

Abstract
An LC-MS/MS method for the determination of isoniazid in human plasma was developed and validated. Human plasma aliquots of 100 μL were used for analysis. The assay used nialamide as the internal standard. The calibration curve concentration range was 50 to 10,000 ng/mL. Sample preparation utilized protein precipitation, and the supernatant was directly injected onto silica column without reconstitution. The recovery was over 90% and matrix effect was negligible. The method is simple and fast, which is advantageous in respect to instability of isoniazid in human plasma and loss on reconstitution due to its low molecular weight.
Keywords: LC-MS/MS, isoniazid, nialamide, human plasma, method development, silica column
1. Introduction
Isoniazid (INH) is a first-line anti-tuberculosis drug. It has been widely used since its introduction in 1952 due to its efficient treatment of tuberculosis, primarily as part of combination therapy [1, 2]. Isoniazid (Figure 1), chemically named 4-pyridinecarboxylic acid hydrazide or isonicotinic acid hydrazide, is a white, odorless, crystalline powder that is soluble in water (14%) and has a melting point of 171.4 °C [3]. Due to its wide use and side effects, many HPLC methods for quantitation of INH have been reported [4-7]. These methods utilized UV [4, 5], fluorometry [6], or electrochemical [7] methods to detect INH. The sensitivity and selectivity of these methods were not as good as a liquid chromatography-mass spectrometry (LC-MS) method.
Figure 1.
Chemical structures of isoniazid and nialamide (IS).
Recently, several LC-MS methods have been reported to determine INH in plasma [8-10]. All of these methods used a C18 column to separate INH. However, due to its high polarity, retention of INH on C18 column is limited. Li and coworkers reported determination of INH using a silica HPLC column coupled with tandem mass spectrometry [11], which resulted in good retention for INH. However, a limitation of this method is that it was developed to quantitate INH in animal plasma and the investigators failed to present any information regarding drug recovery. With implementation and application of this method to human plasma samples, we found that the solid phase extraction used for sample preparation resulted in low and variable recovery.
We currently report a method developed to determine INH in human plasma using hydrophilic interaction liquid chromatography (HILIC) coupled with tandem mass spectrometry (MS/MS). Sample preparation is simple, using protein precipitation with acetonitrile followed by centrifugation. Supernatants are directly injected onto a silica column without reconstitution. This is especially important as INH is not stable in plasma and loss of drug occurs easily during evaporation and reconstitution due to the low molecular weight of the compound.
2. Experimental
2.1. Chemicals and reagents
INH and the internal standard nialamide (NLM) (Figure 1) were purchased from Sigma-Aldrich. (St. Louis, MO, USA). Acetonitrile, water, ammonium acetate, and acetic acid were obtained from Fisher Scientific (Fair Lawn, NJ, USA). All chemicals were of HPLC grade. Water is distilled water if not mentioned specifically. Human plasma was purchased from Biological Specialty Co. (Colmar, PA, USA).
