

Abstract
BCR/ABL-transformed chronic myeloid leukemia (CML) cells accumulate numerous DNA double-strand breaks (DSBs) induced by reactive oxygen species (ROS) and genotoxic agents. To repair these lesions BCR/ABL stimulate unfaithful DSB repair pathways, homologous recombination repair (HRR), non-homologous end-joining (NHEJ) and single-strand annealing (SSA). Here we show that BCR/ABL enhances the expression and increase nuclear localization of WRN (mutated in Werner syndrome), which is required for processing DSB ends during the repair. Other fusion tyrosine kinases (FTKs) such as TEL/ABL, TEL/JAK2, TEL/PDGFβR, and NPM/ALK also elevate WRN. BCR/ABL induces WRN mRNA and protein expression in part by c-MYC -mediated activation of transcription and Bcl-xL –dependent inhibition of caspase-dependent cleavage, respectively. WRN is in complex with BCR/ABL resulting in WRN tyrosine phosphorylation and stimulation of its helicase and exonuclease activities. Activated WRN protects BCR/ABL-positive cells from the lethal effect of oxidative and genotoxic stresses, which causes DSBs. In addition, WRN promotes unfaithful recombination-dependent repair mechanisms HRR and SSA, and enhances the loss of DNA bases during NHEJ in leukemia cells. In summary, we postulate that BCR/ABL-mediated stimulation of WRN modulates the efficiency and fidelity of major DSB repair mechanisms to protect leukemia cells from apoptosis and to facilitate genomic instability.
Keywords: BCR/ABL, WRN, CML, genomic instability, survival
INTRODUCTION
Chromosomal translocations are responsible for the appearance of oncogenes encoding fusion tyrosine kinases (FTKs), such as BCR/ABL, TEL/ABL, TEL/JAK2, TEL/PDGFβR, and NPM/ALK, which induce hematological malignancies such as chronic myeloproliferative disorders (MPDs), acute leukemias and lymphomas (1). For example, BCR/ABL kinase transforms hematopoietic stem cells to induce chronic myeloid leukemia in chronic phase (CML-CP) (2). However, CML-CP eventually progresses to highly malignant blast phase (CML-BP) due to enhanced chromosomal instability (3).
FTKs also enhance DNA damage caused by reactive oxygen species (ROS) and genotoxic agents, and modulate the efficiency and fidelity of DNA repair mechanisms to promote resistance to genotoxic stress and genomic instability (3). We, and others found that leukemia cells expressing BCR/ABL (including CML-CP stem/progenitor cells enriched population) and other FTKs may contain elevated numbers of DNA double-strand breaks (DSBs) caused by reactive oxygen species (ROS) and genotoxic treatment; repair of these DSBs is accelerated but the fidelity of repair mechanisms (homologous recombination repair = HRR, non-homologous end-joining = NHEJ and single-strand annealing = SSA) is compromised (4–9).
Unwinding and exonucleolytic degradation of DSBs by RecQ-like helicase family members is required for the initiation and resolution of the DSBs repair intermediates (10). The RecQ-like helicase family consists of five known proteins: BLM, WRN, RTS, RECQL1 and RECQL5, which aberrant regulation has been detected in tumors.
WRN helicase/exonuclease appears to play a role in carcinogenesis and promotion of tumor cell growth (10). Werner syndrome (WS) is caused by WRN mutations generating truncated proteins that either fail to localize to the nucleus or destabilize the protein, thus affecting DNA repair. WS is associated with premature senescence and higher incidence of cancers probably due to accumulation of DNA lesions and chromosomal instability. On the other hand up-regulation of non-mutated WRN in numerous types of tumors (fibrosarcoma, ovarian cancer, glioma, glioblastoma, fibrosarcoma, colon carcinoma, cervical carcinoma, bladder carcinoma, renal cell carcinoma) and in cells transformed by oncogenic viruses such as SV40 T-antigen or Epstein-Barr virus, promoted proliferation and survival after genotoxic treatment (11–13). Therefore, WRN can exert a tumor-suppressor and tumor-promoting function. In concordance, WRN may change the balance between carcinogenesis versus senescence caused by c-Myc oncogenic stimulation (14).
Here we report that BCR/ABL-mediated regulation of WRN stimulates all three unfaithful DSB repair mechanisms, HRR, NHEJ and SSA, which promote survival and genomic instability in leukemia cells. Therefore, WRN may play a key role in accumulation of additional chromosomal aberrations to facilitate malignant progression of CML.
METHODS
Cells
FL5.12, 32D and BaF3 murine parental hematopoietic cells, MO7 and UT7 human megakaryoblastic cell lines, and their counterparts transformed with p210BCR/ABL, TEL/JAK2, TEL/PDGFRB, TEL/ABL and NPM/ALK were used before (5, 15, 16) and maintained in Iscove’s modified defined medium (IMDM) supplemented with 1 mM glutamine, 10% fetal bovine serum (FBS) and interleukin 3 (IL-3) -conditioned medium (murine cells) or stem cell factor (SCF) –conditioned medium and recombinant granulocyte-macrophagecolony-stimulating factor (GM-CSF) (PeproTech Inc., Rocky Hill, NJ, USA) (human cells) in the concentrations necessary to support cell proliferation. 32D cells overexpressing Bcl-xL were described before (5). CML-CP patient cells were obtained from the Stem Cell and Leukemia Core Facility of the University of Pennsylvania, Philadelphia, PA, USA, after receiving informed consent. CD34+ CML cells were obtained using human CD34+ selection cocktail (StemCell Technologies, Inc., Vancouver, BC, Canada). CD34+ cells from healthy volunteers (NBMC) were obtained from Cambrex Bio Science Walkersville (Walkersville, MD, USA). The research activities involving human samples were approved by the Institutional Review Board. Phoenix Amphotropic packaging cell line (ATCC, Manassas, VA, USA) was maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% FCS.
Semi-quantitative RT-PCR
WRN expression was examined by semi-quantitative RT-PCR simultaneously detecting WRN and GADPH (internal reaction control) in total RNA as described before (17) with modifications (Supplemental Methods).
DNA constructs
pSRα-p210BCR/ABL and pMig retroviral plasmids containing IRES-GFP only, BCR/ABLwild-type-IRES-GFP, BCR/ABL kinase deficient mutant (K1172R)-IRES-GFP or BCR/ABL-IRES-YFP were obtained from Drs: Charles Sawyers (UCLA, Los Angeles, CA, USA), Warren Pear (University of Pennsylvania, Philadelphia, PA, USA) and Bruno Calabretta (Thomas Jefferson University, Philadelphia, PA, USA). LXSN retroviral construct containing the full-length wild-type human WRN cDNA was from Dr. Junko Oshima (University of Washington, Seattle, WA, USA). pBABEpuro-shWRN retroviral plasmid encoding for anti-WRN harpin corresponding to human WRN cDNA bps 391–415 was kindly provided by Dr. Raymond J. Monnat (University of Washington, Seattle, WA, USA) and modified by replacing the puromycin resistance gene with a zeocin cDNA between HindIII and ClaI sites (18). WRN promoter region (nucleotides -573 to +160 of WRN mRNA; GenBank/EMBL Data Bank accession No. AB003173) (18) was amplified by PCR and cloned at the MluI and BglII sites of the pGL3 luciferase reporter vector (Promega, Madison, WI, USA) to generate pGL3-WRN-luc reporter plasmid. pSRαMSVtkneo empty plasmid or that containing two different dominant negative mutants (DNMs) of c-Myc: (Δ106–143) or In373 were obtained from Dr. Sawyers. pMXpuro STAT5B-DNM was described before (16).
