

Abstract
In this mini-review/opinion article we describe evidence that multiple cellular and molecular alterations in Alzheimer’s disease (AD) pathogenesis involve perturbed cellular calcium regulation, and that alterations in synaptic calcium handling may be early and pivotal events in the disease process. With advancing age neurons encounter increased oxidative stress and impaired energy metabolism, which compromise the function of proteins that control membrane excitability and subcellular calcium dynamics. Altered proteolytic cleavage of the β-amyloid precursor protein (APP) in response to the aging process in combination with genetic and environmental factors results in the production and accumulation of neurotoxic forms of amyloid β-peptide (Aβ ). Aβ undergoes a self-aggregation process and concomitantly generates reactive oxygen species that can trigger membrane-associated oxidative stress which, in turn, impairs the functions of ion-motive ATPases and glutamate and glucose transporters thereby rendering neurons vulnerable to excitotoxicity and apoptosis. Mutations in presenilin-1 that cause early-onset AD increase Aβ production, but also result in an abnormal increase in the size of endoplasmic reticulum calcium stores. Some of the events in the neurodegenerative cascade can be counteracted in animal models by manipulations that stabilize neuronal calcium homeostasis including dietary energy restriction, agonists of glucagon-like peptide 1 receptors and drugs that activate mitochondrial potassium channels. Emerging knowledge of the actions of calcium upstream and downstream of Aβ provides opportunities to develop novel preventative and therapeutic interventions for AD.
Keywords: amyloid, BDNF, calcium channels, cognitive impairment, endoplasmic reticulum, GLP-1, mitochondria, oxidative stress
1. Cellular and Molecular Landscape of the Brain in Alzheimer’s Disease (AD)
To understand if and how alterations in neuronal Ca2+ handling contribute to the symptoms of AD (cognitive impairment and emotional disturbances), it is important to peer inside the brains of AD patients and age-matched neurologically normal control subjects using a range of technologies including magnetic resonance imaging (MRI) in living subjects, and microscopy- and molecular biology-based analyses of postmortem brain samples. Several major abnormalities have been described that typify the brain in AD. MRI image-based measurements of the sizes of different brain structures in patients with mild cognitive impairment (MCI) who later develop AD have revealed progressive and profound reductions in the size of the hippocampus, a brain region critical for the acquisition of memories [1]. Gyri in the frontal, parietal and temporal lobes are also reduced in size as the disease advances [2]. The results of (indirect) measurements of regional cellular energy metabolism using positron emission tomography to image relative levels of radiolabeled 2-deoxyglucose uptake, or functional MRI-based imaging of cerebral blood flow, have also proven informative. Reductions in energy utilization in the temporal and frontal lobes occur early in AD patients, even before evidence of cognitive impairment [3]
Examination of brain tissue sections from AD patients and age-matched control subjects have revealed several abnormalities in AD, most notably the presence of so-called neurofibrillary tangles (NFT) in neurons in the same brain regions that shrink as the disease progresses. At the molecular level, NFT have been shown to be comprised of filamentous accumulations of the microtubule-associated protein tau. In contrast to tau in healthy neurons, the tau in NFT is hyperphosphorylated and hyperacetylated [4]. A second major feature of AD is excessive extracellular accumulation, in the form of diffuse and compact ‘plaques’ of amorphous and fibrillar aggregates of amyloid β-peptide (Aβ ). Aβ is a 40–42 amino acid peptide cleaved from a much larger integral membrane precursor protein (APP); the enzymes that liberate Aβ are called β-secretase and γ-secretase. Excessive production of the longer 42 amino acid form of Aβ (Aβ 42) is strongly implicated as a key event in AD pathogenesis because most, if not all, mutations in APP and presenilin-1 (the enzymatic component of the γ-secretase protein complex) that cause early-onset dominantly inherited familial AD (FAD) increase the production of Aβ 42 [5]. Moreover, Aβ 42 has been shown to be toxic to neurons, especially when it is in the form of small peptide oligomers that are in an active state of self-aggregation [6,7]. Unfortunately, treatment strategies for AD based on inhibiting γ-secretase to reduce Aβ production or immunotherapy to stimulate removal of Aβ [8] have failed in clinical trials.
Other features in the Aβ- and tau-riddled landscape of the AD brain are a reduction in the number of synapses and the death of neurons. There are several lines of evidence that point to synapses as the sites where the neurodegenerative process begins in AD. APP is axonally transported and accumulates in presynaptic terminals, and data suggest that Aβ is produced in high amounts in synaptic terminals [9,10]. Synapses are particularly vulnerable to dysfunction and permanent damage caused by Aβ as demonstrated in electrophysiological experiments in which exposure of brain slices to Aβ impairs synaptic plasticity [11,12], and studies of cultured neurons and isolated synaptic terminals which have shown that Aβ can impair synaptic membrane ion and glucose transporters, and can perturb mitochondrial bioenergetics [13, 14]. Recent high resolution in vivo two-photon microscopy imaging studies have clearly shown an intimate physical association between Aβ aggregates and degeneration of neurites and synapses [15]. In addition, Aβ impairs axonal transport which may contribute to intraneuronal accumulation of Aβ , neuronal network dysfunction, and transneuronal spread of the neurodegenerative process in AD [16, 17].
2. The Aging Brain I: Oxidative and Metabolic Stress Compromise Neuronal Ca2+ Handling
The evidence that damage to cellular components by free radicals contributes to the aging process throughout the body including the brain is substantial. Levels of oxidatively-modified proteins, DNA and lipids are elevated with advancing age in multiple brain regions of humans and rodents [18–20]. The same oxidative modifications are even more profound in vulnerable brain regions of AD patients compared to age-matched control subjects [21–24]. Interestingly, several genes encoding proteins involved in the regulation of neuronal excitability accumulate oxidative DNA lesions [25], which may contribute to disruption of neuronal Ca2+ homeostasis and neuronal network dysfunction in aging and AD [26]. Studies of animal models of AD, principally transgenic mice that overexpress mutant human APP (alone or in combination with mutant presenilin-1 and/or tau) have provided evidence that increased oxidative stress occurs in neurons in association with the earliest stages of development of discernable Aβ and tau pathologies [27]. Oxidative stress may promote Aβ production [28] by increasing APP cleavage by both β- and γ-secretases [29]. Elevated intracellular Ca2+ levels resulting from age-related increases in oxidative stress and Aβ toxicity (see [30] and section on Aβ and Ca2+ homeostasis below) may contribute to the increased amyloidogenic processing of APP in AD [31].
Accompanying oxidative damage to neurons during brain aging is a progressive impairment of the function of mitochondria. Imaging studies of regional brain energy metabolism in human subjects have demonstrated hypometabolism in the hippocampus and frontal cortex during aging, which is predictive of subsequent development of cognitive impairment and AD [32,33]. Studies of mitochondria isolated from the brains of rodents of different ages have provided evidence that the ability of mitochondria to generate ATP is compromised with advancing age and that mitochondria from old brain cells exhibit increased free radical-mediated damage [34]. Impaired cellular energy metabolism may render neurons vulnerable to excitotoxic damage [35], particularly when neurons are faced with the additional stresses of Aβ and tau accumulations [36].
Neurons utilize many of the same mechanisms for Ca2+ signaling and restoration of transmembrane Ca2+ gradients as do other cell types including: voltage-gated Ca2+ channels and a Ca2+-ATPase in the plasma membrane; receptors for various ligands that are coupled to inositol phospholipid hydrolysis and Ca2+ release from IP3-sensitive endoplasmic reticulum (ER) Ca2+ stores; ER ryanodine receptor channels that mediate Ca2+-induced Ca2+ release; and mitochondrial Ca2+ uptake and release systems (see [37] and [38] for review). In addition, neurons also possess unique systems for local Ca2+ signaling at synapses including; presynaptic voltage-gated Ca2+ channels coupled to the synaptic vesicle membrane fusion machinery [39]; postsynaptic excitatory glutamate receptor channels which flux either Na+ (AMPA receptors) or Ca2+ (NMDA receptors) [40,41]; and Ca2+ -binding proteins [42].
Studies of animal and cell culture models have clearly shown that the ability of neurons to regulate cellular Ca2+ levels and dynamics properly is compromised by both oxidative stress and impaired cellular energy metabolism [43]. Membrane lipid peroxidation has particularly disruptive effects on neuronal Ca2+ homeostasis. Lipid peroxidation typically occurs when levels of cellular superoxide anion radical and hydrogen peroxide are increased in the presence of even trace amounts of Fe2+ or Cu+, resulting in the production of hydroxyl radical [23]. Hydroxyl radical attacks double bonds in membrane lipids, thereby producing a range of aldehydes. The aldehydic product of lipid peroxidation 4-hydroxynonenal (HNE) may play a particularly prominent role in the disruption of neuronal Ca2+ homeostasis in aging and AD because of its ability to covalently modify proteins on cysteine, lysine and histidine residues. It has been demonstrated in experimental models that HNE impairs the function of at least 4 proteins that are known to play major roles in neuronal Ca2+ signaling: the plasma membrane Na +/K+-ATPase; the plasma membrane Ca2+-ATPase; the neuronal glucose transporter GLUT3; voltage-gated Ca2+ channels [44–46]; and the glutamate transporter in astrocytes [47].