2.2. LC-MS/MS conditions
The PE Sciex API 2000 triple quadrupole mass spectrometer with TurboIonSpray sample inlet was purchased from Perkin-Elmer-Sciex (Concord, Ont., Canada). Perkin-Elmer (Norwalk, CT, USA) supplied the PE Biosystems 200 series autosampler and twin PE Biosystems series 200 micro HPLC pumps. Chromatographic separation was achieved on a Hypersil silica analytical column (50 × 2.1 mm, 3 μm; Thermo Fisher, Bellefonte, PA, USA) equipped with a guard column (10 × 2.1 mm, 3 μM; Thermo Fisher, Bellefonte, PA, USA) and Pre-column filter (MAC-Mod Analytical, Chadds Ford, PA). The LC setting is as follows: solvent A was water (HPLC grade) containing 0.1% HOAC and 2.5 mM NH4AC. Solvent B was acetonitrile with 0.1% HOAC. From 0-4.0 min was isocratic elution with 10% A at a flow rate of 200 μL/min. The retention times for INH and I.S. were 1.9 and 1.7 min, respectively. The column was washed with 30% A from 4.1-4.9 min and 10% A from 5.0 – 10 min at a flow rate of 600 μL/min. Injection volume was 10 μL. The divert valve was set to direct LC eluent to MS source at 1.2 min and to waste line at 3.9 min. Before each injection, the needle was cleaned with one pre-injection wash and after injection with three post-injection washes each with 500 μL of ACN- water (85-15, v/v) containing 0.1% HOAC. The MS conditions for INH and the internal standard were optimized by separate infusion of 1 μg/mL INH or I.S. into the MS at a flow rate of 10 μL/min constantly while adjusting MS parameters to achieve maximal signal. Electro-spray ionization in positive ion mode (ESI+) was used for ionization and multiple reaction monitoring (MRM) mode was chosen for detection. The precursor-product ion pairs were m/z 138 → 121 for INH and m/z 299 → 91 for the internal standard nialamide. The optimized acquisition parameters were as follows: Turbo (Heater) set at 400 °C; Curtain gas (CUR), 25 psi (99.999% Nitrogen); Nebulizer Gas (Gas 1), 10 psi (99.999% Nitrogen); Auxiliary (turbo) Gas (Gas2), 70 psi (99.999% Nitrogen); Collision-Activated Dissociation (CAD) Gas: 4; IonSpray Voltage (IS),5500 v. The parameters for INH and Nialamide (IS) were listed in Table 1. The scan time was set at 250 ms for both INH and IS. Data was processed with Analyst 1.3.1. (Applied Biosystem, Foster City, CA, USA).
Table 1.
MS parameters for isoniazid and IS.
| DP | FP | EP | CE | CEP | CXP | |
|---|---|---|---|---|---|---|
| Isoniazid, 138/121 | 16 | 320 | 9 | 20 | 7 | 4 |
| Nialamide, 299/91 | 21 | 370 | 8 | 45 | 16 | 6 |
DP: declustering potential, applied to the orifice plate. FP: focusing potential, helps to focus the ions through the skimmer. EP: entrance potential. CE: collision energy. CEP: collision cell entrance potential. CXP: collision cell exit potential
2.3. Preparation of INH standard and quality control samples
Two sets of INH stock solutions at 10 mg/mL were prepared in 0.1% aqueous HOAC with separately weighed INH. One solution was used to spike standards and the other for quality control (QC) samples. Calibration standard concentrations of 50, 100, 250, 500, 750, 100, 2500, 5000, and 10000 ng/mL were used. QC samples consisted of 150, 1500, and 8000 ng/mL. They were prepared by spiking INH stock solution into blank human plasma. The internal standard solution was prepared by dissolving nialamide in ACN-water (1:1) to produce a final concentration of 1000 ng/mL. The stock solutions, standards, QC samples, and the internal standard solution were stored at -70°C freezer between uses.
2.4. Sample preparation
To 100 μL aliquot of each standard, QC sample, and blank plasma was added 50 μL internal standard (1000 ng/mL nialamide). After briefly mixing on a vortex shaker for 2 sec, the samples were deproteinized by adding 350 μL ACN and vortex-mixing for 20 sec. Then samples were centrifuged at 15000 g for 5 min. A 200 μL aliquot of the clear supernatant was transferred into autosampler vial and 10 μL was injected into the LC-MS/MS system. All plasma samples needed to be processed immediately once they were thawed at room temperature.
2.5. Method validation procedure
2.5.1. Acceptance criteria
The method validation was conducted according to the guidelines of the AIDS Clinical Trials Group (ACTG) [12], which were based on Food and Drug Administration (FDA) guidelines. The assay was considered acceptable if precision, expressed as relative standard deviation (RSD) or coefficient of variation (%CV), was less than 15% for intra and inter-day variation. RSD for lower limit of quantitation (LLOQ) was set at <20%. The accuracy compared to the nominal value (% deviation) should be within 15% for intra- and inter-day comparison (% deviation for LLOQ was set at <20%). The calibration curve should have a correlation coefficient R of 0.995 or better. The back-calculated values for standards should be within 15% of target (20% at LLOQ). If points were removed from the standard curve, it must be recalculated. At least six (6) nonzero concentration points must be used to derive the standard curve, and >75% of points must be within 15% of the targeted value (LLOQ must be within 20%).