Inhibitors
Imatinib was obtained from Novartis Pharma AG (Switzerland), Z-VAD-FMK was purchased from Bachem AG (Switzerland) and epoxomycin was from Biomol (Plymouth Meeting, PA, USA).
WRN transactivation assay
The assay was performed as previously described (5). Briefly, 293T cells were transiently co-transfected with 10 μg of pMig plasmids containing IRES-GFP, BCR/ABL-IRES-GFP, or BCR/ABL[K1172R]-IRES-GFP, as well as 10 μg of pGL3-WRN-luc reporter plasmid and 5 μg of β-galactosidase plasmid. When indicated cells were also transfected with 10 μg of pSRαMSVtkneo encoding DNMs of c-Myc and pMXpuro-STAT5B-DNM. Transactivation activity was quantified by the Luciferase Assay System (Promega). Transfection efficiency was normalized by β-galactosidase activity.
Immunoprecipitation and Western analysis
Total and nuclear cell lysates were obtained as described before (16, 19). To detect WRN phosphorylation cells were pre-incubated with 100 μM pervanadate for 5 min in serum-free medium (20). Chromatin fractions were prepared by sonication of the insoluble nuclear pellets. Protein lysates and immunoprecipitates were resolved by SDS-PAGE followed by Western analysis using the antibodies specific for: WRN (Abcam Inc., Cambridge, MA, USA and Santa Cruz, Santa Cruz, CA, USA), c-ABL (EMD Chemicals, Inc., Gibbstown, NJ, USA), BCR/ABL (16), P-Tyr, RAD51, RPA70 and RPA32 (Milipore, Temecula, CA, USA), β-actin, XRCC4, Ligase IV, Artemis, XLF, XRCC1, lamin B1 (Abcam Inc.), Caspase-3 and Ligase III (BD Biosciences, Mississauga, ON, Canada), RAD52 and PARP-1 (Cell Signaling Technology, Inc., Danvers, MA, USA), Ku70 and Ku80 (AbD Serotec, Raleigh, NC, USA), tubulin (Novus Biologicals, Littletown, CO, USA) and DNA-PKcs(Thermo Scientific Inc., Fremont, CA, USA). Densitometry analysis was performed using Quantity One software (Bio-Rad, Hercules, CA, USA).
WRN enzymatic assays
Helicase and exonuclease activities were determined as described previously with modifications (21, 22) (Supplemental Methods). Reaction samples were subjected to electrophoresis in 14% non-denaturing (helicase) and denaturing (exonuclease) gels, and visualized by autoradiography. The products were quantified using Quantity One software (Bio-Rad, Hercules, CA, USA).
Retroviral infection
To produce viral supernatants, retroviral DNAs were transiently transfected into Phoenix Amphotropic cells. The viral supernatants were collected 36h later, filtered and concentrated by centrifugation at 20,000rpm for 2 hours at 14°C. Cells (0.5 × 106cells/ml) were infected with 1:1 mixture of freshly concentrated virus and medium, twice a day for two days in the presence of 4 μg/ml of polybrene(Sigma, St. Louis, MO, USA) as described before (15).
Genotoxic treatment
Downregulation of WRN was achieved as described by Grandori et al. (18). Briefly, BCR/ABL-positive UT7 and MO7 cells were infected with pBABEpuro-shWRN or with pBABEpuro and selected in puromycin. Furthermore, cells expressing pBABEpuro-shWRN were co-transfected with LXSN-WRN cDNA construct and selected in G418. Downregulation and restoration of WRN expression was confirmed by Western analysis. Cells were treated with increasing doses of H2O2 or γ-radiation (137Cs) and examined 48 hours and 5 days later by trypan blue exclusion test and clonogenic assay in methylcellulose, respectively, as described before (5, 16, 21).
Examination of DSB repair
The activities of HRR, NHEJ and SSA were measured as described before (7, 16, 23) with modifications (Supplemental Methods).
RESULTS
BCR/ABL and other FTKs stimulate the expression of WRN
Our genome-wide array suggested that WRN is overexpressed in CML mononuclear cells by 2.7 ± 1.1 -fold in comparison to normal counterparts (21). To confirm this observation total RNA was isolated from 32D and 32D-BCR/ABL (32D-B/A) cell lines, and CD34+ bone marrow cells obtained from healthy donors (NBMC) or CML-CP patients (CML) and WRN expression was examined by semi-quantitative RT-PCR. BCR/ABL-positive 32D cells and CD34+ CML cells contained more WRN mRNA in comparison to their BCR/ABL-negative counterparts (Figure 1A).
Figure 1. BCR/ABL enhances the expression of WRN.
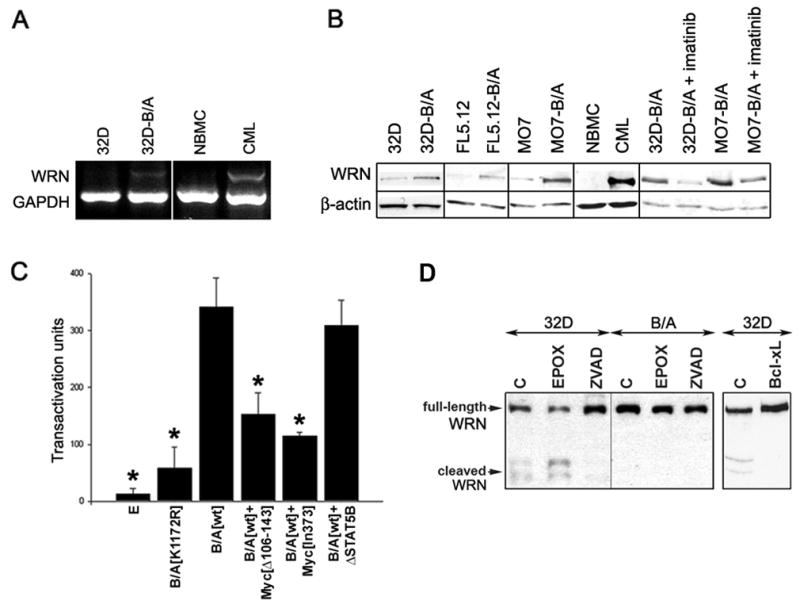
(A) Total RNA was isolated from 32D parental (32D) and 32D-BCR/ABL cells (B/A) and CD34+ bone marrow cells obtained from healthy donors (NBMC) or CML patients (CML). WRN expression was examined by semi-quantitative RT-PCR simultaneously detecting WRN and GADPH (internal reaction control). The results are representative for 5 32D-BCR/ABL clones, 4 CML-CP and one CML-BP patients. (B) Cellular lysates were obtained from parental and BCR/ABL-transformed (B/A) murine (32D, FL5.12) and human (M07) cell lines, and from CD34+ bone marrow cells from healthy donor (NBMC) and CML-CP patient after incubation for 12h in the absence of growth factors. 1μM imatinib was added when indicated. WRN expression was examined by Western analysis; β-actin is shown as loading control. (C) Transacrivation assay was performed with the use of WRN promoter sequence linked to the luciferase gene. 293T cells were co-transfected with the WRN promoter-luciferase reporter plasmid and empty plasmid [E], or plasmids encoding BCR/ABL kinase-dead mutant [K1172R], BCR/ABL kinase [wt], BCR/ABL kinase + c-Myc[Δ106–143] dominant-negative mutant (B/A[wt]+Myc [Δ106–143]), BCR/ABL kinase + c-Myc[In373] dominant-negative mutant (B/A[wt]+Myc[In373]), or BCR/ABL kinase +ΔSTAT5B dominant-negative mutant (B/A[wt]+ΔSTAT5B). Results represent 4 independent experiments; * p<0.001, p=0.001, p<0.01 and p<0.001; 1, 2, 4 and 5 compared to 3, respectively. (D) 32D parental cells (32D) and BCR/ABL-transformed counterparts (B/A) were untreated (C) or treated with 20μM Z-VAD-FMK (ZVAD) and 1μM epoxomycin (EPOX) in the absence of IL-3 for 12h (left box). In addition, 32D cells and those overexpressing Bcl-xL were incubated in the absence of IL-3 for 12h (right box). Full-length and cleaved WRN was analyzed by Western blotting in total cell lysates.