Decrements in levels of ATP and NAD+, the primary energy substrates in neurons, are implicated in age-related cognitive dysfunction and AD. These energy substrates are particularly critical for the function and survival of neurons because neurons must consume large amounts of energy to rapidly restore ion gradients after synaptic activation and action potential generation. When cellular energy levels are reduced in neurons, as occurs dramatically during an ischemic stroke and more insidiously during aging and in AD, the intracellular Ca2+ levels remain elevated as the result of sustained influx through glutamate- and voltage-gated channels in combination with impaired ion-motive ATPase activities [48–50]. Depletion of NAD+ can be prevented by administering nicotinamide, thereby enabling neurons to maintain intracellular Ca2+ levels low enough to prevent damage and death [51].
3. The Aging Brain II: Impaired Abilities to Prevent and Repair Cellular Damage
While the bulk of the research on brain aging and AD has focused on factors that damage neurons (oxidative stress, energy impairment, Aβ , etc.), less emphasis has been placed on the possible failure of molecular and cellular mechanisms that may protect neurons against dysfunction and degeneration. That such intrinsic protective mechanisms exist and can determine whether or not a given person develops late onset AD is suggested by epidemiological data demonstrating associations between lifestyle factors and the risk of AD. Two examples are regular exercise and moderation in dietary energy intake, both of which may reduce the risk of AD. For example a population-based study provided evidence that regular exercise in mid- or late-life is associated with reduced risk of mild cognitive impairment; this beneficial effect of exercise was dose-dependent [52]. Another study demonstrated that a 6-month exercise program resulted in significant improvement in cognitive function in elderly women [53]. Accumulating evidence suggests that overeating/obesity and diabetes increase the risk of cognitive impairment in AD. For example, an epidemiological study found that individuals on the lower end of the calorie intake spectrum are at reduced risk for AD [54], caloric restriction improves cognitive performance in the elderly [55] and more individuals with diabetes exhibit cognitive impairment and develop AD compared to those without diabetes [56,57]. Studies of animal models of AD support the notion that exercise and moderation in energy intake can retard the molecular and cellular alterations underlying cognitive impairment and AD. Long-term voluntary exercise decreases the accumulation of Aβ in the brains of APP mutant mice [58], and short-term (3 weeks) voluntary wheel running improves cognitive performance in aged APP mutant AD mice [59]. When transgenic mice that express mutant human APP, presenilin-1 and tau (3xTgAD mice) were maintained on either alternate day fasting or limited daily feeding (40% caloric restriction) for 1 year, their learning and memory performance was superior to that of 3xTgAD mice maintained on the usual ad libitum diet [60]. Diabetic mice exhibit several abnormalities in their hippocampus that are associated with impaired cognitive function including reduced long-term potentiation of synaptic strength and a decrement in neurogenesis [61]. Collectively, the evidence suggests that a chronic positive energy balance and diabetes adversely affect neuroplasticity and cognitive function during aging, whereas a negative energy balance in overweight subjects or a neutral energy balance in low weight subjects promotes successful brain aging.
What is the mechanism(s) by which exercise and dietary energy restriction protect the brain against age-related cognitive impairment and AD? Several studies have shown that exercise [62] and energy restriction [63,64] increase the production of brain-derived neurotrophic factor (BDNF). This likely contributes to the beneficial effects of exercise and energy restriction on cognition because BDNF plays important roles in synaptic plasticity and neurogenesis, and BDNF can protect neurons against oxidative and metabolic insults (see [65, 66] for review). Dietary energy restriction, exercise and BDNF can protect neurons against dysfunction and death in experimental models in which the damage to neurons is Ca2+-mediated including excitotoxic seizures, ischemic stroke and AD [60, 67–69]. Evidence that BDNF signaling is impaired in AD includes: reduced levels of BDNF mRNA and protein in vulnerable brain regions of AD patients compared to age-matched control subjects [70]; a negative association between cerebrospinal fluid BDNF concentration and cognitive function in elderly subjects [71]; reduced BDNF levels associated with Aβ aggregation state in the brain in transgenic mouse models of AD [72]; and Aβ impairs retrograde BDNF trafficking/signaling [73].
Several different neurotrophic factors have been shown to prevent cellular Ca2+ overload in cultured neurons in experimental models of excitotoxic and metabolic stress. For example, fibroblast growth factor 2 (FGF2) protected cultured hippocampal neurons against death induced by exposure to glutamate [74] or glucose deprivation [75] by a mechanism involving suppression of Ca2+ overload. FGF2 also protected cultured hippocampal neurons against Aβ-induced death, again by stabilizing intracellular Ca2+ levels [76]. Insulin-like growth factor 1 (IGF1) is another example of a neurotrophic factor that promotes neuronal plasticity and survival, at least in part, by modifying cellular systems that regulate Ca2+ dynamics. For example, IGF1 protected cultured rat hippocampal and septal neurons against death induced by glucose deprivation, and this protection was associated with prevention of excessive elevation of intracellular Ca2+ levels [77]. A decrement in IGF1 signaling may play a role in AD pathogenesis because levels of IGF1 expression and signaling are reduced in brain tissue samples from AD patients compared to age-matched control subjects [78]. Because IGF1 can protect neurons against Aβ toxicity [79], a deficit in IGF1 would be expected to render neurons more vulnerable to being damaged by Aβ in AD.
A particularly interesting recent series of findings provide evidence for a novel endo-neurocrine system that simultaneously enhances peripheral glucose metabolism and engages a signaling pathway in neurons that promotes their survival and adaptive plasticity. Glucagon-like peptide 1 (GLP1) is produced in enteroendocrine cells in the gut, from which it is released in response to food (particularly carbohydrate) ingestion. GLP1 increases insulin production and release from pancreatic β-cells and increases insulin sensitivity of muscle and liver cells. The latter actions of GLP1 led to the development of a longer-lasting synthetic peptide analog of GLP1 called Exendin-4, which is now used to treat many patients with type 2 diabetes [80]. We found that Exendin-4 reduces neuronal damage and improves functional outcome in animal models of stroke, Parkinson’s disease and Huntington’s disease [81,82]. GLP1 receptors are coupled to Gs and cyclic AMP production resulting in the activation of the transcription factor CREB, which then induces the expression of a range of genes involved in neuronal plasticity and survival including BDNF and certain DNA repair enzymes [83, 84]. Cell culture studies and in vivo studies have shown that GLP1 receptor activation can prevent aberrant elevations of intracellular Ca2+ levels in models of excitotoxicity and Aβ toxicity [85–87]. Finally, two recent studies have demonstrated beneficial effects of GLP1 receptor agonists on Aβ pathology and synaptic plasticity in mouse models of AD [88, 89].
AD and other major age-related neurodegenerative disorders involve abnormal intracellular accumulations of proteins which differ somewhat amongst the disorders: tau and Aβ in AD, α-synuclein in Parkinson’s disease, and huntingtin in Huntington’s disease. Increasing evidence suggests that impaired cellular “garbage disposal” mechanisms contribute to the build-up of the abnormal proteins. The two major garbage disposal systems, the proteasome and the autophagy/lysosome apparatus, become dysfunctional in AD [90, 91]. Excessive elevations of intracellular Ca2+ levels may impair proteasome function and autophagy [92, 93] on the one hand, while impaired proteasome and autophagy activity can result in dysregulation of cellular Ca2+ homeostasis [94, 95] on the other hand.
A final example of a cytoprotective regulatory system within neurons that recent findings suggest is compromised during aging and AD is the plasma membrane (PM) redox system (PMRS). The PM contains redox enzymes that provide electrons for energy metabolism and recycling of antioxidants such as coenzyme Q and vitamin E. PMRS enzymes include NADH-ascorbate free radical reductase, NADH-quinone oxidoreductase 1, NADH-ferrocyanide reductase, NADH-coenzyme Q10 reductase and NADH-cytochrome c reductase. In addition, the antioxidants α-tocopherol and coenzyme Q10 are important components of the PMRS. We found that caloric restriction, a manipulation that protects the brain against aging and disease, increases activities of PMRS enzymes and antioxidant levels (α-tocopherol and coenzyme Q10) in brain PMs during aging [96]. Age-related increases in PM lipid peroxidation, protein carbonyls, and nitrotyrosine were attenuated by caloric restriction, and levels of PMRS enzyme activities were higher and markers of oxidative stress were lower in cultured neuronal cells treated with serum from calorie-restricted animals compared with those treated with serum from ad libitum-fed control animals. These findings suggest important roles for the PMRS in protecting brain cells against age-related increases in oxidative and metabolic stress. By rendering mitochondria dysfunctional, we found that the PMRS can sustain cellular redox state and viability [97]. The activity levels of multiple PMRS enzymes was found to be reduced in brain tissue samples from AD patients and 3xTgAD mice, suggesting a role for compromised PMRS function in AD pathogenesis [98].
