mice, PON1 activity was maintained at levels comparable to WT and G2A-/- mice (Figure 1 and Table 1). Although this may appear incongruous in light of the cumulative data pointing to reductions in HDL particle number in gld mice, HDL-C and ApoA1 reduction in G2A-/-gld mice was significantly less pronounced compared to that in their gld counterparts (Figures 1 and 2A, Table 1). Furthermore, unlike cholesterol, PON1 is not necessarily present on all HDL particles and it is possible that disruption of critical ApoA1/PON1 interactions (9,10) due to anti-ApoA1 autoantibody binding may result in dissociation of PON1 prior to immune complex-mediated HDL clearance, followed by its incorporation onto other HDL particles. Indeed, there was a preferential loss of PON1 activity on larger HDL particles in gld and G2A-/-gld mice relative to that in WT and G2A-/- mice (indicated by a rightward shift of the corresponding PON1 activity profiles in Figure 1C). However, this loss of PON1 activity on large HDL particles could also be reflective of compositional changes in their phospholipids elicited by HDL oxidation and enzymatic hydrolysis that inhibit PON1 activity (44,45), a possibility we are currently investigating. Indeed, the impact of autoimmunity and systemic inflammation on HDL is complex and multi-factorial, involving modifications of lipid and protein composition as well as cholesterol metabolism that may contribute to reducing cholesterol levels and PON1 activity on HDL particles (46). Acute phase enzymes such as group II secretory phospholipase (sPLA2-IIA) may also contribute to the modifications of HDL associated with SLE by generating lipid-poor ApoA1 and promoting HDL catabolism (47,48). Mechanisms other than anti-ApoA1 autoantibodies may therefore contribute significantly to HDL-C and PON1 reduction in gld mice. Indeed, although HDL-C levels were not reduced in gld mice between 4 and 12 weeks of age, they were nevertheless lower in 12 week-old gld mice compared to their age-matched WT counterparts (Figure 1A) despite the absence of an increase in anti-ApoA1 autoantibodies (Figure 5A). It will therefore be important to determine the exact contribution of anti-ApoA1 autoantibodies to HDL-C reduction in normolipidemic SLE-prone mice and to distinguish their effects on specific functional properties of HDL from those mediated by enzymatic and oxidative modifications of HDL.
Supplementary Material
Acknowledgments
Supported by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS P30 AR048311) and the National Heart, Lung, and Blood Institute (NHLBI RO1 HL088642).
References
- 1.Lewis GF, Rader DJ. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ Res. 2005 Jun 24;96(12):1221–32. doi: 10.1161/01.RES.0000170946.56981.5c. [DOI] [PubMed] [Google Scholar]
- 2.Shih DM, Lusis AJ. The roles of PON1 and PON2 in cardiovascular disease and innate immunity. Curr Opin Lipidol. 2009 Aug;20(4):288–92. doi: 10.1097/MOL.0b013e32832ca1ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cuchel M, Rader DJ. Macrophage reverse cholesterol transport: key to the regression of atherosclerosis? Circulation. 2006 May 30;113(21):2548–55. doi: 10.1161/CIRCULATIONAHA.104.475715. [DOI] [PubMed] [Google Scholar]
- 4.Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999 Feb 11;340(6):448–54. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 5.Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, Feingold KR, et al. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res. 2004 Jul;45(7):1169–96. doi: 10.1194/jlr.R300019-JLR200. [DOI] [PubMed] [Google Scholar]
- 6.Van Lenten BJ, Hama SY, de Beer FC, Stafforini DM, McIntyre TM, Prescott SM, et al. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J Clin Invest. 1995 Dec;96(6):2758–67. doi: 10.1172/JCI118345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feingold KR, Memon RA, Moser AH, Grunfeld C. Paraoxonase activity in the serum and hepatic mRNA levels decrease during the acute phase response. Atherosclerosis. 1998 Aug;139(2):307–15. doi: 10.1016/s0021-9150(98)00084-7. [DOI] [PubMed] [Google Scholar]