Western analysis of BCR/ABL-transformed hematopoietic cell lines and their parental counterparts, and also CD34+ cells from CML-CP patient and healthy donor indicated that in the absence of growth factors BCR/ABL kinase stimulates the expression of total cellular WRN protein (Figure 1B) by 6.9 ± 3.7 -fold as determined by densitometry analysis. Abrogation of BCR/ABL kinase activity by imatinib caused downregulation of WRN. Moreover, other FTKs such as: TEL/ABL, TEL/JAK2, TEL/PDGFβR, and NPM/ALK also elevated WRN protein expression (Supplemental Figure 1).
To investigate the mechanism of WRN up-regulation the transactivation assay was performed with the use of WRN promoter sequence linked to the luciferase gene. WRN transactivation required BCR/ABL kinase activity; the BCR/ABL[K1172R] kinase-dead mutant did not exert significant activity (Figure 1C). In addition, BCR/ABL kinase-mediated transactivation of WRN was inhibited by c-Myc dominant-negative mutants (DNMs), but not by STAT5 DNM. Altogether, it appears that WRN transactivation is mediated at least in part by BCR/ABL kinase - c-Myc pathway.
WRN was degraded in parental cells undergoing apoptosis due to growth factor starvation (Figure 1D, left box) (24). The process is probably caspase(s)-dependent and does not involve proteasomal activity because it was prevented by the pan-caspase inhibitor Z-VAD-FMK but not by the proteasome inhibitor epoxomycin. Expression of BCR/ABL kinase and overexpression of Bcl-xL, a known inhibitor of caspase-3 activation downstream of BCR/ABL kinase prevented WRN degradation (Figure 1D, left and right box, respectively). Thus, we hypothesize that BCR/ABL kinase – Bcl-xL pathway protects WRN protein from caspase-3 -dependent degradation.
Although total cellular WRN levels were stimulated also by IL-3 signaling in normal cells (Figure 2A), BCR/ABL in comparison to IL-3 enhanced nuclear and chromatin localization of WRN by >3.5 –fold as determined by densitometry analysis (Figure 2B).
Figure 2. Enhanced nuclear and chromatin localization of WRN in BCR/ABL-positive cells.
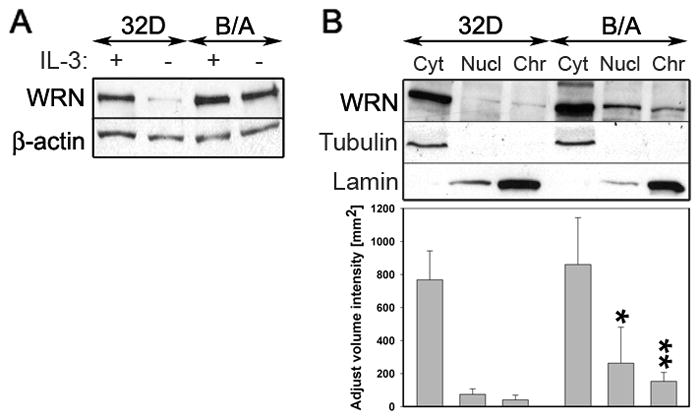
(A) Western analysis of WRN expression in total cellular lysates obtained from 32D parental cells (32D) and 32D-BCR/ABL-transformed counterparts (B/A). Cells were maintained in the presence (+) or absence (−) of IL-3 for 12h. β-actin was detected for loading control. (B, upper panel) Representative Western analysis of WRN expression in cytoplasmic (Cyt), nuclear (Nucl) and chromatin (Chr) fractions were obtained from 32D parental cells (32D) and 32D-BCR/ABL-transformed counterparts (B/A) maintained in the presence of IL-3. β-actin, tubulin and lamin were detected for loading and purity control. (B, lower panel) Densitometry analysis of WRN expression in cytoplasmic, nuclear and chromatin fractions; results represent mean ± SD from three experiments; *p=0.02 and **p=0.03 in comparison to corresponding samples from parental cells.
BCR/ABL kinase interacts with WRN resulting in its tyrosine phosphorylation and stimulation of the helicase and exonuclease activity
To determine if WRN interacts with BCR/ABL, WRN was immunoprecipitated from 32D-BCR/ABL cells. p210BCR/ABL protein was readily detected in anti-WRN specific immunoprecipitates (Figure 3A); on the other hand WRN was present in anti-BCR/ABL specific immunoprecipitates (Figure 3B). WRN immunoprecipitated from 32D-BCR/ABL cells was phosphorylated on tyrosine residue(s) (Figure 3C). This effect was abrogated when the cells were treated with the ABL kinase inhibitor imatinib suggesting that WRN might be a substrate for BCR/ABL kinase.
Figure 3. WRN interacts with BCR/ABL kinase and is phosphorylated on tyrosine(s) in vivo.

(A) WRN and (B) p210BCR/ABL were immunoprecipitated from total cell lysates (Input, 10%) obtained from 32D-BCR/ABL cells using anti-WRN and anti-p210BCR/ABL antibodies, respectively, or non-specific immunoglobulins (ns). The presence of p210BCR/ABL and WRN in the immunoprecipitates was detected by Western analysis. (C) WRN was immunoprecipitated from the total cell lysates of 32D-BCR/ABL (32D-B/A) cells incubated (+) or not (−) with 0.5 μM imatinib for 12h in the presence of IL-3. The immunocomplexes were analyzed by Western blotting using anti-P.Tyr (upper box) and anti-WRN (lower box) antibody. The band in the upper box represents tyrosine-phosphorylated WRN.