4. Disruption of Neuronal Ca2+ Homeostasis by Amyloid β-Peptide
Exposure of cultured human or rodent neurons to Aβ (1–40 or 1–42) can result in an elevation of the resting intracellular Ca2+ concentration and can render the neurons vulnerable to excitotoxicity [99]. The cytotoxic actions of Aβ appear to require that the peptide is in the process of self-aggregation because small oligomeric forms of Aβ are particularly damaging to neurons [12, 36]. The plasma membrane appears to be the major site at which neurons are damaged by Aβ . Two major mechanisms for such damaging effects of Aβ have been described and both involve Ca2+ as a central mediator of Aβ ‘s pathological actions. First, when aggregating at the cell surface Aβ generates reactive oxygen species (hydrogen peroxide and hydroxyl radical) resulting in membrane lipid peroxidation [100,101]. Membrane lipid peroxidation results in the generation of the toxic aldehyde 4-hydroxynonenal which impairs the function of membrane ion-motive ATPases and glucose and glutamate transporters thereby promoting membrane depolarization, Ca2+ influx and cellular energy depletion [13, 44, 45, 47]. That lipid peroxidation and 4-hydroxynonenal are pivotal events in the toxic action of Aβ is suggested by the ability of antioxidants that inhibit membrane lipid peroxidation and molecules that scavenge 4-hydroxynonenal to protect neurons from being damaged and killed by Aβ [13, 101–103]. A second mechanism by which neuronal Ca2+ homeostasis might be disrupted by Aβ is that Aβ oligomers may form Ca2+-permeable pores in the plasma membrane [104].
Studies of postmortem brain tissue samples from AD patients, and of animal models of AD, support a role for disruption of neuronal (particularly synaptic) Ca2+ regulation in the neurotoxic action of Aβ . Evidence for hyperactivation of calpains (Ca2+-dependent proteases) in neurons undergoing neurfibrillary degeneration in AD has been reported [105, 106]. Imaging of Aβ deposits and intracellular Ca2+ levels in neurons in the brains of APP mutant mice have provided convincing evidence that Aβ causes an aberrant elevation of Ca2+ levels in neurites [107]. A subsequent study provided evidence that activation of the Ca2+-dependent phosphatase calcineurin mediates Aβ-induced spine loss and dendritic degeneration [108]. Glutamate receptor-mediated Ca2+ elevations have been reported to cause changes in tau similar to those seen in neurofibrillary tangles [109], suggesting a pivotal role for aberrant neuronal Ca2+ regulation in the neurodegenerative process in AD.
5. The α-Secretase-Derived Secreted Form of APP Stabilizes Neuronal Calcium Homeostasis
While sequential cleavages of APP by β- and γ-secretases generate Aβ , cleavage of APP in the middle of the Aβ sequence by α-secretase generates a secreted form of APP called sAPPα [110]. sAPPα is believed to be released from presynaptic terminals in response to electrical activity and Ca2+ influx [111]. However, hyper-excitation of neurons can result in increased Aβ production [112], and presumably reduced sAPPα production. When recombinant sAPPα is applied to cultured hippocampal neurons, a rapid hyperpolarization of the membrane occurs and Ca2+ responses to glutamate are dampened [113,114]. Whole cell patch clamp recordings demonstrated that sAPPα suppresses spontaneous action potential firing, and that this results from activation of high conductance charybdotoxin-sensitive K+ channels [113]. The mechanism by which sAPPα prevents cellular Ca2+ overload and protects neurons against excitotoxic and metabolic insults, and Aβ toxicity, may involve activation of a membrane-associated guanylate cyclase which generates cyclic GMP [115]. The latter signaling pathway results in activation of cyclic GMP-dependent protein kinase which can activate K+ channels, and may also enhance glucose and glutamate transport in synaptic compartments [116]. Finally, sAPPα can protect neurons against the cell death-promoting actions of presenilin-1 mutations (see section 6 below) by stabilizing intracellular Ca2+ levels [117]. A major impediment to further research on the biological functions of sAPPα is that a receptor for sAPPα has not yet been identified. The discovery of such a receptor would therefore be an important advance in the field.
6. Presenilin-1 Mutations and Perturbed Endoplasmic Reticulum Ca2+ Release in AD
Numerous families have been identified in which dominantly inherited early-onset AD is caused by a missense mutation in the presenilin-1 gene [118]. Affected individuals typically become symptomatic when they are in their 40s or 50s. Presenilin-1 is the enzymatic component of the γ-secretase enzyme complex that cleaves APP to generate Aβ , and AD-causing mutations in presenilin-1 increases the production of Aβ 42 [119]. Presenilin-1 mutations can adversely affect neurons by increasing the production of Aβ 42 which then perturbs neuronal Ca2+ regulation as described in section 4 above. However, considerable evidence suggests that AD-causing mutations in presenilin-1 may disrupt a different function of presenilin-1, a function in regulating endoplasmic reticulum (ER) Ca2+ homeostasis.
In 1996 and 1997 we reported the results of experiments in which we investigated different aspects of cellular Ca2+ homeostasis, and cellular vulnerability to various insults, in cultured neural cells expressing either mutant or wild-type presenilin-1 [120, 121]. We found that when challenged with agonists that release Ca2+ from IP3-sensitive ER stores, cells expressing mutant presenilin-1 release much more Ca2+ compared to cells expressing wild-type presenilin-1. Further analysis indicated that, for an unknown reason, the presenilin-1 mutations caused the ER to accumulate abnormally large amounts of Ca2+ [120,121]. To better understand the consequences of presenilin-1 mutations on neuronal Ca2+ regulation we generated and characterized presenilin-1 mutant (M146V mutation) knockin (PS1KI) mice. The mice exhibit no overt phenotypes and do not exhibit learning and memory deficits. However, neurons in the hippocampus and cerebral cortex of the PS1KI mice exhibit increased vulnerability to excitotoxic and ischemic injury [122, 123]. Enhanced Ca2+ release from the ER plays a pivotal role in the neuro-endangering actions of mutant presenilin-1 because drugs that block Ca2+ release through ryanodine receptors and overexpression of the Ca2+-binding protein calbindin protect neurons against the adverse effects of the presenilin-1 mutation [122,124,125]. Levels of ryanodine receptor are increased in neurons expressing mutant presenilin-1 [126] and, interestingly, though ER Ca2+ content is increased as the result of presenilin-1 mutations, capacitative Ca2+ entry is impaired [127].
Synaptic Ca2+ dynamics are perturbed by presenilin-1 mutations. For example, synapses of presenilin-1 mutant mice exhibit enhanced elevations of cytoplasmic Ca2+ levels during exposure to depolarizing agents, Aβ and a mitochondrial toxin compared with synaptosomes from nontransgenic mice and mice overexpressing wild-type presenilin-1 [128]. Treatments that buffer cytoplasmic Ca2+ or that prevent Ca2+ release from the ER protected synapses against the adverse effect of presenilin-1 mutations on mitochondrial function. Interestingly, PS1KI mice exhibit enhanced long-term potentiation of synaptic transmission at hippocampal CA1 synapses [129]. In a recent study of 3xTgAD mice, it was found that young (presymptomatic) mice exhibit exaggerated ryanodine receptor-mediated Ca2+ release in synaptic areas in the CA1 region of the hippocampus which was apparently due to increased expression of the ryanodine receptor 2 isoform [130]. Therefore, presenilin-1 mutations may actually enhance synaptic Ca2+ responses early in the process of AD, while at the same time rendering neurons vulnerable to excessive Aβ production and excitotoxic damage. On the other hand, the perturbed Ca2+ regulation caused by presenilin-1 mutations may impair responses to neurotransmitters that normally employ Ca2+ as part of their signaling mechanism. Thus, activation of muscarinic receptors impairs LTP in hippocampal slices from PS1KI mice, whereas acetylcholine enhances LTP in slices from wild type mice [131]. The latter study also provided evidence for a reduction in NMDA currents in CA1 neurons of PS1KI mice, suggesting a potential mechanism whereby presenilin-1 mutations may impair synaptic plasticity.
The molecular basis of the perturbed ER Ca2+ handling caused by presenilin-1 mutations is not yet established, but recent findings suggest that at least some mutations result in the loss of a normal Ca2+ handling function of wild type presenilin-1. Tu et al. [132] reported that wild type presenilin-1 forms Ca2+ leak channels in the ER membrane, and that presenilin-1 mutations disrupt this proposed function of presenilin-1. The latter finding would seem to provide an explanation for the previous evidence that the ER Ca2+ pool is abnormally increased in neurons expressing mutant presenilin-1. Other studies have provided evidence that presenilin-1 interacts with IP3 receptor in the ER and that presenilin-1 mutations alter the gating activity of the receptor so as to enhance its opening and release of Ca2+ from the ER [133]. The results of electrophysiological recordings of IP3 receptor currents in lymphoblasts derived from people with AD-causing presenilin-1 mutations or cortical neurons from presenilin-1 mutant mice demonstrated that the presenilin-1 mutations increase the time that the IP3 receptor channels were in an open Ca2+ burst mode [134].