- 8.van der Westhuyzen DR, de Beer FC, Webb NR. HDL cholesterol transport during inflammation. Curr Opin Lipidol. 2007 Apr;18(2):147–51. doi: 10.1097/MOL.0b013e328051b4fe. [DOI] [PubMed] [Google Scholar]
- 9.Coetzee GA, Strachan AF, van der Westhuyzen DR, Hoppe HC, Jeenah MS, de Beer FC. Serum amyloid A-containing human high density lipoprotein 3. Density, size, and apolipoprotein composition. J Biol Chem. 1986 Jul 25;261(21):9644–51. [PubMed] [Google Scholar]
- 10.Sorenson RC, Bisgaier CL, Aviram M, Hsu C, Billecke S, La Du BN. Human serum Paraoxonase/Arylesterase's retained hydrophobic N-terminal leader sequence associates with HDLs by binding phospholipids : apolipoprotein A-I stabilizes activity. Arterioscler Thromb Vasc Biol. 1999 Sep;19(9):2214–25. doi: 10.1161/01.atv.19.9.2214. [DOI] [PubMed] [Google Scholar]
- 11.Kindy MS, de Beer MC, Yu J, de Beer FC. Expression of mouse acute-phase (SAA1.1) and constitutive (SAA4) serum amyloid A isotypes: influence on lipoprotein profiles. Arterioscler Thromb Vasc Biol. 2000 Jun;20(6):1543–50. doi: 10.1161/01.atv.20.6.1543. [DOI] [PubMed] [Google Scholar]
- 12.Khovidhunkit W, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR. Regulation of scavenger receptor class B type I in hamster liver and Hep3B cells by endotoxin and cytokines. J Lipid Res. 2001 Oct;42(10):1636–44. [PubMed] [Google Scholar]
- 13.McGillicuddy FC, de la Llera Moya M, Hinkle CC, Joshi MR, Chiquoine EH, Billheimer JT, et al. Inflammation impairs reverse cholesterol transport in vivo. Circulation. 2009 Mar 3;119(8):1135–45. doi: 10.1161/CIRCULATIONAHA.108.810721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilhelm AJ, Zabalawi M, Grayson JM, Weant AE, Major AS, Owen J, et al. Apolipoprotein A-I and its role in lymphocyte cholesterol homeostasis and autoimmunity. Arterioscler Thromb Vasc Biol. 2009 Jun;29(6):843–9. doi: 10.1161/ATVBAHA.108.183442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell. 2008 Jul 11;134(1):97–111. doi: 10.1016/j.cell.2008.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith JD. Dysfunctional HDL as a diagnostic and therapeutic target. Arterioscler Thromb Vasc Biol. 2010 Feb;30(2):151–5. doi: 10.1161/ATVBAHA.108.179226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruce IN. ‘Not only…but also’: factors that contribute to accelerated atherosclerosis and premature coronary heart disease in systemic lupus erythematosus. Rheumatology (Oxford) 2005 Dec;44(12):1492–502. doi: 10.1093/rheumatology/kei142. [DOI] [PubMed] [Google Scholar]
- 18.Delgado Alves J, Ames PR, Donohue S, Stanyer L, Nourooz-Zadeh J, Ravirajan C, et al. Antibodies to high-density lipoprotein and beta2-glycoprotein I are inversely correlated with paraoxonase activity in systemic lupus erythematosus and primary antiphospholipid syndrome. Arthritis Rheum. 2002 Oct;46(10):2686–94. doi: 10.1002/art.10542. [DOI] [PubMed] [Google Scholar]
- 19.Batuca JR, Ames PR, Isenberg DA, Alves JD. Antibodies toward high-density lipoprotein components inhibit paraoxonase activity in patients with systemic lupus erythematosus. Ann N Y Acad Sci. 2007 Jun;1108:137–46. doi: 10.1196/annals.1422.016. [DOI] [PubMed] [Google Scholar]
- 20.McMahon M, Grossman J, FitzGerald J, Dahlin-Lee E, Wallace DJ, Thong BY, et al. Proinflammatory high-density lipoprotein as a biomarker for atherosclerosis in patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 2006 Aug;54(8):2541–9. doi: 10.1002/art.21976. [DOI] [PubMed] [Google Scholar]