WRN helicase and exonuclease activities were analyzed in 32D-BCR/ABL cells treated or not treated with imatinib. WRN immunoprecipitates obtained from the cells incubated with 0.5μM imatinib in the presence of IL-3 displayed 2–3 –fold reduction of helicase and exonuclease activity in comparison to these obtained from untreated counterparts (Figure 4A and 4B, respectively). Thus, BCR/ABL kinase activity stimulated both helicase and exonuclease activities of WRN. Similar amounts of immunoprecipitated WRN from the cells treated or not with imatinib were used for these assays. Although BLM, which may co-precipitate with WRN and exert helicase activity was not detectable in WRN immunoprecipitates (data not shown) we cannot exclude the potential role of another helicase and/or exonuclease.
Figure 4. BCR/ABL kinase stimulates WRN helicase and exonuclease activity.
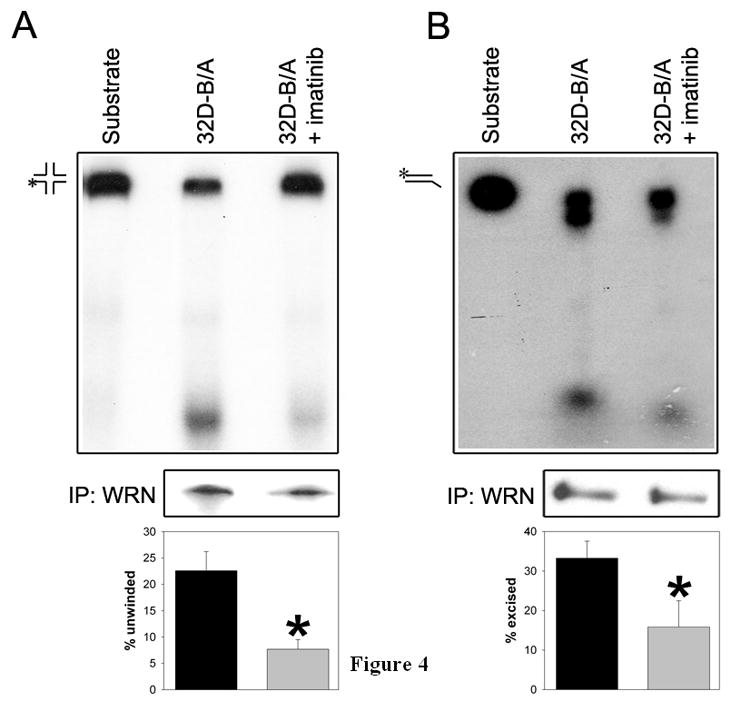
WRN was immunoprecipitated from BCR/ABL-transformed 32D cells cultured in the presence of IL-3 and treated (32D-B/A+imatinib) or not (32D-B/A) with 0.5 μM imatinib for 24h. The immunoprecipitates were used for (A) helicase assay with the synthetic four-way junction DNA substrate, and for (B) exonuclease activity using double-stranded DNA substrate. Representative reactions are shown in upper boxes. The substrate and product positions are indicated; asterisks indicate radionucleotides. Similar amounts of immunoprecipitated WRN were used for the reactions (middle boxes). The mean ± SD reaction values from 3–5 experiments are presented in the bottom boxes; *p<0.05 in comparison to 32D-B/A group.
WRN protects BCR/ABL-positive cells after DNA damage caused by genotoxic and oxidative stress
WRN was downregulated in MO7 parental cells and MO7-BCR/ABL cells by transfection with the plasmid encoding human WRN-specific shRNA as confirmed by Western analysis (Figure 5A). WRN-specific shRNA did not significantly affect the proliferation of transfected cells (data not shown). Genotoxic and oxidative stress were induced by γ-irradiation and H2O2 and cell survival and proliferation were monitored by trypan blue exclusion and clonogenic assays. As expected MO7-BCR/ABL cells were less sensitive to γ-irradiation and H2O2 treatment than parental counterparts (Figure 5B and 5C). Downregulation of WRN increased the sensitivity of MO7-BCR/ABL cells to H2O2 (Figure 5B) and γ-irradiation (Figure 5C). Similar effect was also observed in UT7-BCR/ABL leukemia cells (Supplemental Figure 2). Partial restoration of WRN protein levels in MO7-BCR/ABL cells expressing shWRN by co-transfection with a retroviral construct expressing WRN cDNA (Figure 5D) diminished their sensitivity to H2O2 (Figure 5E) and γ-irradiation (Figure 5F). Incomplete restoration of WRN expression was probably due to a balance between simultaneous action of the plasmids encoding for overexpression (LXSN-WRN cDNA) and downregulation (pBABEpuro-shWRN) of the protein.
Figure 5. Role of WRN in BCR/ABL-mediated inhibition of oxidative and genotoxic stress-induced apoptosis.

(A) WRN expression in MO7 parental cells and MO7-BCR/ABL cells transfected with shWRN (MO7 +shWRN and MO7-B/A + shWRN, respectively) or with empty plasmid (MO7 and MO7-B/A, respectively); β-actin served as loading control. Cells were (B) incubated with H2O2 or (C) γ-irradiated. Cell viability and proliferation were assessed by trypan blue exclusion test (left panels) and clonogenic assay (right panels). (D) WRN expression in MO7-BCR/ABL cells transfected with empty plasmid (MO7-B/A), shWRN (MO7-B/A + shWRN), and shWRN + WRN cDNA (MO7-B/A + shWRN + WRN); β-actin served as loading control. Cells were (E) incubated with H2O2 or (F) γ-irradiated. Cell viability and proliferation were assessed as described in B and C. Results represent mean percentage ± SD of the viability and clonogenicity of treated versus untreated cells; *p<0.05 (Student t test) in (B, C) MO7 + shWRN and MO7-B/A + shWRN cells versus MO7 and MO7-B/A cells, respectively, and (E, F) MO7-B/A + shWRN + WRN cells versus MO7-B/A + shWRN cells.
WRN modulates the efficiency of recombination-dependent repair (HRR and SSA) and the fidelity of NHEJ in BCR/ABL-positive cells
We first examined the expression levels of HRR- and SSA- related proteins in MO7 parental cells and MO7-BCR/ABL leukemia cells treated or not with imatinib. Cells were incubated with GM-CSF in concentrations necessary to stimulate proliferation of parental cells. In these conditions BCR/ABL-positive leukemia cells overexpressed HRR- related nuclear RAD51 and imatinib reduced the level of nuclear RAD51 in leukemia cells (Figure 6A, left box) in concordance with previous report (16). Expression of HRR-related RPA70 and RPA32 and SSA-related RAD52 and ERCC1 were not affected by BCR/ABL kinase (Figure 6B, left box).
Figure 6. WRN facilitates recombination-dependent HRR and SSA in BCR/ABL-positive cells.