7. Involvement of Mitochondrial Disturbances in Aberrant Neuronal Ca2+ Handling in AD
As described in Section 2, there is considerable evidence that neurons suffer from an energy deficit for some time period before they become dysfunctional and die in AD. Several alterations in mitochondria have been documented in studies of: 1) postmortem brain tissue samples from AD patients; 2) ‘cybrid’ cell lines derived by fusing fibroblasts from AD patients with tumor cells; and 3) experimental cell culture and animal models of AD (see [135, 136] for review). Activity levels of several enzymes in the mitochondrial tricarboxylic acid (TCA) cycle are reduced in brain tissue samples from AD patients compared to samples from age-matched control subjects including α-ketoglutarate dehydrogenase complex, pyruvate dehydrogenase complex, isocitrate dehydrogenase [137]. Interestingly, levels of two other TCA cycle enzymes, malate dehydrogenase and succinate dehydrogenase, were increased, perhaps as an attempt at an adaptive response. Studies of cybrid cells provides evidence for multiple structural and functional alterations in mitochondria from AD patients [138]. In another study, AD cybrids exhibited a major decrease in mitochondrial complex IV activity and increased oxyradical production, without a change in complex I activity [139]. Moreover, basal cytosolic Ca2+ concentration was elevated in AD cybrid cells, and the cells with AD mitochondria also restored Ca2+ levels more slowly when challenged with carbachol, a muscarinic receptor agonist that induces Ca2+ release from IP3-sensitive ER stores.
Alterations in mitochondria in AD may occur, in part, as an indirect result of the toxic actions of Aβ at the plasma membrane. Multiple studies have documented alterations in the mitochondria of neurons exposed to Aβ 40 or Aβ 42 including membrane depolarization, accumulation of Ca2+, superoxide production, reduced ATP production, increased fission, opening of membrane permeability transition pores and triggering of apoptosis [140–142]. Mitochondrial superoxide production is believed to play an important role in the neurotoxic actions of Aβ because exposure of neurons to Aβ results in increased mitochondrial oxyradical production, and mitochondrial superoxide dismutase (SOD2) protects neurons from being damaged and killed by Aβ [143]. The cell death-promoting effects of presenilin-1 mutations also involve increased mitochondrial superoxide production [144] and associated aberrant elevations of intracellular Ca2+ levels [128]. Direct actions of Aβ on mitochondria have also been reported. In a recent study the authors found that mitochondria lacking cyclophilin D, a key protein of the mitochondrial permeability transition pore, are resistant to several adverse effects of Aβ including mitochondrial swelling, calcium accumulation and oxyradical production [145]. Cyclophilin D-deficient cells are resistant to being killed by Aβ . Interestingly, a recent study has linked transient openings of permeability transition pores to bursts of superoxide production [146]. Perhaps mitochondria in neurons subjected to the (oxidative, metabolic and Ca2+-related) stresses of aging and Aβ accumulation become unable to control permeability pore opening resulting in uncontrolled superoxide production, further disruption of Ca2+ homeostasis and cell degeneration.
8. Conclusions and Implications for Novel Approaches for the Prevention and Treatment of AD
There are multiple molecular and cellular changes that occur in the brain during normal aging, and perhaps changes specific for AD as well, that tend to destabilize Ca2+-handling systems in neurons. Increased oxyradical production and accumulation of oxidative damage to proteins, lipids and DNA, and impaired mitochondrial function, are among such major age-related alterations. Excessive oxidative stress, particularly membrane lipid peroxidation, impairs the function of synaptic ion-motive ATPases, glucose transporters and glutamate transporters which may predispose neurons to Ca2+ overload, synaptic failure and degeneration of axons and dendrites [23, 36]. Similarly, compromised mitochondrial function may occur in neurons during aging resulting in reduced levels of cellular ATP and NAD+, both of which are critical for the maintenance of neuronal Ca2+ homeostasis [136]. Increased production and self-aggregation of Aβ 42, resulting from aging, genetic factors and/or environmental factors can have particularly abrasive effects on Ca2+ handling systems in neurons and in some individuals Aβ 42 may be the pivotal factor that accelerates synaptic dysfunction and neuronal death. Studies of presenilin-1 mutations that cause early-onset AD have kindled an interest in ER Ca2+ regulation in AD pathogenesis, implicating an aberrantly large accumulation of Ca2+ in the ER in the disease process [147].
Cholinesterase inhibitors that can provide temporary improvement in cognitive function are the most widely prescribed drug for AD patients. Currently, the major focus of drug development programs throughout the pharmaceutical industry and academia is on preventing Aβ production by targeting γ- and β-secretases, or enhancing Aβ clearance using immunotherapy approaches [8, 148]. Unfortunately, however, a recent phase III clinical trial of a γ-secretase inhibitor not only did not have a beneficial effect, but instead accelerated the cognitive decline in the AD patients [149]. Aβ immunotherapy trials have also proven disappointing with severe side effects in many AD patients undergoing active immunization [150], and in a recent passive immunization trial in which a monoclonal Aβ antibody was administered to AD patients there was no significant effect of the Aβ antibody in the primary efficacy analysis [151].
The only drug that has been shown to slow the progression of AD targets neuronal Ca2+ homeostasis. Memantine blocks Ca2+ influx through NMDA-responsive glutamate receptor channels, but only when the membrane is depolarized and the channel is open [152]. A controlled clinical trial of memantine in patients with moderate to severe AD demonstrated its efficacy in retarding disease progression [153]. The results of preclinical studies suggest the potential benefit of drugs that target other Ca2+-regulating systems. For example, it was recently reported that long-term treatment with diazoxide can ameliorate learning and memory deficits and Aβ and tau pathologies in the 3xTgAD mouse model [154]. Diazoxide opens mitochondrial ATP-sensitive K+ channels at low concentrations and plasma membrane K+ channels at somewhat higher concentrations. Via these two-site actions diazoxide reduces neuronal excitability, decreases mitochondrial oxyradical production and protects neurons against Ca2+ overload. Another approach is to stimulate the production of endogenous neurotrophic factors (or administer exogenous factors), such as BDNF, NGF and fibroblast growth factor which activate signaling pathways that modulate the expression of a range of proteins involved in cellular Ca2+ regulation [155 – 158].
Finally, emerging evidence suggests that manipulations that induce adaptive stress responses in brain cells may protect against cognitive impairment in aging and AD. For example, exercise and dietary energy restriction have been shown to stimulate the production in brain cells of several proteins known to play important roles in cellular responses to stress including BDNF, protein chaperones, antioxidant enzymes and mitochondrial uncoupling proteins [159–161]. Several transcription factors known to be responsive to oxidative and metabolic stress are activated in brain cells in response to exercise and/or dietary energy restriction including CREB. It may be possible to mimic the effects of exercise and dietary energy restriction on neuroplasticity and resistance to disease using pharmacological agents [161]. Calcium likely plays roles in mediating some of the beneficial actions of exercise and dietary energy restriction on brain cells. We suggest that increased synaptic activity is involved in the upregulation of BDNF that occurs in response to exercise and dietary energy restriction. Some drugs that have been shown to protect neurons against dysfunction and degeneration in animal models of AD may also act by a hormesis (adaptive stress response) mechanism. For example, the mitochondrial K+ channel opener diazoxide is known to cause energetic stress [162], and an adaptive response to such stress may explain its beneficial effects in AD mice [154]. Targeting cellular Ca2+ handling systems, either directly or indirectly through adaptive stress response mechanisms, may prove effective in preventing neuronal degeneration and sustaining neuronal function during aging and in AD.
Figure 1.
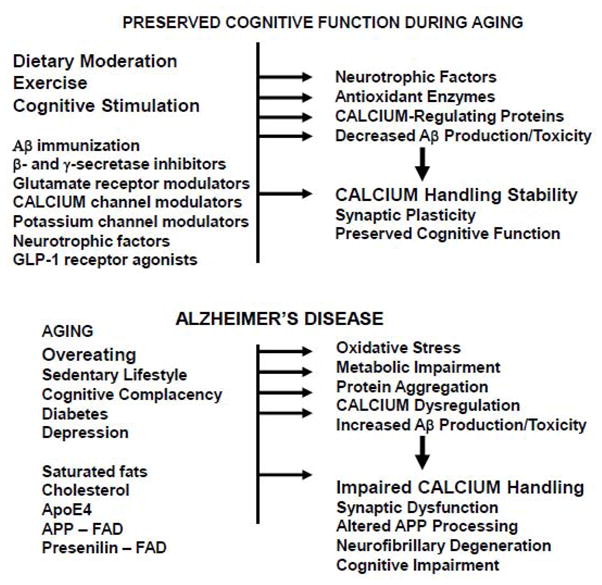
Factors and mechanisms that may preserve cognitive function during aging, or that may promote the development of Alzheimer’s disease. A. Increasing evidence suggests that moderation in dietary energy intake, regular exercise and a cognitively challenging lifestyle can promote maintenance of cognitive function during aging. Pharmacological interventions that target one or more regulatory systems, such as those listed, are being tested in translational research studies. These lifestyle and pharmacological agents may act by inducing the expression of neurotrophic factors, antioxidants, calcium-regulating proteins, and/or by modifying Aβ production and clearance. In these ways, neuronal calcium regulation is maintained resulting in the preservation of synaptic plasticity and cognitive function. B. Factors that may lead to cognitive impairment and AD include excessive calorie intake, a sedentary and cognitively impoverished lifestyle, diabetes and depression. Diets high in saturated fats and cholesterol, and genetic factors (ApoE-e4 genotype), may increase the risk for late-onset AD. Mutations in APP or presenilin-1 cause early-onset AD, and the disease process in such individuals may be particularly difficult to modify. AD develops when levels of oxidative stress, cellular energy deficits, dysregulation of calcium homeostasis and Aβ accumulation become excessive. Evidence described in the text suggest that perturbed neuronal (synaptic) calcium handling plays a pivotal role in the synaptic dysfunction, neuronal degeneration and cell death that underlies cognitive impairment in AD. Modified from reference 30.