2.5.2. Calibration curve
Calibration curves were obtained by linear regression of the peak area ratio of INH to internal standard (Y-axis) versus the nominal INH concentrations (X-axis) with a weighting factor of 1/x.
2.5.3. Lower limit of quantitation (LLOQ)
The LLOQ was established using six samples independent of standards to determine accuracy and precision. The accuracy should be within 20% of the nominal concentration and precision should be <20%. The signal intensity of the LLOQ should be ≥ 5-fold blank response.
2.5.4. Intra- and inter-day precision and accuracy
Intra-day precision and accuracy were determined by analysis of six replicates of each QC sample (n=6) at low (150 ng/mL), medium (1500 ng/mL), and high (8000 ng/mL) concentration levels extracted with a set of standards in one batch. The same procedure was repeated on five (5) different days with new samples to determine inter-day precision and accuracy (total: n = 30 per concentration level). Precision was reported as RSD and accuracy as percent of the nominal concentration (% deviation).
2.5.5. Recovery and matrix effect
The recovery of INH from plasma following sample preparation was assessed by comparing the peak area of INH from plasma extracts (pre-precipitation-spiked INH, set 3) to the peak area of the same concentration of INH spiked into blank plasma extracts after precipitation (post-precipitation-spiked INH, set 2). Matrix effects were evaluated by comparing the peak area of INH from post-precipitation-spiked samples (set 2) to clean samples (set 1). Process efficiency was evaluated by comparing set 3 to set 1. Clean samples consist of water spiked with INH at low (150 ng/mL) and high (8000 ng/mL) levels in 5 replicates (n = 5) and were processed as normal samples. Five different lots of human plasma were used. For set 2, a 100 μL aliquot from each lot of plasma (with EDTA as anticoagulant) was processed by ACN precipitation and centrifuged, then a 180 μL aliquot of each supernatant was spiked with INH and IS (10 μL each) to make the final concentration the same level as that of the clean samples. For set 3, a 100 μL aliquot from each lot of plasma spiked with INH at the same concentration levels was processed. The recovery, matrix effect and process efficiency were calculated with the following formulae:
Potential interference of possible concomitant HIV drugs was also evaluated by spiking the medium QC (1500 ng/mL) with each of the tested drugs to make a final concentration of 5000 ng/mL (n = 3). The following HIV drugs were tested: efavirenz (EFV), zidovudine (ZDV), tenofovir (TFV), atazanavir (ATV), lopinavir (LPV), ritonavir (RTV), nelfinavir (NFV), nevirapine (NVP), indinavir (IDV), amprenavir (APV), abacavir (ABV), and saquinavir (SQV).
2.5.6. Stability
The stability of INH in human plasma was evaluated at these conditions: 2 freeze-thaw cycles, storage at -70 °C for 27 days, room temperature (22 °C) for 1, 3, and 20 hours. Each condition was tested with QC samples at low and high concentration levels in triplicates. Fresh samples were used as reference. Stability of processed samples was evaluated after standing on the bench for 3 days. INH stock aqueous solution in 0.1 % HOAC was tested up to 6 months for storage at − 70 °C.