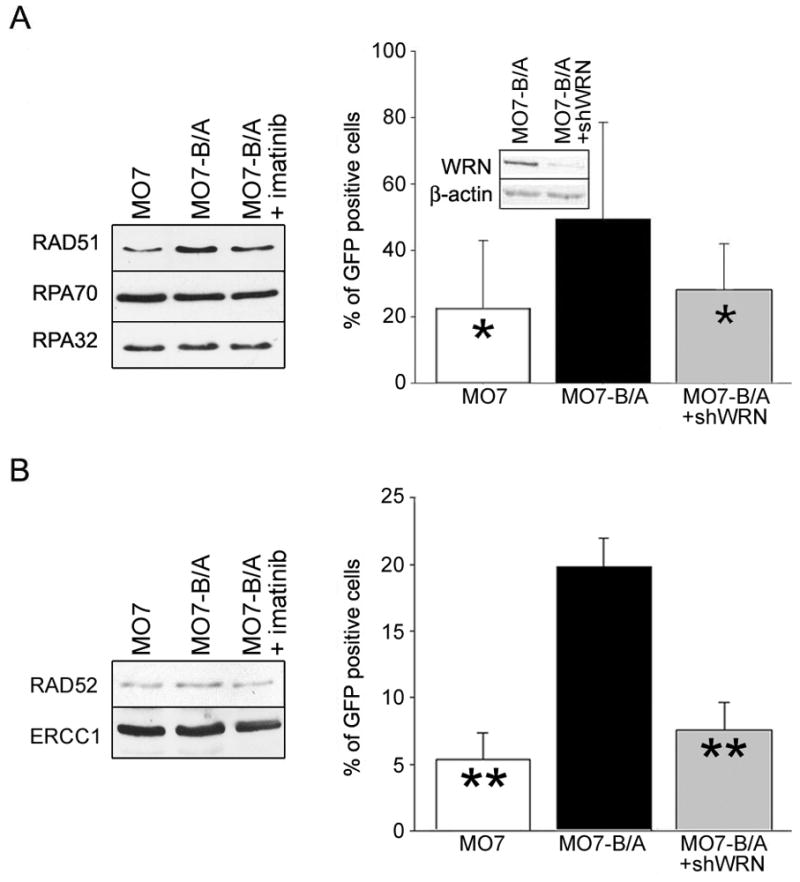
(A, B - left boxes) Western analysis of the nuclear lysates from parental MO7 cells (MO7), BCR/ABL-positive counterparts (MO7-B/A) and B/A cells treated with 1 μM of imatinib for 24h (MO7-B/A + imatinib). Cells were incubated with minimal concentrations of GM-CSF necessary to stimulate proliferation of parental cells. (A, B - right boxes) MO7, MO7-B/A and MO7-B/A cells in which WRN was downregulated (inset) by shWRN (MO7-B/A+shWRN) contained DR-GFP (A) and SA-GFP (B) reporter cassettes. Cells were transfected with I-SceI (to induced DSB) and pDsRed1-Mito (to control transfection efficiency). HRR (A) and SSA (B) efficiency was determined by examining the percentage of GFP+ cells in Red1+ cell population. Results represent three independent experiments (*p<0.05 and **p<0.01 in comparison to MO7-B/A).
To examine HRR and SSA activity, DR-GFP and SA-GFP recombination cassettes containing inactivated GFP gene due to introduction of the unique I-SceI restriction site with two stop codons and a truncated version of the gene containing BcgI restriction site was integrated into the genome of MO7 parental and MO7-BCR/ABL cells. A DSB is generated in DR-GFP and SA-GFP cassettes upon transient transfection with I-SceI expression plasmid, which if repaired by HRR and SSA, respectively, could restore a functional GFP gene and GFP protein expression. Cells were transfected with I-SceI and pDsRed1-Mito (transfection efficiency control) expression plasmids and the efficiency of HRR and SSA was measured 48h later by scoring the percentage of double-positive GFP+Red1+ cells in all transfected cells (Red1+).
As expected, BCR/ABL stimulated HRR and SSA (Figure 6A and 6B, right boxes) (7, 16). Downregulation of WRN in MO7-BCR/ABL cells (Figure 6A; inset) by previously validated shRNA complementary to human WRN caused ~2 and ~3 -fold inhibition of HRR and SSA, respectively (Figure 6A and 6B, right). Similar effect was observed in BCR/ABL-positive Daoy medulloblastoma cells (Supplemental Figure 3).
NHEJ usually occurs via DNA-PKcs –dependent (D-NHEJ) classical pathway involving Ku70, Ku80, DNA-PKcs, Artemis, XLF, ligase IV-XRCC4 proteins, and via backup pathway (B-NHEJ) exerted by PARP-1, XRCC1 and Ligase III (25). Although BCR/ABL-positive leukemia cells and parental counterparts display similar levels of nuclear Ku70, Ku80, XRCC1 and XRCC4 proteins in the presence of growth factors, DNA-PKcs was downregulated, and Ligase IV, Artemis, XLF, PARP-1 and Ligase III were upregulated (Figure 7A).
Figure 7. WRN does not facilitate NHEJ, but modifies nucleotide loss in BCR/ABL-positive cells.

(A) Western analysis of the nuclear lysates from parental 32D cells (32D), BCR/ABL-positive counterparts (32D-B/A) and B/A cells treated with 1 μM of imatinib for 24h (32D-B/A + imatinib). Cells were incubated with minimal concentrations of IL-3 necessary to stimulate proliferation of parental cells. (B) NHEJ activity was measured in the nuclear extracts from 32D and 32D-B/A cells with pBlueScript II(SK) digested with XhoI and XbaI. Results represent mean percentages of end-joined substrate ± SD; *p<0.01 in comparison to other groups. WRN was immunodepleted (−WRN) from the extracts as confirmed by Western analysis (inset). (C) Loss of DNA bases per individual reaction is shown; p=0.037, when an average nucleotide loss in 32D-B/A -WRN group was compared to that in 32D-B/A group.
To study the role of WRN in NHEJ we used the in vitro assay as described before (26). The in vivo assay measuring NHEJ activity and fidelity with the use of DR-GFP cassette should not be used here because downregulation of WRN may affect also Mre11, an exonuclease upregulated by BCR/ABL kinase which promotes deletional NHEJ (27–29). Because the majority of DSBs generated by ROS and γ-irradiation do not have ligatable termini, pBluescript plasmid linearized by XhoI+XbaI digestion creating non-complementary 3′ and 5′ overhangs was used as the substrate to assess the activity of NHEJ. The synapsed DNA ends must be processed before ligation during NHEJ to generate multimers of plasmid. We used this substrate because WRN interacted with it (30). The substrate was added to nuclear cell lysates from 32D parental and 32D-BCR/ABL cells, in which WRN was immunodepleted or not by specific antibody (Figure 7B, inset). 32D-BCR/ABL cells were used here because of the very efficient immunodepletive capability of the anti-WRN antibody in murine cell lysates.
As expected (23) BCR/ABL stimulated NHEJ by ~ 3-fold and immunodepletion of WRN did not affect NHEJ activity in lysates from 32D-BCR/ABL cells (Figure 7B). In concordance with our previous report (23) the presence of BCR/ABL promoted larger deletions in some NHEJ products (3/8 products averaged loss of 29 ± 4.35 bases; 5/8 products averaged loss of 3.8 ± 2.17 bases) (Figure 7C; Supplemental Figure 4). Overall, NHEJ products catalyzed by 32D-BCR/ABL lysates lost on average 13.25 ± 13.35 bases. Large deletions were not detected in NHEJ product catalyzed by 32D parental cells (average loss of 3.29 ± 1.9 bases). Immunodepletion of WRN prevented larger deletions in NHEJ products catalyzed by 32D-BCR/ABL lysates, which averaged 1.43 ± 1.27 bases lost.