- 21.Feng X, Li H, Rumbin AA, Wang X, La Cava A, Brechtelsbauer K, et al. ApoE-/-Fas-/- C57BL/6 mice: a novel murine model simultaneously exhibits lupus nephritis, atherosclerosis, and osteopenia. J Lipid Res. 2007 Apr;48(4):794–805. doi: 10.1194/jlr.M600512-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.Stanic AK, Stein CM, Morgan AC, Fazio S, Linton MF, Wakeland EK, et al. Immune dysregulation accelerates atherosclerosis and modulates plaque composition in systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2006 May 2;103(18):7018–23. doi: 10.1073/pnas.0602311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daugherty A. Mouse models of atherosclerosis. Am J Med Sci. 2002 Jan;323(1):3–10. doi: 10.1097/00000441-200201000-00002. [DOI] [PubMed] [Google Scholar]
- 24.Mahley RW, Rall SC., Jr Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–37. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 25.Parks BW, Lusis AJ, Kabarowski JH. Loss of the Lysophosphatidylcholine Effector, G2A, Ameliorates Aortic Atherosclerosis in Low-Density Lipoprotein Receptor Knockout Mice. Arterioscler Thromb Vasc Biol. 2006 Sep 21;26(12):2703–9. doi: 10.1161/01.ATV.0000246774.02426.71. [DOI] [PubMed] [Google Scholar]
- 26.Parks BW, Srivastava R, Yu S, Kabarowski JH. ApoE-dependent modulation of HDL and atherosclerosis by G2A in LDL receptor-deficient mice independent of bone marrow-derived cells. Arterioscler Thromb Vasc Biol. 2009 Apr;29(4):539–47. doi: 10.1161/ATVBAHA.108.179937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parks BW, Gambill GP, Lusis AJ, Kabarowski JH. Loss of G2A promotes macrophage accumulation in atherosclerotic lesions of low-density lipoprotein receptor-deficient mice. J Lipid Res. 2005 July;46:1405–15. doi: 10.1194/jlr.M500085-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Kabarowski JH. G2A and LPC: regulatory functions in immunity. Prostaglandins Other Lipid Mediat. 2009 Sep;89(3-4):73–81. doi: 10.1016/j.prostaglandins.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le LQ, Kabarowski JH, Weng Z, Satterthwaite AB, Harvill ET, Jensen ER, et al. Mice lacking the orphan G protein-coupled receptor G2A develop a late-onset autoimmune syndrome. Immunity. 2001 May;14(5):561–71. doi: 10.1016/s1074-7613(01)00145-5. [DOI] [PubMed] [Google Scholar]
- 30.Rozenberg O, Shih DM, Aviram M. Human serum paraoxonase 1 decreases macrophage cholesterol biosynthesis: possible role for its phospholipase-A2-like activity and lysophosphatidylcholine formation. Arterioscler Thromb Vasc Biol. 2003 Mar 1;23(3):461–7. doi: 10.1161/01.ATV.0000060462.35946.B3. [DOI] [PubMed] [Google Scholar]
- 31.Hahn BH. Should antibodies to high-density lipoprotein cholesterol and its components be measured in all systemic lupus erythematosus patients to predict risk of atherosclerosis? Arthritis Rheum. 2010 Jan 7;62(3):639–42. doi: 10.1002/art.27298. [DOI] [PubMed] [Google Scholar]
- 32.Qiao JH, Xie PZ, Fishbein MC, Kreuzer J, Drake TA, Demer LL, et al. Pathology of atheromatous lesions in inbred and genetically engineered mice. Genetic determination of arterial calcification. Arterioscler Thromb. 1994 Sep;14(9):1480–97. doi: 10.1161/01.atv.14.9.1480. [DOI] [PubMed] [Google Scholar]
- 33.Gu L, Johnson MW, Lusis AJ. Quantitative trait locus analysis of plasma lipoprotein levels in an autoimmune mouse model : interactions between lipoprotein metabolism, autoimmune disease, and atherogenesis. Arterioscler Thromb Vasc Biol. 1999 Feb;19(2):442–53. doi: 10.1161/01.atv.19.2.442. [DOI] [PubMed] [Google Scholar]
- 34.Kelley VE, Roths JB. Interaction of mutant lpr gene with background strain influences renal disease. Clin Immunol Immunopathol. 1985 Nov;37(2):220–9. doi: 10.1016/0090-1229(85)90153-9. [DOI] [PubMed] [Google Scholar]