3. Results
3.1. LC-MS/MS optimization
Due to its high polarity, INH is easily dissolved in water, with minimal retention on a reverse phase column even with a highly aqueous mobile phase. The capacity factor (or retention factor, k) is a measurement of retention on HPLC column and calculated as follows (11): k = (tr-t0)/t0. Ideally k = 2 – 10. A low k value will cause potential matrix effect as many other components in the samples may elute out together with the analyte. Among the columns tested, C18, C8, and RP polar columns all yielded very short retention times (k<1). The C2 column had a capacity factor of ∼1.5 while the silica column gave ∼2. Therefore, we chose the silica column for further analytical development. Broad peaks were observed if using ACN/water/acetic acid/TFA, 70:30:1:0.025 as the mobile phase. Since NH4 AC is able to sharpen the peaks but also reduce the retention time, a low amount (2.5 mM in solvent A) was used in the mobile phase. Under these conditions, the retention time for INH was (1.9 ± 0.2) min. The dead volume (V0) for the silica column (50 × 2.1 mm) was 0.11 mL (0.5 × 0.212 × 5 = 0.11) and the dead time was 0.6 min (0.11/0.2). Thus the k value was estimated as 2.
For internal standard selection, isoniazid-d4 would be our first choice. However, we realized that the deuterated isoniazid was not commercially available immediately in respect to our required timeline for method development. Therefore, five pyridine or hydrazide derivatives were tested: isonipecotamide, isonicotinamide, indole-3-acetic hydrazide, 4-nitrobenzoic hydrazide, and nialamide. Nialamide was chosen because its retention time was close to INH (their peaks were partially overlapped) and ionization was similar with INH.
Positive ion mode was chosen because INH and IS contain nitrogen atoms. Representative product ion mass spectra of INH and IS were shown in Figure 2. The precursor-product ion pairs 138/121 for INH and 299/91 for IS were monitored in MRM mode for quantification of INH.
Figure 2.
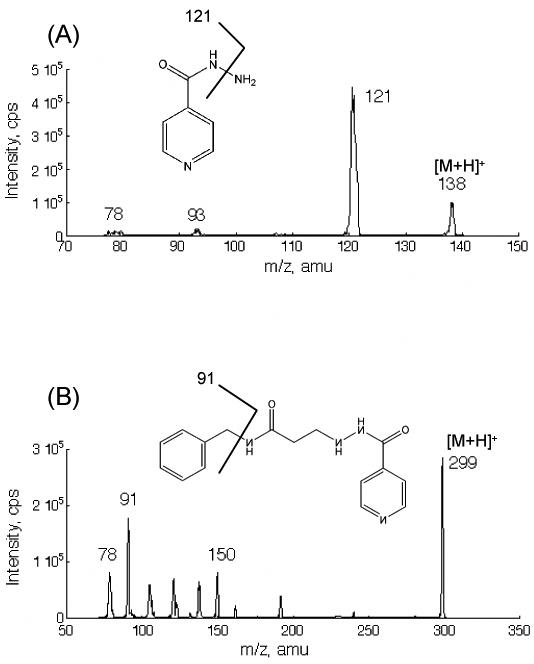
MS/MS product ion spectra of the precursor ions [M+H]+ of isoniazid and nialamide (IS).
3.2. Sample preparation
During optimization of sample preparation, several SPE columns were tested. Waters OASIS® HLB 1cc SPE column gave very low recovery. INH wasn't retained well in the column. Although recovery could be increased by increasing pH with NH4OH during loading and washing steps, the recovery was still below 80% and large variation was observed. MCX SPE column (Waters OASIS®) was expected to absorb INH as INH is a base. However, the recovery was not acceptable either. Finally, protein precipitation with ACN was chosen for sample preparation. To check the matrix effect, the supernatant from blank plasma precipitation was injected into LC while INH was infused into the LC column elute via a “T” connector constantly. Besides ion suppression near the dead time (t0 = 0.6 min), another significant ion suppression occurred in ∼15 minutes, this is probably due to phospholipids from plasma. To reduce the run time, we tried to use silica SPE column (Thermo Hypersep) to remove the phospholipids. Extract from protein precipitation was loaded onto silica SPE column, and washed with equal volume of ACN. The combined elute still contained the phospholipids. The phospholipids were not retained in the silica SPE column probably due to more complicated matrix in the plasma extract than HPLC mobile phase. Finally the protein precipitation method was adopted even though a longer HPLC run time was needed. This was proven to be beneficial because INH was found to be unstable in plasma. Once samples thawed, fast sample processing was required.