DISCUSSION
To protect the leukemia cells from excessive ROS- and genotoxic agent- induced DSBs, BCR/ABL kinase stimulates HRR, NHEJ and SSA at the cost of repair fidelity (6, 7, 23). These repair mechanisms require the presence of WRN, which plays a critical role in optimizing DSB repair mechanisms due to its DSB end-processing helicase and exonuclease activities (30).
Our results suggest that BCR/ABL and other FTKs stimulate WRN expression in part by c-Myc -induced transactivation and Bcl-xL -dependent inhibition of caspase-mediated cleavage. The involvement of c-Myc is supported by the presence of several non-canonical Myc-Max binding sites in WRN promoter (18). WRN contains putative caspase-3 binding sites (31) thus explaining the potential protective role of caspase inhibitor Z-VAD-FMK and anti-apoptotic protein Bcl-xL, which prevents caspase-3 activation in BCR/ABL-positive cells (32). More abundant stimulation of WRN protein (6.9-fold) than mRNA (2.7-fold) expression is also consistent with BCR/ABL-dependent induction of transactivation followed by protection from caspase-mediated degradation of WRN.
BCR/ABL kinase forms a complex with WRN protein resulting in its constitutive tyrosine phosphorylation and also activates WRN helicase and exonuclease activity. In addition, elevated expression of WRN in leukemia cells was associated with enhanced nuclear and chromatin localization, which is essential in response to DNA damage (33, 34).
Cheng et al. reported that WRN formed a complex with c-ABL and that ABL-mediated tyrosine phosphorylation of WRN resulted in inhibition of its helicase and exonuclease activities (22). Since c-ABL interacts directly with the exonuclease and helicase domains of WRN, its excessive presence in the in vitro reaction mixture might promote prolonged interaction, which will physically block the domains of WRN responsible for its enzymatic activities. In fact, results from stoichiometric experiments demonstrated that inhibition of WRN helicase and exonuclease activities in vitro is c-ABL protein –dosage dependent, and not phosphorylation dependent phenomenon. However, the in vivo stoichiometric conditions may promote BCR/ABL-WRN complex formation and WRN phosphorylation followed by complex disassembly and migration of pre-activated WRN to the nucleus.
BCR/ABL-mediated regulation of WRN expression, localization and activity may exert a profound impact on the response to elevated levels of ROS- and genotoxic agent- induced DSBs in CML CD34+ stem/progenitor cells (7). Using shRNA to downregulate WRN we showed that WRN promotes the survival of BCR/ABL-positive leukemia cells under oxidative and genotoxic stress, in concordance with other reports from solid tumors (11, 12, 14, 35). This effect may depend on WRN’s capability to facilitate the repair of numerous DBSs in leukemia cells (6, 7, 16). Since BCR/ABL stimulates unfaithful DSB repair mechanisms (6–9, 23, 36), WRN, in opposite to normal cells (10) may promote genomic instability in CML-CP.
Our studies showed that BCR/ABL and other FTKs, such as TEL/ABL, TEL/JAK2, TEL/PDGFβR, NPM/ALK stimulate recombination-dependent DSB repair: RAD51-mediated
HRR and RAD52-mediated SSA (5, 7). Although HRR normally act to maintain genetic stability, if over-stimulated, it can produce intra- and inter-chromosomal deletions, chromosomal translocations and aneuploidity (37). In addition, point-mutations can be accumulated in HRR products (6). When sequence repeats (for example Alu) are present near DSBs, they can undergo SSA, resulting in intra-chromosomal deletions and chromosomal translocations (38).
Results presented here suggest that WRN is essential for enhanced activity of unfaithful HRR and SSA in BCR/ABL-transformed cells thus implicating WRN in genomic instability in CML. In support to these findings, WRN interacts with RAD51, a key mediator of HRR responsible for initial pairing and strand exchange, which is upregulated by BCR/ABL (16, 39). Moreover, WRN interacts with RAD52, which enhances its helicase activity and increases the efficiency of RAD52-mediated strand annealing (40).
We and others also reported that BCR/ABL kinase facilitates NHEJ and promote more extensive deletions in about 35% of repair products (6, 8, 23). NHEJ has an important function in maintaining genomic integrity, but not necessarily the fidelity because it may re-join DSBs in a non-conservative manner, and usually some genetic information is lost (41). Accordingly, stimulation of NHEJ may increase the risk of accumulation of genetic aberrations including these in quiescent cells (42).
We show that WRN did not regulate the efficiency of NHEJ in BCR/ABL-positive leukemia cells but it caused excessive loss of bases in the repair products. In normal cells WRN did not affect, or even reduced deletions (depending on the DSB ends) in NHEJ products (30). However, the properties of WRN and the intracellular environment in CML cells are substantially altered in comparison to normal cells. Not only BCR/ABL kinase stimulates the expression, tyrosine phosphorylation and nuclear localization of WRN, it also enhances WRN helicase and exonuclease activity. In addition, WRN exerts its function in the context of modified expression of other proteins involved in NHEJ, which may have a substantial impact on WRN properties.
It appears that proteins responsible for D-NHEJ are deregulated by BCR/ABL: nuclear DNA-PKcs was downregulated, whereas Ligase IV, Artemis and XLF were upregulated by BCR/ABL kinase, but XRCC4, Ku70 and Ku80 were not changed, which may affect the activity of WRN. Ku70-Ku80 and XRCC4-Ligase IV complexes activate WRN exonuclease activity (Ku enables the WRN exonuclease to digest through regions of DNA containing 8-oxoA and 8-oxoG, which are frequent in CML cells (4, 6)), whereas DNA-PKcs phosphorylates WRN to inhibit its exonuclease and helicase activity (43–45). Thus we postulate that a combination of BCR/ABL-mediated tyrosine phosphorylation of WRN in the presence of Ku70-Ku80 and/or XRCC4-Ligase IV and the absence of DNA-PKcs may be responsible for prolonged WRN helicase and/or exonuclease activity causing large deletions during NHEJ. Our hypothesis is supported by the reports that downregulation of DNA-PKcs in CML cells was also reported by others (46) and that extensive deletions were associated with inhibition of DNA-PKcs in advanced bladder carcinoma and chemotherapy resistant B-cell chronic lymphocytic leukemia (47, 48).
On the other hand, PARP-1 and Ligase III are overexpressed in BCR/ABL cells implicating enhanced activity of a backup pathway (B-NHEJ) in concordance with the report by Sallmyr et al. (49). WRN interacts with PARP-1 and appears to play a role in B-NHEJ (50). Thus, we cannot exclude the possibility that the absence of DNA-PKcs promotes a switch from Ku70-Ku80 –initiated D-NHEJ to B-NHEJ on the DSBs which were already extensively processed by WRN thus resulting in larger deletions. This hypothesis is supported by the report that NHEJ-mediated large deletions in CML-CP cells might depend on the presence of Ku70-Ku80, but not DNA-PKcs (8).
However, extensive secondary genetic and epigenetic aberrations in more advanced CML-BC cells may affect WRN properties (22, 49). For example, although WRN expression was stimulated, its enzymatic activities were inhibited, and its role in NHEJ was altered in CML-BP cell line K562.