Acknowledgments
This article was written as an original review/opinion article and was not intended to be a comprehensive review. We therefore acknowledge those scientists who have contributed to this field of investigation, many of which are cited in other recent review articles (see review articles cited below). This work was supported by the Intramural Research Program of the National Institute on Aging, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jack CR, Jr, Shiung MM, Weigand SD, O’Brien PC, Gunter JL, Boeve BF, Knopman DS, Smith GE, Ivnik RJ, Tangalos EG, Petersen RC. Brain atrophy rates predict subsequent clinical conversion in normal elderly and amnestic MCI. Neurology. 2005;65:1227–1231. doi: 10.1212/01.wnl.0000180958.22678.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McDonald CR, McEvoy LK, Gharapetian L, Fennema-Notestine C, Hagler DJ, Jr, Holland D, Koyama A, Brewer JB, Dale AM. Alzheimer’s Disease Neuroimaging Initiative, Regional rates of neocortical atrophy from normal aging to early Alzheimer disease. Neurology. 2009;73:457–465. doi: 10.1212/WNL.0b013e3181b16431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mosconi L, Mistur R, Switalski R, Tsui WH, Glodzik L, Li Y, Pirraglia E, De Santi S, Reisberg B, Wisniewski T, de Leon MJ. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2009;36:811–822. doi: 10.1007/s00259-008-1039-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mattson MP. Acetylation unleashes protein demons of dementia. Neuron. 2010;67:900–902. doi: 10.1016/j.neuron.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 5.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 6.Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashe KH, Zahs KR. Probing the biology of Alzheimer’s disease in mice. Neuron. 2010;66:631–645. doi: 10.1016/j.neuron.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Citron M. Alzheimer’s disease: strategies for disease modification. Nat Rev Drug Discov. 2010;9:387–398. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- 9.Buxbaum JD, Thinakaran G, Koliatsos V, O’Callahan J, Slunt HH, Price DL, Sisodia S. Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci. 1998;18:9629–9637. doi: 10.1523/JNEUROSCI.18-23-09629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lazarov O, Lee M, Peterson DA, Sisodia SS. Evidence that synaptically released beta-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci. 2002;22:9785–9793. doi: 10.1523/JNEUROSCI.22-22-09785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL. Soluble oligomers of beta amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924:133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- 12.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 13.Keller JN, Pang Z, Geddes JW, Begley JG, Germeyer A, Waeg G, Mattson MP. Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid beta-peptide: role of the lipid peroxidation product 4-hydroxynonenal. J Neurochem. 1997;69:273–284. doi: 10.1046/j.1471-4159.1997.69010273.x. [DOI] [PubMed] [Google Scholar]
- 14.Mattson MP, Partin J, Begley JG. Amyloid beta-peptide induces apoptosis-related events in synapses and dendrites. Brain Res. 1998;807:167–176. doi: 10.1016/s0006-8993(98)00763-x. [DOI] [PubMed] [Google Scholar]
- 15.Spires-Jones TL, Meyer-Luehmann M, Osetek JD, Jones PB, Stern EA, Bacskai BJ, Hyman BT. Impaired spine stability underlies plaque-related spine loss in an Alzheimer’s disease mouse model. Am J Pathol. 2007;171:1304–1311. doi: 10.2353/ajpath.2007.070055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pigino G, Morfini G, Pelsman A, Mattson MP, Brady ST, Busciglio J. Alzheimer’s presenilin 1 mutations impair kinesin-based axonal transport. J Neurosci. 2003;23:4499–4508. doi: 10.1523/JNEUROSCI.23-11-04499.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pigino G, Morfini G, Atagi Y, Deshpande A, Yu C, Jungbauer L, LaDu M, Busciglio J, Brady S. Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc Natl Acad Sci U S A. 2009;106:5907–5912. doi: 10.1073/pnas.0901229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Floyd RA, Carney JM. Free radical damage to protein and DNA: mechanisms involved and relevant observations on brain undergoing oxidative stress. Ann Neurol. 1992;32:S22–S27. doi: 10.1002/ana.410320706. [DOI] [PubMed] [Google Scholar]
- 19.Mecocci P, MacGarvey U, Kaufman AE, Koontz D, Shoffner JM, Wallace DC, Beal MF. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann Neurol. 1993;34:609–616. doi: 10.1002/ana.410340416. [DOI] [PubMed] [Google Scholar]
- 20.Bruce-Keller AJ, Li YJ, Lovell MA, Kraemer PJ, Gary DS, Brown RR, Markesbery WR, Mattson MP. 4-Hydroxynonenal, a product of lipid peroxidation, damages cholinergic neurons and impairs visuospatial memory in rats. J Neuropathol Exp Neurol. 1998a;57:257–267. doi: 10.1097/00005072-199803000-00007. [DOI] [PubMed] [Google Scholar]
- 21.Lovell MA, Ehmann WD, Mattson MP, Markesbery WR. Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer’s disease. Neurobiol Aging. 1997;18:457–461. doi: 10.1016/s0197-4580(97)00108-5. [DOI] [PubMed] [Google Scholar]
- 22.Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 23.Mattson MP. Metal-catalyzed disruption of membrane protein and lipid signaling in the pathogenesis of neurodegenerative disorders. Ann N Y Acad Sci. 2004a;1012:37–50. doi: 10.1196/annals.1306.004. [DOI] [PubMed] [Google Scholar]
- 24.Weissman L, Jo DG, Sørensen MM, de Souza-Pinto NC, Markesbery WR, Mattson MP, Bohr VA. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007;35:5545–5555. doi: 10.1093/nar/gkm605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 26.Gleichmann M, Zhang Y, Wood WH, Becker KG, Mughal MR, Pazin MJ, van Praag H, Kobilo T, Zonderman AB, Troncoso JC, Markesbery WR, Mattson MP. Molecular changes in brain aging and Alzheimer’s disease are mirrored in experimentally silenced cortical neuron networks. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2010.08.012. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdul HM, Sultana R, St Clair DK, Markesbery WR, Butterfield DA. Oxidative damage in brain from human mutant APP/PS-1 double knock-in mice as a function of age. Free Radic Biol Med. 2008;45:1420–1425. doi: 10.1016/j.freeradbiomed.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li F, Calingasan NY, Yu F, Mauck WM, Toidze M, Almeida CG, Takahashi RH, Carlson GA, Beal MF, Lin MT, Gouras GK. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J Neurochem. 2004;89:1308–1312. doi: 10.1111/j.1471-4159.2004.02455.x. [DOI] [PubMed] [Google Scholar]
- 29.Jo DG, Arumugam TV, Woo HN, Park JS, Tang SC, Mughal M, Hyun DH, Park JH, Choi YH, Gwon AR, Camandola S, Cheng A, Cai H, Song W, Markesbery WR, Mattson MP. Evidence that gamma-secretase mediates oxidative stress-induced beta-secretase expression in Alzheimer’s disease. Neurobiol Aging. 2010;31:917–925. doi: 10.1016/j.neurobiolaging.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang B, Duan BY, Zhou XP, Gong JX, Luo ZG. Calpain activation promotes BACE1 expression, amyloid precursor protein processing, and amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J Biol Chem. 2010;285:27737–27744. doi: 10.1074/jbc.M110.117960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jagust W, Gitcho A, Sun F, Kuczynski B, Mungas D, Haan M. Brain imaging evidence of preclinical Alzheimer’s disease in normal aging. Ann Neurol. 2006;59:673–681. doi: 10.1002/ana.20799. [DOI] [PubMed] [Google Scholar]
- 33.Mosconi L, De Santi S, Li J, Tsui WH, Li Y, Boppana M, Laska E, Rusinek H, de Leon MJ. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol Aging. 2008;29:676–692. doi: 10.1016/j.neurobiolaging.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toescu EC, Myronova N, Verkhratsky A. Age-related structural and functional changes of brain mitochondria. Cell Calcium. 2000;28:329–338. doi: 10.1054/ceca.2000.0167. [DOI] [PubMed] [Google Scholar]
- 35.Beal MF. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann Neurol. 1992;31:119–130. doi: 10.1002/ana.410310202. [DOI] [PubMed] [Google Scholar]
- 36.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004b;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 38.Bardo S, Cavazzini MG, Emptage N. The role of the endoplasmic reticulum Ca2+ store in the plasticity of central neurons. Trends Pharmacol Sci. 2006;27:78–84. doi: 10.1016/j.tips.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 39.Catterall WA, Few AP. Calcium channel regulation and presynaptic plasticity. Neuron. 2008;59:882–901. doi: 10.1016/j.neuron.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 40.Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- 41.McKinney RA. Excitatory amino acid involvement in dendritic spine formation, maintenance and remodelling. J Physiol. 2010;588:107–116. doi: 10.1113/jphysiol.2009.178905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dumas TC, Powers EC, Tarapore PE, Sapolsky RM. Overexpression of calbindin D(28k) in dentate gyrus granule cells alters mossy fiber presynaptic function and impairs hippocampal-dependent memory. Hippocampus. 2004;14:701–709. doi: 10.1002/hipo.10210. [DOI] [PubMed] [Google Scholar]