3.3. Method validation
The LLOQ of INH in this assay was set at 50 ng/mL in human plasma. The calibration curve was linear over the concentration range of 50 – 10000 ng/mL with a mean correlation coefficient r of 0.9991 ± 0.00065 (Table 2). No carry over was observed in blank plasma. Representative MRM ion chromatograms of blank human plasma and plasma spiked with I.S. and INH at LLOQ were shown in Figure 3.
Table 2.
Inter-day average back-calculated standard concentrations (n = 5)
| Theoretic cor ng/ml | Mean ng/ml | SD | Precision (RSD,%) | Accuracy (% dev) |
|---|---|---|---|---|
| 50 | 54.2 | 5.0 | 9.2 | 8.4 |
| 100 | 98.6 | 6.5 | 6.6 | -1.4 |
| 250 | 256 | 17 | 6.7 | 2.5 |
| 500 | 503 | 43 | 8.5 | 0.6 |
| 750 | 714 | 23 | 3.3 | -4.8 |
| 1000 | 956 | 47 | 4.9 | -4.4 |
| 2500 | 2434 | 186 | 7.6 | 2.6 |
| 5000 | 5032 | 136 | 2.7 | 0.6 |
| 10000 | 10100 | 255 | 2.5 | 1.0 |
| Slope | 0.00305 | 0.00045 | ||
| Y-intercept | 0.00806 | |||
| R | 0.9991 | 0.00065 |
Figure 3.
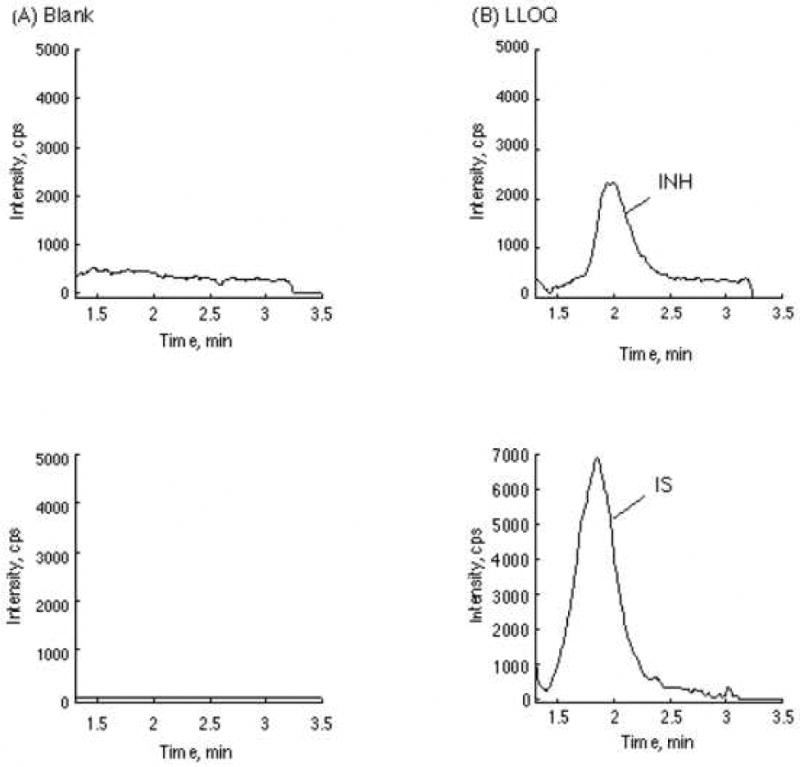
Representative chromatograms of blank human plasma (A) and lower limit of quantification (LLOQ) (B). The upper panel is for INH Channel. Lower panel is the IS channel.
The intra-day precisions (n = 6) over 5 days were ranged from 1.1 to 7.2 at the three concentration levels, and inter-day precisions were ranged from 4.7 to 7.2, all of them within 15%. The intra-day and inter-day precisions for LLOQ were within 20%. Intra-day and inter-day accuracies were also within the acceptance limit, except for an outlier for LLOQ in the 1st run day (Table 3).