In conclusion, we postulate that BCR/ABL kinase –dependent stimulation of the expression, nuclear localization, phosphorylation and helicase/exonuclease activity of WRN affects all major DSB repair pathways: enhances the activity of unfaithful recombination repair
(HRR and SSA) and increases the infidelity of NHEJ. These effects may have a profound impact on leukemia cell survival and accumulation of additional genetic aberrations in CML cells containing numerous DSBs induced by ROS and genotoxic treatment (6, 26, 36), thus implicating WRN in malignant progression of the disease. BCR/ABL-WRN interaction may become an attractive target for therapeutic intervention preventing malignant progression of CML-CP to CML-BP.
Supplementary Material
Acknowledgments
Financial support: This work was supported by NIH/NCI CA89052 and CA123014 (T.S.), and by the Leukemia Research Foundation New Investigator Grant (A.S.).
References
- 1.Turner SD, Alexander DR. Fusion tyrosine kinase mediated signalling pathways in the transformation of haematopoietic cells. Leukemia. 2006;20:572–82. doi: 10.1038/sj.leu.2404125. [DOI] [PubMed] [Google Scholar]
- 2.Marley SB, Gordon MY. Chronic myeloid leukaemia: stem cell derived but progenitor cell driven. Clin Sci (Lond) 2005;109:13–25. doi: 10.1042/CS20040336. [DOI] [PubMed] [Google Scholar]
- 3.Perrotti D, Jamieson C, Goldman J, Skorski T. Chronic myeloid leukemia: mechanisms of blastic transformation. J Clin Invest. 2010;120:2254–64. doi: 10.1172/JCI41246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koptyra M, Falinski R, Nowicki MO, et al. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood. 2006;108:319–27. doi: 10.1182/blood-2005-07-2815. Epub 2006 Mar 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slupianek A, Hoser G, Majsterek I, et al. Fusion tyrosine kinases induce drug resistance by stimulation of homology-dependent recombination repair, prolongation of G(2)/M phase, and protection from apoptosis. Mol Cell Biol. 2002;22:4189–201. doi: 10.1128/MCB.22.12.4189-4201.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nowicki MO, Falinski R, Koptyra M, et al. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood. 2004;104:3746–53. doi: 10.1182/blood-2004-05-1941. Epub 2004 Aug 10. [DOI] [PubMed] [Google Scholar]
- 7.Cramer K, Nieborowska-Skorska M, Koptyra M, et al. BCR/ABL and other kinases from chronic myeloproliferative disorders stimulate single-strand annealing, an unfaithful DNA double-strand break repair. Cancer Res. 2008;68:6884–8. doi: 10.1158/0008-5472.CAN-08-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaymes TJ, Mufti GJ, Rassool FV. Myeloid leukemias have increased activity of the nonhomologous end-joining pathway and concomitant DNA misrepair that is dependent on the Ku70/86 heterodimer. Cancer Res. 2002;62:2791–7. [PubMed] [Google Scholar]
- 9.Fernandes MS, Reddy MM, Gonneville JR, et al. BCR-ABL promotes the frequency of mutagenic single-strand annealing DNA repair. Blood. 2009;114:1813–9. doi: 10.1182/blood-2008-07-172148. Epub 2009 Jul 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bohr VA. Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance. Trends Biochem Sci. 2008;33:609–20. doi: 10.1016/j.tibs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Opresko PL, Calvo JP, von Kobbe C. Role for the Werner syndrome protein in the promotion of tumor cell growth. Mech Ageing Dev. 2007;128:423–36. doi: 10.1016/j.mad.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Futami K, Ishikawa Y, Goto M, Furuichi Y, Sugimoto M. Role of Werner syndrome gene product helicase in carcinogenesis and in resistance to genotoxins by cancer cells. Cancer Sci. 2008;99:843–8. doi: 10.1111/j.1349-7006.2008.00778.x. Epub 2008 Feb 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shiratori M, Sakamoto S, Suzuki N, et al. Detection by epitope-defined monoclonal antibodies of Werner DNA helicases in the nucleoplasm and their upregulation by cell transformation and immortalization. J Cell Biol. 1999;144:1–9. doi: 10.1083/jcb.144.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grandori C, Robinson KL, Galloway DA, Swisshelm K. Functional link between Myc and the Werner gene in tumorigenesis. Cell Cycle. 2004;3:22–5. [PubMed] [Google Scholar]
- 15.Nieborowska-Skorska M, Hoser G, Kossev P, Wasik MA, Skorski T. Complementary functions of the antiapoptotic protein A1 and serine/threonine kinase pim-1 in the BCR/ABL-mediated leukemogenesis. Blood. 2002;99:4531–9. doi: 10.1182/blood.v99.12.4531. [DOI] [PubMed] [Google Scholar]
- 16.Slupianek A, Schmutte C, Tombline G, et al. BCR/ABL regulates mammalian RecA homologs, resulting in drug resistance. Mol Cell. 2001;8:795–806. doi: 10.1016/s1097-2765(01)00357-4. [DOI] [PubMed] [Google Scholar]
- 17.Nowicki MO, Pawlowski P, Fischer T, Hess G, Pawlowski T, Skorski T. Chronic myelogenous leukemia molecular signature. Oncogene. 2003;22:3952–63. doi: 10.1038/sj.onc.1206620. [DOI] [PubMed] [Google Scholar]
- 18.Grandori C, Wu KJ, Fernandez P, et al. Werner syndrome protein limits MYC-induced cellular senescence. Genes Dev. 2003;17:1569–74. doi: 10.1101/gad.1100303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu A, Sciacca L, Baserga R. Nuclear translocation of insulin receptor substrate-1 by the insulin receptor in mouse embryo fibroblasts. J Cell Physiol. 2003;195:453–60. doi: 10.1002/jcp.10261. [DOI] [PubMed] [Google Scholar]
- 20.Bennett PA, Dixon RJ, Kellie S. The phosphotyrosine phosphatase inhibitor vanadyl hydroperoxide induces morphological alterations, cytoskeletal rearrangements and increased adhesiveness in rat neutrophil leucocytes. J Cell Sci. 1993;106 (Pt 3):891–901. doi: 10.1242/jcs.106.3.891. [DOI] [PubMed] [Google Scholar]
- 21.Slupianek A, Gurdek E, Koptyra M, et al. BLM helicase is activated in BCR/ABL leukemia cells to modulate responses to cisplatin. Oncogene. 2005;24:3914–22. doi: 10.1038/sj.onc.1208545. [DOI] [PubMed] [Google Scholar]
- 22.Cheng WH, von Kobbe C, Opresko PL, et al. Werner syndrome protein phosphorylation by abl tyrosine kinase regulates its activity and distribution. Mol Cell Biol. 2003;23:6385–95. doi: 10.1128/MCB.23.18.6385-6395.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slupianek A, Nowicki MO, Koptyra M, Skorski T. BCR/ABL modifies the kinetics and fidelity of DNA double-strand breaks repair in hematopoietic cells. DNA Repair (Amst) 2006;5:243–50. doi: 10.1016/j.dnarep.2005.10.005. Epub 2005 Nov 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho RL, Johnson DE. Characterization of caspase proteases in cytokine-dependent myeloid progenitor cells using enzyme affinity labeling. J Cell Biochem. 1999;73:79–89. [PubMed] [Google Scholar]