- 43.Gleichmann M, Mattson M. Neuronal calcium homeostasis and dysregulation. Antioxid Redox Signal. 2010 doi: 10.1089/ars.2010.3386. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mark RJ, Pang Z, Geddes JW, Uchida K, Mattson MP. Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. J Neurosci. 1997a;17:1046–1054. doi: 10.1523/JNEUROSCI.17-03-01046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mark RJ, Keller JN, Kruman I, Mattson MP. Basic FGF attenuates amyloid beta-peptide-induced oxidative stress, mitochondrial dysfunction, and impairment of Na+/K+-ATPase activity in hippocampal neurons. Brain Res. 1997b;756:205–214. doi: 10.1016/s0006-8993(97)00196-0. [DOI] [PubMed] [Google Scholar]
- 46.Lu C, Chan SL, Fu W, Mattson MP. The lipid peroxidation product 4-hydroxynonenal facilitates opening of voltage-dependent Ca2+ channels in neurons by increasing protein tyrosine phosphorylation. J Biol Chem. 2002;277:24368–24375. doi: 10.1074/jbc.M201924200. [DOI] [PubMed] [Google Scholar]
- 47.Blanc EM, Keller JN, Fernandez S, Mattson MP. 4-hydroxynonenal, a lipid peroxidation product, impairs glutamate transport in cortical astrocytes. Glia. 1998;22:149–160. doi: 10.1002/(sici)1098-1136(199802)22:2<149::aid-glia6>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 48.Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium. 2003;34:385–397. doi: 10.1016/s0143-4160(03)00128-3. [DOI] [PubMed] [Google Scholar]
- 49.Liu D, Pitta M, Mattson MP. Preventing NAD(+) depletion protects neurons against excitotoxicity: bioenergetic effects of mild mitochondrial uncoupling and caloric restriction. Ann N Y Acad Sci. 2008;1147:275–282. doi: 10.1196/annals.1427.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nicholls DG. Oxidative stress and energy crises in neuronal dysfunction. Ann N Y Acad Sci. 2008;1147:53–60. doi: 10.1196/annals.1427.002. [DOI] [PubMed] [Google Scholar]
- 51.Liu D, Gharavi R, Pitta M, Gleichmann M, Mattson MP. Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neuromolecular Med. 2009;11:28–42. doi: 10.1007/s12017-009-8058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Geda YE, Roberts RO, Knopman DS, Christianson TJ, Pankratz VS, Ivnik RJ, Boeve BF, Tangalos EG, Petersen RC, Rocca WA. Physical exercise, aging, and mild cognitive impairment: a population-based study. Arch Neurol. 2010;67:80–86. doi: 10.1001/archneurol.2009.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baker LD, Frank LL, Foster-Schubert K, Green PS, Wilkinson CW, McTiernan A, Plymate SR, Fishel MA, Watson GS, Cholerton BA, Duncan GE, Mehta PD, Craft S. Effects of aerobic exercise on mild cognitive impairment: a controlled trial. Arch Neurol. 2010;67:71–79. doi: 10.1001/archneurol.2009.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luchsinger JA, Tang MX, Shea S, Mayeux R. Caloric intake and the risk of Alzheimer disease. Arch Neurol. 2002;59:1258–1263. doi: 10.1001/archneur.59.8.1258. [DOI] [PubMed] [Google Scholar]
- 55.Witte AV, Fobker M, Gellner R, Knecht S. A. Flöel, Caloric restriction improves memory in elderly humans. Proc Natl Acad Sci U S A. 2009;106:1255–1260. doi: 10.1073/pnas.0808587106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Craft S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol. 2009;66:300–305. doi: 10.1001/archneurol.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ahtiluoto S, Polvikoski T, Peltonen M, Solomon A, Tuomilehto J, Winblad B, Sulkava R, Kivipelto M. Diabetes, Alzheimer disease, and vascular dementia. A population-based neuropathologic study. Neurology. 2010 doi: 10.1212/WNL.0b013e3181f4d7f8. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 58.Adlard PA, Perreau VM, Pop V, Cotman CW. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25:4217–4221. doi: 10.1523/JNEUROSCI.0496-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nichol KE, Parachikova AI, Cotman CW. Three weeks of running wheel exposure improves cognitive performance in the aged Tg2576 mouse. Behav Brain Res. 2007;184:124–132. doi: 10.1016/j.bbr.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Halagappa VK, Guo Z, Pearson M, Matsuoka Y, Cutler RG, Laferla FM, Mattson MP. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple–transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. 2007;26:212–220. doi: 10.1016/j.nbd.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 61.Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci. 2008;11:309–317. doi: 10.1038/nn2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Neeper SA, Gómez-Pinilla F, Choi J, Cotman CW. Physical activity increases mRNA for brain-derived neurotrophic factor and nerve growth factor in rat brain. Brain Res. 1996;726:49–56. [PubMed] [Google Scholar]
- 63.Lee J, Duan W, Mattson MP. Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J Neurochem. 2002;82:1367–1375. doi: 10.1046/j.1471-4159.2002.01085.x. [DOI] [PubMed] [Google Scholar]
- 64.Stranahan AM, Lee K, Martin B, Maudsley S, Golden E, Cutler RG, Mattson MP. Voluntary exercise and caloric restriction enhance hippocampal dendritic spine density and BDNF levels in diabetic mice. Hippocampus. 2009;19:951–961. doi: 10.1002/hipo.20577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann N Y Acad Sci. 2008;1144:97–112. doi: 10.1196/annals.1418.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gottmann K, Mittmann T, Lessmann V. BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Exp Brain Res. 2009;199:203–234. doi: 10.1007/s00221-009-1994-z. [DOI] [PubMed] [Google Scholar]
- 67.Bruce-Keller AJ, Umberger G, McFall R, Mattson MP. Food restriction reduces brain damage and improves behavioral outcome following excitotoxic and metabolic insults. Ann Neurol. 1999;45:8–15. [PubMed] [Google Scholar]
- 68.Chen YW, Chen SH, Chou W, Lo YM, Hung CH, Lin MT. Exercise pretraining protects against cerebral ischaemia induced by heat stroke in rats. Br J Sports Med. 2007;41:597–602. doi: 10.1136/bjsm.2006.033829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arumugam TV, Phillips TM, Cheng A, Morrell CH, Mattson MP, Wan R. Ann Neurol. 2010;67:41–52. doi: 10.1002/ana.21798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron. 1991;7:695–702. doi: 10.1016/0896-6273(91)90273-3. [DOI] [PubMed] [Google Scholar]
- 71.Li G, Peskind ER, Millard SP, Chi P, Sokal I, Yu CE, Bekris LM, Raskind MA, Galasko DR, Montine TJ. Cerebrospinal fluid concentration of brain-derived neurotrophic factor and cognitive function in non-demented subjects. PLoS One. 2009;4:e5424. doi: 10.1371/journal.pone.0005424. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peng S, Garzon DJ, Marchese M, Klein W, Ginsberg SD, Francis BM, Mount HT, Mufson EJ, Salehi A, Fahnestock M. Decreased brain-derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer’s disease. J Neurosci. 2009;29:9321–9329. doi: 10.1523/JNEUROSCI.4736-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Poon WW, Blurton-Jones M, Tu CH, Feinberg LM, Chabrier MA, Harris JW, Jeon NL, Cotman CW. beta-Amyloid impairs axonal BDNF retrograde trafficking. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.05.012. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mattson MP, Murrain M, Guthrie PB, Kater SB. Fibroblast growth factor and glutamate: opposing roles in the generation and degeneration of hippocampal neuroarchitecture. J Neurosci. 1989;9:3728–3740. doi: 10.1523/JNEUROSCI.09-11-03728.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cheng B, Mattson MP. NGF and bFGF protect rat hippocampal and human cortical neurons against hypoglycemic damage by stabilizing calcium homeostasis. Neuron. 1991;7:1031–1041. doi: 10.1016/0896-6273(91)90347-3. [DOI] [PubMed] [Google Scholar]
- 76.Mattson MP, Tomaselli KJ, Rydel RE. Calcium-destabilizing and neurodegenerative effects of aggregated beta-amyloid peptide are attenuated by basic FGF. Brain Res. 1993;621:35–49. doi: 10.1016/0006-8993(93)90295-x. [DOI] [PubMed] [Google Scholar]
- 77.Cheng B, Mattson MP. IGF-I and IGF-II protect cultured hippocampal and septal neurons against calcium-mediated hypoglycemic damage. J Neurosci. 1992;12:1558–1566. doi: 10.1523/JNEUROSCI.12-04-01558.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: link to brain reductions in acetylcholine. J Alzheimers Dis. 2005;8:247–268. doi: 10.3233/jad-2005-8304. [DOI] [PubMed] [Google Scholar]