Table 3.
Intra-day and inter-day precision (%RSD) and accuracy (% dev) for analysis of INH in human plasma.
| Nominal, ng/ml | Intra-day | Inter-day | ||||||
|---|---|---|---|---|---|---|---|---|
| 50 | 150 | 1500 | 8000 | 50 | 150 | 1500 | 8000 | |
| mean, ng/ml | 51.2 - 54.1 | 138 - 151 | 1305 - 1573 | 7478 - 8035 | 51.7 | 145 | 1442 | 7791 |
| SD | 0.9 - 4.1 | 4.1 - 9.1 | 16.4 - 102 | 82.3 - 524 | 2.9 | 8.0 | 103 | 367 |
| RSD,% | 1.7 - 8.0 | 3.0 - 6.5 | 1.3 - 7.2 | 1.1 - 6.7 | 5.7 | 5.5 | 7.2 | 4.7 |
| %dev | -1.2 - 8.9 | -8.3 - 0.4 | -13.0 - 4.9 | -6.5 - 0.4 | 3.3 | -3.1 | -3.9 | -2.6 |
| n | 6 (5) | 6 | 6 | 6 | 29 | 30 | 30 | 30 |
The results for recovery, matrix effect, and process efficiency were shown in Table 4. The recovery of INH was very high because of simple sample preparation. The matrix effect was evaluated with 5 different lots of human plasma. Matrix effect for INH and the internal standard was negligible. Process efficiency reflects both recovery and matrix effect. The process efficiency for both INH and the internal standard were above 90%.
Table 4.
Recovery (RE), matrix effect (ME), and process efficiency (PE).
| concentration | Peak area (×10e4), n = 5 | RE | ME | PE | ||
|---|---|---|---|---|---|---|
| clean sample (in ACN-water) | postextraction spiked | preextraction spiked | ||||
| Low (150 ng/ml) | 9.82 ± 0.97 | 10.0 ± 0.89 | 9.42 ± 0.70 | 94.2 | 102 | 95.9 |
| High (8000 ng/ml) | 580 ± 21 | 578 ± 28 | 567 ± 61 | 98.1 | 99.7 | 97.8 |
| IS (1000 ng/ml) | 14.0 ± 0.3 | 14.5 ± 1.1 | 12.8 ± 1.6 | 88 | 104 | 91.4 |
Results for stability of INH stock solution and INH in human plasma were shown in Table 5. INH stock solution in 0.1% aqueous HOAC was stable under the tested conditions. INH in plasma was stable during storage in deep freezer (-70 °C) for the tested period of time. Stability for longer storage time at -70 °C was ongoing. Noteworthily, INH was not stable in plasma once the samples were thawed. We tested 1-, 3-, and 20-hour stability at room temperature (22 °C). No significant degradation of INH in plasma was found in 1 hour. However, fair amount of INH was degraded after 3 hours, and about 40% degradation was observed after 20 hours at room temperature. We also tested 4 °C storage for 20 hours (data not shown), over 15 % degradation was observed. Therefore, once plasma samples were thawed, immediate processing was required. After processing, INH was stable even if left on bench for 3 days.
Table 5.
Stability of INH stock and plasma samples.