- 25.Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. Epub 2007 Nov 12. [DOI] [PubMed] [Google Scholar]
- 26.Nieborowska-Skorska M, Stoklosa T, Datta M, et al. ATR-Chk1 Axis Protects BCR/ABL Leukemia Cells from the Lethal Effect of DNA Double-Strand Breaks. Cell Cycle. 2006;5:9. doi: 10.4161/cc.5.9.2722. [DOI] [PubMed] [Google Scholar]
- 27.Rink L, Slupianek A, Stoklosa T, et al. Enhanced phosphorylation of Nbs1, a member of DNA repair/checkpoint complex Mre11-RAD50-Nbs1, can be targeted to increase the efficacy of imatinib mesylate against BCR/ABL-positive leukemia cells. Blood. 2007;110:651–60. doi: 10.1182/blood-2006-08-042630. Epub 2007 Apr 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhuang J, Jiang G, Willers H, Xia F. Exonuclease function of human Mre11 promotes deletional nonhomologous end joining. J Biol Chem. 2009;284:30565–73. doi: 10.1074/jbc.M109.059444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turaga RV, Paquet ER, Sild M, et al. The Werner syndrome protein affects the expression of genes involved in adipogenesis and inflammation in addition to cell cycle and DNA damage responses. Cell Cycle. 2009;8:2080–92. doi: 10.4161/cc.8.13.8925. [DOI] [PubMed] [Google Scholar]
- 30.Chen L, Huang S, Lee L, et al. WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell. 2003;2:191–9. doi: 10.1046/j.1474-9728.2003.00052.x. [DOI] [PubMed] [Google Scholar]
- 31.Bischof O, Galande S, Farzaneh F, Kohwi-Shigematsu T, Campisi J. Selective cleavage of BLM, the bloom syndrome protein, during apoptotic cell death. J Biol Chem. 2001;276:12068–75. doi: 10.1074/jbc.M006462200. [DOI] [PubMed] [Google Scholar]
- 32.Amarante-Mendes GP, Naekyung Kim C, Liu L, et al. Bcr-Abl exerts its antiapoptotic effect against diverse apoptotic stimuli through blockage of mitochondrial release of cytochrome C and activation of caspase-3. Blood. 1998;91:1700–5. [PubMed] [Google Scholar]
- 33.Boulikas T. Nuclear import of DNA repair proteins. Anticancer Res. 1997;17:843–63. [PubMed] [Google Scholar]
- 34.Vaitiekunaite R, Butkiewicz D, Krzesniak M, et al. Expression and localization of Werner syndrome protein is modulated by SIRT1 and PML. Mech Ageing Dev. 2007;128:650–61. doi: 10.1016/j.mad.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 35.Agrelo R, Cheng WH, Setien F, et al. Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proc Natl Acad Sci U S A. 2006;103:8822–7. doi: 10.1073/pnas.0600645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brady N, Gaymes TJ, Cheung M, Mufti GJ, Rassool FV. Increased error-prone NHEJ activity in myeloid leukemias is associated with DNA damage at sites that recruit key nonhomologous end-joining proteins. Cancer Res. 2003;63:1798–805. [PubMed] [Google Scholar]
- 37.Richardson C, Stark JM, Ommundsen M, Jasin M. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene. 2004;23:546–53. doi: 10.1038/sj.onc.1207098. [DOI] [PubMed] [Google Scholar]
- 38.Elliott B, Richardson C, Jasin M. Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol Cell. 2005;17:885–94. doi: 10.1016/j.molcel.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 39.Machwe A, Xiao L, Groden J, Matson SW, Orren DK. RecQ Family Members Combine Strand Pairing and Unwinding Activities to Catalyze Strand Exchange. J Biol Chem. 2005;280:23397–407. doi: 10.1074/jbc.M414130200. Epub 2005 Apr 20. [DOI] [PubMed] [Google Scholar]
- 40.Baynton K, Otterlei M, Bjoras M, von Kobbe C, Bohr VA, Seeberg E. WRN interacts physically and functionally with the recombination mediator protein RAD52. J Biol Chem. 2003;278:36476–86. doi: 10.1074/jbc.M303885200. Epub 2003 May 15. [DOI] [PubMed] [Google Scholar]
- 41.Jasin M. Chromosome breaks and genomic instability. Cancer Invest. 2000;18:78–86. doi: 10.3109/07357900009023065. [DOI] [PubMed] [Google Scholar]
- 42.Heidenreich E, Novotny R, Kneidinger B, Holzmann V, Wintersberger U. Non-homologous end joining as an important mutagenic process in cell cycle-arrested cells. file. 2003;22:2274–83. doi: 10.1093/emboj/cdg203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yannone SM, Roy S, Chan DW, et al. Werner syndrome protein is regulated and phosphorylated by DNA-dependent protein kinase. J Biol Chem. 2001;276:38242–8. doi: 10.1074/jbc.M101913200. [DOI] [PubMed] [Google Scholar]
- 44.Kusumoto R, Dawut L, Marchetti C, et al. Werner protein cooperates with the XRCC4-DNA ligase IV complex in end-processing. Biochemistry. 2008;47:7548–56. doi: 10.1021/bi702325t. Epub 2008 Jun 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Orren DK, Machwe A, Karmakar P, Piotrowski J, Cooper MP, Bohr VA. A functional interaction of Ku with Werner exonuclease facilitates digestion of damaged DNA. Nucleic Acids Res. 2001;29:1926–34. doi: 10.1093/nar/29.9.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deutsch E, Dugray A, AbdulKarim B, et al. BCR-ABL down-regulates the DNA repair protein DNA-PKcs. Blood. 2001;97:2084–90. doi: 10.1182/blood.v97.7.2084. [DOI] [PubMed] [Google Scholar]
- 47.Bentley J, Diggle CP, Harnden P, Knowles MA, Kiltie AE. DNA double strand break repair in human bladder cancer is error prone and involves microhomology-associated end-joining. Nucleic Acids Res. 2004;32:5249–59. doi: 10.1093/nar/gkh842. Print 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deriano L, Merle-Beral H, Guipaud O, Sabatier L, Delic J. Mutagenicity of non-homologous end joining DNA repair in a resistant subset of human chronic lymphocytic leukaemia B cells. Br J Haematol. 2006;133:520–5. doi: 10.1111/j.1365-2141.2006.06071.x. [DOI] [PubMed] [Google Scholar]
- 49.Sallmyr A, Tomkinson AE, Rassool FV. Up-regulation of WRN and DNA ligase IIIalpha in chronic myeloid leukemia: consequences for the repair of DNA double-strand breaks. Blood. 2008;112:1413–23. doi: 10.1182/blood-2007-07-104257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.von Kobbe C, Harrigan JA, Schreiber V, et al. Poly(ADP-ribose) polymerase 1 regulates both the exonuclease and helicase activities of the Werner syndrome protein. Nucleic Acids Res. 2004;32:4003–14. doi: 10.1093/nar/gkh721. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.