- 79.Doré S, Kar S, Quirion R. Insulin-like growth factor I protects and rescues hippocampal neurons against beta-amyloid- and human amylin-induced toxicity. Proc Natl Acad Sci U S A. 1997;94:4772–4777. doi: 10.1073/pnas.94.9.4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim W, Egan JM. The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol Rev. 2008;60:470–512. doi: 10.1124/pr.108.000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li Y, Perry T, Kindy MS, Harvey BK, Tweedie D, Holloway HW, Powers K, Shen H, Egan JM, Sambamurti K, Brossi A, Lahiri DK, Mattson MP, Hoffer BJ, Wang Y, Greig NH. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci U S A. 2009b;106:1285–1290. doi: 10.1073/pnas.0806720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martin B, Golden E, Carlson OD, Pistell P, Zhou J, Kim W, Frank BP, Thomas S, Chadwick WA, Greig NH, Bates GP, Sathasivam K, Bernier M, Maudsley S, Mattson MP, Egan JM. Exendin-4 improves glycemic control, ameliorates brain and pancreatic pathologies, and extends survival in a mouse model of Huntington’s disease. Diabetes. 2009;58:318–328. doi: 10.2337/db08-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tardito D, Perez J, Tiraboschi E, Musazzi L, Racagni G, Popoli M. Signaling pathways regulating gene expression, neuroplasticity, and neurotrophic mechanisms in the action of antidepressants: a critical overview. Pharmacol Rev. 2006;58:115–134. doi: 10.1124/pr.58.1.7. [DOI] [PubMed] [Google Scholar]
- 84.Yang JL, Takahashi T, Keijzers G, Mattson MP, Bohr VA. Neurons efficiently repair glutamate-induced oxidative DNA damage by a process involving CREB-mediated Up-regulation of APE1. J Biol Chem. 2010;285:28191–28199. doi: 10.1074/jbc.M109.082883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Perry T, Haughey NJ, Mattson MP, Egan JM, Greig NH. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J Pharmacol Exp Ther. 2002;302:881–888. doi: 10.1124/jpet.102.037481. [DOI] [PubMed] [Google Scholar]
- 86.Perry T, Lahiri DK, Sambamurti K, Chen D, Mattson MP, Egan JM, Greig NH. Glucagon-like peptide-1 decreases endogenous amyloid-beta peptide (Abeta) levels and protects hippocampal neurons from death induced by Abeta and iron. J Neurosci Res. 2003;72:603–612. doi: 10.1002/jnr.10611. [DOI] [PubMed] [Google Scholar]
- 87.Gilman CP, Perry T, Furukawa K, Grieg NH, Egan JM, Mattson MP. Glucagon-like peptide 1 modulates calcium responses to glutamate and membrane depolarization in hippocampal neurons. J Neurochem. 2003;87:1137–1144. doi: 10.1046/j.1471-4159.2003.02073.x. [DOI] [PubMed] [Google Scholar]
- 88.Li Y, Duffy KB, Ottinger MA, Ray B, Bailey JA, Holloway HW, Tweedie D, Perry T, Mattson MP, Kapogiannis D, Sambamurti K, Lahiri DK, Greig NH. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J Alzheimers Dis. 2010;19:1205–1219. doi: 10.3233/JAD-2010-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gengler S, McClean PL, McCurtin R, Gault VA, Hölscher C. Val(8)GLP-1 rescues synaptic plasticity and reduces dense core plaques in APP/PS1 mice. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2010.02.014. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 90.Cecarini V, Ding Q. J.N. Keller Oxidative inactivation of the proteasome in Alzheimer’s disease. Free Radic Res. 2007;41:673–680. doi: 10.1080/10715760701286159. [DOI] [PubMed] [Google Scholar]
- 91.Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen X, Kintner DB, Baba A, Matsuda T, Shull GE, Sun D. Protein aggregation in neurons following OGD: a role for Na+ and Ca2+ ionic dysregulation. J Neurochem. 2010;112:173–182. doi: 10.1111/j.1471-4159.2009.06438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Green DR, Wang R. Calcium and energy: making the cake and eating it too? Cell. 2010;142:200–202. doi: 10.1016/j.cell.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 94.Li X, Yang D, Li L, Peng C, Chen S, Le W. Proteasome inhibitor lactacystin disturbs the intracellular calcium homeostasis of dopamine neurons in ventral mesencephalic cultures. Neurochem Int. 2007;50:959–965. doi: 10.1016/j.neuint.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 95.Wu S, Hyrc KL, Moulder KL, Lin Y, Warmke T, Snider BJ. Cellular calcium deficiency plays a role in neuronal death caused by proteasome inhibitors. J Neurochem. 2009;109:1225–1236. doi: 10.1111/j.1471-4159.2009.06037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hyun DH, Emerson SS, Jo DG, Mattson MP, de Cabo R. Calorie restriction up-regulates the plasma membrane redox system in brain cells and suppresses oxidative stress during aging. Proc Natl Acad Sci U S A. 2006;103:19908–19912. doi: 10.1073/pnas.0608008103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hyun DH, Hunt ND, Emerson SS, Hernandez JO, Mattson MP, de Cabo R. Up-regulation of plasma membrane-associated redox activities in neuronal cells lacking functional mitochondria. J Neurochem. 2007;10:1364–1374. doi: 10.1111/j.1471-4159.2006.04411.x. [DOI] [PubMed] [Google Scholar]
- 98.Hyun DH, Mughal MR, Yang H, Lee JH, Ko EJ, Hunt ND, de Cabo R, Mattson MP. The plasma membrane redox system is impaired by amyloid beta-peptide and in the hippocampus and cerebral cortex of 3xTgAD mice. Exp Neurol. 2010 doi: 10.1016/j.expneurol.2010.07.020. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–89. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem. 1997c;68:255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- 102.Bruce-Keller AJ, Begley JG, Fu W, Butterfield DA, Bredesen DE, Hutchins JB, Hensley K, Mattson MP. Bcl-2 protects isolated plasma and mitochondrial membranes against lipid peroxidation induced by hydrogen peroxide and amyloid beta-peptide. J Neurochem. 1998b;70:31–39. doi: 10.1046/j.1471-4159.1998.70010031.x. [DOI] [PubMed] [Google Scholar]
- 103.Guiotto A, Calderan A, Ruzza P, Osler A, Rubini C, Jo DG, Mattson MP, Borin G. Synthesis and evaluation of neuroprotective alpha,beta-unsaturated aldehyde scavenger histidyl-containing analogues of carnosine. J Med Chem. 2005;48:6156–6161. doi: 10.1021/jm050507q. [DOI] [PubMed] [Google Scholar]
- 104.Zhu YJ, Lin H, Lal R. Fresh and nonfibrillar amyloid beta protein (1–40) induces rapid cellular degeneration in aged human fibroblasts: evidence for AbetaP-channel-mediated cellular toxicity. FASEB J. 2000;14:1244–1254. doi: 10.1096/fasebj.14.9.1244. [DOI] [PubMed] [Google Scholar]
- 105.Iwamoto N, Thangnipon W, Crawford C, Emson PC. Localization of calpain immunoreactivity in senile plaques and in neurones undergoing neurofibrillary degeneration in Alzheimer’s disease. Brain Res. 1991;561:177–180. doi: 10.1016/0006-8993(91)90766-o. [DOI] [PubMed] [Google Scholar]
- 106.Nixon RA, Saito KI, Grynspan F, Griffin WR, Katayama S, Honda T, Mohan PS, Shea TB, Beermann M. Calcium-activated neutral proteinase (calpain) system in aging and Alzheimer’s disease. Ann N Y Acad Sci. 1994;747:77–91. doi: 10.1111/j.1749-6632.1994.tb44402.x. [DOI] [PubMed] [Google Scholar]
- 107.Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, Bacskai BJ, Hyman BT. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mattson MP. Antigenic changes similar to those seen in neurofibrillary tangles are elicited by glutamate and Ca2+ influx in cultured hippocampal neurons. Neuron. 1990;4:105–117. doi: 10.1016/0896-6273(90)90447-n. [DOI] [PubMed] [Google Scholar]
- 110.Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- 111.Nitsch RM, Farber SA, Growdon JH, Wurtman RJ. Release of amyloid beta-protein precursor derivatives by electrical depolarization of rat hippocampal slices. Proc Natl Acad Sci U S A. 1993;90:5191–5193. doi: 10.1073/pnas.90.11.5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 113.Furukawa K, Barger SW, Blalock EM, Mattson MP. Activation of K+ channels and suppression of neuronal activity by secreted beta-amyloid-precursor protein. Nature. 1996a;379:74–78. doi: 10.1038/379074a0. [DOI] [PubMed] [Google Scholar]
- 114.Furukawa K, Sopher BL, Rydel RE, Begley JG, Pham DG, Martin GM, Fox M, Mattson MP. Increased activity-regulating and neuroprotective efficacy of alpha-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J Neurochem. 1996b;67:1882–1896. doi: 10.1046/j.1471-4159.1996.67051882.x. [DOI] [PubMed] [Google Scholar]
- 115.Barger SW, Mattson MP. The secreted form of the Alzheimer’s beta-amyloid precursor protein stimulates a membrane-associated guanylate cyclase. Biochem J. 1995;311:45–47. doi: 10.1042/bj3110045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mattson MP, Guo ZH, Geiger JD. Secreted form of amyloid precursor protein enhances basal glucose and glutamate transport and protects against oxidative impairment of glucose and glutamate transport in synaptosomes by a cyclic GMP-mediated mechanism. J Neurochem. 1999;73:532–537. doi: 10.1046/j.1471-4159.1999.0730532.x. [DOI] [PubMed] [Google Scholar]
- 117.Guo Q, Robinson N, Mattson MP. Secreted beta-amyloid precursor protein counteracts the proapoptotic action of mutant presenilin-1 by activation of NF-kappaB and stabilization of calcium homeostasis. J Biol Chem. 1998a;273:12341–12351. doi: 10.1074/jbc.273.20.12341. [DOI] [PubMed] [Google Scholar]
- 118.Cruts M, Van Broeckhoven C. Presenilin mutations in Alzheimer’s disease. Hum Mutat. 1998;11:183–190. doi: 10.1002/(SICI)1098-1004(1998)11:3<183::AID-HUMU1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 119.Sisodia SS, Annaert W, Kim SH, De Strooper B. Gamma-secretase: never more enigmatic. Trends Neurosci. 2001;24:S2–S6. doi: 10.1016/s0166-2236(00)01987-1. [DOI] [PubMed] [Google Scholar]