| storage conditions | INH sample(ng/mL) | % remaining | % RSD |
|---|---|---|---|
| 2 freeze/thaw cycles | Low (180) in plasma | 95.1 | 1.2 |
| High (9000) in plasma | 95.0 | 2.0 | |
| freezing for 27 days | Low (180) in plasma | 97.1 | 0.6 |
| High (9000) in plasma | 92.1 | 9.0 | |
| benchtop 1 hr | Low (180) in plasma | 100.4 | 3.4 |
| High (9000) in plasma | 100 | 0.8 | |
| benchtop 3 hr | Low (180) in plasma | 83.4 | 3.1 |
| High (9000) in plasma | 91.2 | 2.7 | |
| benchtop 20 hr | Low (180) in plasma | 63.2 | 6.6 |
| High (9000) in plasma | 61.7 | 3.0 | |
| autosampler (72 hr) | Low (180) in extract | 93.4 | 6.2 |
| High (9000) in extract | 89.7 | 2.3 | |
| freezing for 6 months benchtop 20 hr | INH Stock solution | 101 | 2.1 |
| INH Stock solution | 99.0 | 5.5 |
Finally, we tested selectivity of the method for INH over other potential concomitant HIV drugs. By comparing the peak area ratios of the HIV drug-spiked samples with that of non-spiked sample (medium QC, 1500 ng/mL INH), no significant interference was observed (Table 6).
Table 6.
Potential concomitant medication interferences
Medium validation sample (1500 ng/mL) was spiked with potential concomitant drugs at 5000 ng/mL and processed as usual.
| Drugs | Peak area ratio (INH/IS) | STD | %CV | %Dev due to matrix effect | |||
|---|---|---|---|---|---|---|---|
| replicate 1 | replicate 2 | replicate 3 | mean | ||||
| control | 3.64 | 3.74 | 3.67 | 3.68 | 0.05 | 1.35 | |
| EFV | 3.64 | 3.44 | 3.68 | 3.59 | 0.13 | 3.55 | -2.49 |
| ZDV | 3.62 | 3.51 | 3.55 | 3.56 | 0.05 | 1.52 | -3.35 |
| TFV | 3.79 | 3.78 | 3.67 | 3.75 | 0.07 | 1.76 | 1.85 |
| ATV | 3.78 | 3.77 | 4.09 | 3.88 | 0.19 | 4.80 | 5.42 |
| LPV | 3.69 | 3.87 | 3.94 | 3.83 | 0.13 | 3.43 | 4.18 |
| RTV | 3.61 | 3.87 | 3.70 | 3.73 | 0.13 | 3.46 | 1.23 |
| NFV | 4.22 | 4.01 | 4.22 | 4.15 | 0.12 | 2.89 | 12.8 |
| NVP | 3.31 | 3.37 | 3.47 | 3.39 | 0.08 | 2.40 | -7.98 |
| IDV | 3.93 | 3.87 | 3.88 | 3.89 | 0.03 | 0.89 | 5.82 |
| APV | 3.88 | 3.67 | 3.66 | 3.74 | 0.13 | 3.39 | 1.52 |
| ABV | 3.52 | 4.03 | 4.00 | 3.85 | 0.29 | 7.45 | 4.67 |
| SQV | 3.73 | 3.88 | 3.76 | 3.79 | 0.08 | 2.07 | 2.94 |
4. Discussion and Conclusion
Numerous LC-MS methods have been reported for INH quantification (8-11). Most of these methods utilized C18 column for separation (8-10). Since retention of INH on C18 column is limited (k<1), significant matrix effects may affect the quantification, especially for an electrospray ionization mode. We found use of a silica column resulted in good retention for INH, which is consistent with the findings of Li and coworkers (11). However, we could not repeat the results with the same mobile phase (ACN/water/acetic acid/TFA, 70:30:1:0.025); instead, a very broad peak was observed. Peak broadening was probably due to the pyridine derivatives being partially charged (some molecules are positively charged, others not), which interact with silica via hydrophilic and also ionic interactions. Since salt suppresses ionic interaction, by adding NH4AC to the mobile phase, a better peak shape was achieved. The final peaks for INH and IS were still fairly broad; this didn't compromise the selectivity of the method, which may be contributed to high selectivity of mass spectrometer as detector. As simple sample preparation was adapted, wash phase was necessary to minimize matrix effect caused by late elutes (possibly phospholipids). We found significant matrix effect at ∼ 15 min when eluted with 80% ACN at 0.2 mL/min: a huge trough due to ion suppression was observed in the infusion experiment. However, when final LC method was applied, we were not able to observe similar trough in infusion experiment. We speculated that it was buried in the signal drop due to flow rate increase (from 0.2 mL/min to 0.6 mL/min). Also of note is that a long equilibration/conditioning time is required for silica column. Otherwise, the retention times for INH and IS may be shifted. Thus, we suggest equilibration be performed by repeating injection of a low standard or QC sample and the gradient elution for 5-10 times until retention times for INH and IS are consistent in 3-5 continuous injections. This also serves as test of system suitability. If the column is new or hasn't been used for long time (such as more than a month), then column conditioning needs longer time (several hours).