- 35.Osmers I, Smith SS, Parks BW, Yu S, Srivastava R, Wohler JE, et al. Deletion of the G2A receptor fails to attenuate experimental autoimmune encephalomyelitis. J Neuroimmunol. 2009 Feb 15;207(1-2):18–23. doi: 10.1016/j.jneuroim.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Annema W, Nijstad N, Tolle M, de Boer JF, Buijs RV, Heeringa P, et al. Myeloperoxidase and serum amyloid A contribute to impaired in vivo reverse cholesterol transport during the acute phase response but not group IIA secretory phospholipase A(2) J Lipid Res. 2010 Apr;51(4):743–54. doi: 10.1194/jlr.M000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aviram M, Rosenblat M, Bisgaier CL, Newton RS, Primo-Parmo SL, La Du BN. Paraoxonase inhibits high-density lipoprotein oxidation and preserves its functions. A possible peroxidative role for paraoxonase. J Clin Invest. 1998 Apr 15;101(8):1581–90. doi: 10.1172/JCI1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilensky RL, Macphee CH. Lipoprotein-associated phospholipase A(2) and atherosclerosis. Curr Opin Lipidol. 2009 Oct;20(5):415–20. doi: 10.1097/MOL.0b013e3283307c16. [DOI] [PubMed] [Google Scholar]
- 39.Tetta C, Bussolino F, Modena V, Montrucchio G, Segoloni G, Pescarmona G, et al. Release of platelet-activating factor in systemic lupus erythematosus. Int Arch Allergy Appl Immunol. 1990;91(3):244–56. doi: 10.1159/000235124. [DOI] [PubMed] [Google Scholar]
- 40.Cederholm A, Svenungsson E, Stengel D, Fei GZ, Pockley AG, Ninio E, et al. Platelet-activating factor-acetylhydrolase and other novel risk and protective factors for cardiovascular disease in systemic lupus erythematosus. Arthritis Rheum. 2004 Sep;50(9):2869–76. doi: 10.1002/art.20432. [DOI] [PubMed] [Google Scholar]
- 41.Gomes RN, Bozza FA, Amancio RT, Japiassu AM, Vianna RC, Larangeira AP, et al. Exogenous platelet-activating factor acetylhydrolase reduces mortality in mice with systemic inflammatory response syndrome and sepsis. Shock. 2006 Jul;26(1):41–9. doi: 10.1097/01.shk.0000209562.00070.1a. [DOI] [PubMed] [Google Scholar]
- 42.Yang J, Xu J, Chen X, Zhang Y, Jiang X, Guo X, et al. Decrease of plasma platelet-activating factor acetylhydrolase activity in lipopolysaccharide induced mongolian gerbil sepsis model. PLoS One. 2010;5(2):e9190. doi: 10.1371/journal.pone.0009190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu R, Svenungsson E, Gunnarsson I, Andersson B, Lundberg I, Schafer Elinder L, et al. Antibodies against lysophosphatidylcholine and oxidized LDL in patients with SLE. Lupus. 1999;8(2):142–50. doi: 10.1191/096120399678847434. [DOI] [PubMed] [Google Scholar]
- 44.Deakin S, Moren X, James RW. HDL oxidation compromises its influence on paraoxonase-1 secretion and its capacity to modulate enzyme activity. Arterioscler Thromb Vasc Biol. 2007 May;27(5):1146–52. doi: 10.1161/ATVBAHA.107.141747. [DOI] [PubMed] [Google Scholar]
- 45.Nguyen SD, Sok DE. Preferable stimulation of PON1 arylesterase activity by phosphatidylcholines with unsaturated acyl chains or oxidized acyl chains at sn-2 position. Biochim Biophys Acta. 2006 Apr;1758(4):499–508. doi: 10.1016/j.bbamem.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 46.McMahon M, Grossman J, Skaggs B, Fitzgerald J, Sahakian L, Ragavendra N, et al. Dysfunctional proinflammatory high-density lipoproteins confer increased risk of atherosclerosis in women with systemic lupus erythematosus. Arthritis Rheum. 2009 Aug;60(8):2428–37. doi: 10.1002/art.24677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jahangiri A, de Beer MC, Noffsinger V, Tannock LR, Ramaiah C, Webb NR, et al. HDL remodeling during the acute phase response. Arterioscler Thromb Vasc Biol. 2009 Feb;29(2):261–7. doi: 10.1161/ATVBAHA.108.178681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Beer FC, Connell PM, Yu J, de Beer MC, Webb NR, van der Westhuyzen DR. HDL modification by secretory phospholipase A(2) promotes scavenger receptor class B type I interaction and accelerates HDL catabolism. J Lipid Res. 2000 Nov;41(11):1849–57. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.