- 120.Guo Q, Furukawa K, Sopher BL, Pham DG, Xie J, Robinson N, Martin GM, Mattson MP. Alzheimer’s PS-1 mutation perturbs calcium homeostasis and sensitizes PC12 cells to death induced by amyloid beta-peptide. Neuroreport. 1996;8:379–383. doi: 10.1097/00001756-199612200-00074. [DOI] [PubMed] [Google Scholar]
- 121.Guo Q, Sopher BL, Furukawa K, Pham DG, Robinson N, Martin GM, Mattson MP. Alzheimer’s presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid beta-peptide: involvement of calcium and oxyradicals. J Neurosci. 1997;17:4212–4222. doi: 10.1523/JNEUROSCI.17-11-04212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999a;5:101–106. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- 123.Mattson MP, Zhu H, Yu J, Kindy MS. Presenilin-1 mutation increases neuronal vulnerability to focal ischemia in vivo and to hypoxia and glucose deprivation in cell culture: involvement of perturbed calcium homeostasis. J Neurosci. 2000;20:1358–1364. doi: 10.1523/JNEUROSCI.20-04-01358.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Guo Q, Christakos S, Robinson N, Mattson MP. Calbindin D28k blocks the proapoptotic actions of mutant presenilin 1: reduced oxidative stress and preserved mitochondrial function. Proc Natl Acad Sci U S A. 1998b;95:3227–3232. doi: 10.1073/pnas.95.6.3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Keller JN, Guo Q, Holtsberg FW, Bruce-Keller AJ, Mattson MP. Increased sensitivity to mitochondrial toxin-induced apoptosis in neural cells expressing mutant presenilin-1 is linked to perturbed calcium homeostasis and enhanced oxyradical production. J Neurosci. 1998b;18:4439–4450. doi: 10.1523/JNEUROSCI.18-12-04439.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chan SL, Mayne M, Holden CP, Geiger JD, Mattson MP. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J Biol Chem. 2000;275:18195–18200. doi: 10.1074/jbc.M000040200. [DOI] [PubMed] [Google Scholar]
- 127.Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J Cell Biol. 2000;149:793–798. doi: 10.1083/jcb.149.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Begley JG, Duan W, Chan S, Duff K, Mattson MP. Altered calcium homeostasis and mitochondrial dysfunction in cortical synaptic compartments of presenilin-1 mutant mice. J Neurochem. 1999;72:1030–1039. doi: 10.1046/j.1471-4159.1999.0721030.x. [DOI] [PubMed] [Google Scholar]
- 129.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 130.Chakroborty S, Goussakov I, Miller MB, Stutzmann GE. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci. 2009;29:9458–9470. doi: 10.1523/JNEUROSCI.2047-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang Y, Greig NH, Yu QS, Mattson MP. Presenilin-1 mutation impairs cholinergic modulation of synaptic plasticity and suppresses NMDA currents in hippocampus slices. Neurobiol Aging. 2009;30:1061–1068. doi: 10.1016/j.neurobiolaging.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cheung KH, Shineman D, üller MM, Cárdenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008;58:871–883. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cheung KH, Mei L, Mak DO, Hayashi I, Iwatsubo T, Kang DE, Foskett JK. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci Signal. 2010;3:ra22. doi: 10.1126/scisignal.2000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. Biochim Biophys Acta. 1998;1366:211–223. doi: 10.1016/s0005-2728(98)00114-5. [DOI] [PubMed] [Google Scholar]
- 136.Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008;60:748–766. doi: 10.1016/j.neuron.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol. 2005;257:695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- 138.Trimmer PA, Keeney PM, Borland MK, Simon FA, Almeida J, Swerdlow RH, Parks JP, Parker WD, Jr, Bennett JP., Jr Mitochondrial abnormalities in cybrid cell models of sporadic Alzheimer’s disease worsen with passage in culture. Neurobiol Dis. 2004;15:29–39. doi: 10.1016/j.nbd.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 139.Sheehan JP, Swerdlow RH, Miller SW, Davis RE, Parks JK, Parker WD, Tuttle JB. Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer’s disease. J Neurosci. 1997;17:4612–4622. doi: 10.1523/JNEUROSCI.17-12-04612.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci U S A. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gibson GE, Karuppagounder SS, Shi Q. Oxidant-induced changes in mitochondria and calcium dynamics in the pathophysiology of Alzheimer’s disease. Ann N Y Acad Sci. 2008;1147:221–232. doi: 10.1196/annals.1427.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 2008a;105:19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Keller JN, Kindy MS, Holtsberg FW, St Clair DK, Yen HC, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J Neurosci. 1998c;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Guo Q, Fu W, Holtsberg FW, Steiner SM, Mattson MP. Superoxide mediates the cell-death-enhancing action of presenilin-1 mutations. J Neurosci Res. 1999b;56:457–470. doi: 10.1002/(SICI)1097-4547(19990601)56:5<457::AID-JNR2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 145.Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Wang W, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell. 2008b;134:279–290. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mattson MP. ER calcium and Alzheimer’s disease: in a state of flux. Sci Signal. 2010 Mar 23;3(114):pe10. doi: 10.1126/scisignal.3114pe10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Evin G, Sernee MF, Masters CL. Inhibition of gamma-secretase as a therapeutic intervention for Alzheimer’s disease: prospects, limitations and strategies. CNS Drugs. 2006;20:351–372. doi: 10.2165/00023210-200620050-00002. [DOI] [PubMed] [Google Scholar]
- 149.http://newsroom.lilly.com/releasedetail.cfm?releaseid=499794.
- 150.Kokjohn TA, Roher AE. Antibody responses, amyloid-beta peptide remnants and clinical effects of AN-1792 immunization in patients with AD in an interrupted trial. CNS Neurol Disord Drug Targets. 2009;8:88–97. doi: 10.2174/187152709787847315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, Sabbagh M, Honig LS, Doody R, van Dyck CH, Mulnard R, Barakos J, Gregg KM, Liu E, Lieberburg I, Schenk D, Black R, Grundman M. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73:2061–2070. doi: 10.1212/WNL.0b013e3181c67808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chen HS, Lipton SA. Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: uncompetitive antagonism. J Physiol. 1997;499:27–46. doi: 10.1113/jphysiol.1997.sp021909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ Memantine Study Group. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med. 2003;348:1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- 154.Liu D, Pitta M, Lee JH, Ray B, Lahiri D, Furukawa K, Mughal M, Jiang H, Villarreal J, Cutler RG, Greig NH, Mattson MP. The KATP channel activator diazoxide ameliorates Aβ and tau pathologies and improves memory in the 3xTgAD mouse model of Alzheimer’s disease. J Alzheimer’s Dis. 2010 doi: 10.3233/JAD-2010-101017. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Guo Q, Sebastian L, Sopher BL, Miller MW, Glazner GW, Ware CB, Martin GM, Mattson MP. Neurotrophic factors [activity-dependent neurotrophic factor (ADNF) and basic fibroblast growth factor (bFGF)] interrupt excitotoxic neurodegenerative cascades promoted by a PS1 mutation. Proc Natl Acad Sci U S A. 1999;96:4125–4130. doi: 10.1073/pnas.96.7.4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Massa SM, Yang T, Xie Y, Shi J, Bilgen M, Joyce JN, Nehama D, Rajadas J, Longo FM. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J Clin Invest. 2010;120:1774–1785. doi: 10.1172/JCI41356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Blurton-Jones M, Kitazawa M, Martinez-Coria H, Castello NA, Müller FJ, Loring JF, Yamasaki TR, Poon WW, Green KN, LaFerla FM. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A. 2009;106:13594–13599. doi: 10.1073/pnas.0901402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Williams BJ, Eriksdotter-Jonhagen M, Granholm AC. Nerve growth factor in treatment and pathogenesis of Alzheimer’s disease. Prog Neurobiol. 2006;80:114–128. doi: 10.1016/j.pneurobio.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 159.Mattson MP, Kapogiannis D, Greig NH. Tweaking energy metabolism to prevent and treat neurological disorders. Clin Pharmacol Ther. 2010;88:437–439. doi: 10.1038/clpt.2010.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mattson MP. Awareness of hormesis will enhance future research in basic and applied neuroscience. Crit Rev Toxicol. 2008;38:633–639. doi: 10.1080/10408440802026406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Stranahan AM, Zhou Y, Martin B, Maudsley S. Pharmacomimetics of exercise: novel approaches for hippocampally-targeted neuroprotective agents. Curr Med Chem. 2009;16:4668–4678. doi: 10.2174/092986709789878292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.O’Rourke B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circ Res. 2004;94:420–432. doi: 10.1161/01.RES.0000117583.66950.43. [DOI] [PMC free article] [PubMed] [Google Scholar]