An interesting finding is that INH is not stable in human plasma at room temperature. Ng and coworkers studied INH stability in rat plasma and reported that significant degradation (>20%) of INH was observed after 6 hours at room temperature (8). Our results from human plasma show similar instability. This is likely caused by an enzymatic reaction (either hydrolysis or oxidation) based on the fact that INH was stable for at least 3 days once the plasma samples were processed by protein precipitation. Further study on INH stability in human plasma is ongoing.
In summary, the application of hydrophilic interaction liquid chromatography (HILIC) and tandem mass spectrometry resulted in a simple, fast and robust method for determination for INH in human plasma to support pharmacokinetic study of INH in patients with tuberculosis. In respect to instability of INH in plasma and reconstitution loss due to its low molecular weight, this current method is advantageous over reported methods due to its simplicity in regards to sample preparation and direct injection into LC-MS/MS without further treatment. This method has been validated according to the ACTG guidelines [12], which were based on FDA guidelines.
Acknowledgments
This work was supported by NIH/NIAID International Maternal Pediatric Adolescent AIDS Clinical Trials (IMPAACT) Group (grant number 1U01AI068632).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weber WW, Hein DW. Clinical Pharmacokinetics. 1979;4:401. doi: 10.2165/00003088-197904060-00001. [DOI] [PubMed] [Google Scholar]
- 2.Grosset JH. Reviews of infectious diseases. 1989;11 2:S347–52. doi: 10.1093/clinids/11.supplement_2.s347. [DOI] [PubMed] [Google Scholar]
- 3.Budavari Susan., editor. The Merck Index. 12th. 1996. p. 5204. [Google Scholar]
- 4.Smith PJ, van Dyk J, Fredericks A. Int J Tuberc Lung Dis. 1999;3:S325. [PubMed] [Google Scholar]
- 5.Unsalan S, Sancar M, Bekce B, Clark PM, Karagoz T, Izzettin FV, Rollas S. Chromatographia. 2005;61:595. [Google Scholar]
- 6.Olson WA, Dayton PG, Israilli ZH, Pruitt AW. Clin Chem. 1977;23:745. [PubMed] [Google Scholar]
- 7.Delahunty T, Lee B, Conte JE. J Chromatogr B. 1998;705:323. doi: 10.1016/s0378-4347(97)00510-0. [DOI] [PubMed] [Google Scholar]
- 8.Ng KY, Zhou HY, Zhang YL, Hybertson B, Randolph T, Christians U. J Chromatogr B. 2007;847:188. doi: 10.1016/j.jchromb.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 9.Chen XY, Song B, Jiang HJ, Yu K, Zhong DF. Rapid Commun Mass Spectrom. 2005;19:2591. doi: 10.1002/rcm.2100. [DOI] [PubMed] [Google Scholar]
- 10.Song SH, Jun SH, Park KU, Yoon Y, Lee JH, Kim JQ, Song J. Rapid Commun Mass Spectrom. 2007;21:1331. doi: 10.1002/rcm.2961. [DOI] [PubMed] [Google Scholar]
- 11.Li AC, Junga H, Shou WZ, Bryant MS, Jiang X, Weng N. Rapid Commun Mass Spectrom. 2004;18:2343. doi: 10.1002/rcm.1623. [DOI] [PubMed] [Google Scholar]
- 12.ACTG guidelines for method development and validation based on (and including) FDA guidelines dated by 2001 Version. 2005;2 [Google Scholar]