

Abstract
Injury, infection and autoimmune triggers increase CNS expression of the chemokine CCL21. Outside the CNS, CCL21 contributes to chronic inflammatory disease and autoimmunity by three mechanisms: recruitment of lymphocytes into injured or infected tissues, organization of inflammatory infiltrates into lymphoid-like structures and promotion of homeostatic CD4+ T-cell proliferation. To test if CCL21 plays the same role in CNS inflammation, we generated transgenic mice with astrocyte-driven expression of CCL21 (GFAP-CCL21 mice). Astrocyte-produced CCL21 was bioavailable and sufficient to support homeostatic CD4+ T-cell proliferation in cervical lymph nodes even in the absence of endogenous CCL19/CCL21. However, lymphocytes and glial-activation were not detected in the brains of uninfected GFAP-CCL21 mice, although CCL21 levels in GFAP-CCL21 brains were higher than levels expressed in inflamed Toxoplasma-infected nontransgenic brains. Following Toxoplasma infection, T-cell extravasation into submeningeal, perivascular and ventricular sites of infected CNS was not CCL21-dependent, occurring even in CCL19/CCL21-deficient mice. However, migration of extravasated CD4+, but not CD8+ T cells from extra-parenchymal CNS sites into the CNS parenchyma was CCL21-dependent. CD4+ T cells preferentially accumulated at perivascular, submeningeal and ventricular spaces in infected CCL21/CCL19-deficient mice. By contrast, greater numbers of CD4+ T cells infiltrated the parenchyma of infected GFAP-CCL21 mice than in wild-type or CCL19/CCL21-deficient mice. Together these data indicate that CCL21 expression within the CNS has the potential to contribute to T cell-mediated CNS pathology via: (a) homeostatic priming of CD4+ T-lymphocytes outside the CNS and (b) by facilitating CD4+ T-cell migration into parenchymal sites following pathogenic insults to the CNS.
Keywords: autoimmunity, glia, lymphopenia, microglia, multiple sclerosis, neuroinflammation, neurodegeneration, T cell trafficking
Introduction
The CNS is an immune privileged site (Carson et al. 2006). Data gathered over the last two decades has clarified that CNS immune privilege is not equivalent to CNS immune isolation. Instead, CNS immune responses are regulated in a tissue-specific manner (Carson et al. 2006, Davis and Carson, 2009). Thus, a high threshold for initiating antigen-specific T cell responses from within the CNS does exist; yet, large numbers of T cells readily enter the CNS in response to a wide variety of insults.
T-cell influx associated with pro-inflammatory pathogen defense functions can be neurotoxic and disruptive of CNS function, as evidenced by the destructive effects of autoreactive T cells in experimental autoimmune encephalomyelitis (EAE) (Baxter 2007). However, in response to chronic infection by natural pathogens, the CNS can tolerate ongoing influx of activated pro-inflammatory T cells. For example, the obligate intracellular protozoan, Toxoplasma gondii (T. gondii) cannot be cleared from the CNS of infected individuals (Lambert and Barragan, 2010; Wilson et al. 2010). Instead, IFNγ producing T cells targeting T. gondii antigens are required to drive T. gondii from the cytotoxic tachyzoite stage of their life cycle to the latent cyst forming bradyzoite stage. Animal models and T cell-deficient individuals have demonstrated that continual influx of IFNγ producing T cells into the CNS is also required to prevent re-emergence of cytotoxic T. gondii tachyzoites from cysts. For example, decreased T-cell influx into the CNS due to AIDS or chemotherapy leads to reactivation of cysts and development of Toxoplasmic encephalitis characterized by cognitive and motor defects associated with necrotic brain lesions. Similarly, persistent CNS presentation of viral antigens to CD4+ and CD8+ T cells and production of IFNγ is critical to maintaining latency of neurotropic viruses and can occur without causing tissue damage, as in the case of persistent Herpes Virus infection (Frank et al. 2010; Lewandowski et al. 1993).
The presence of activated T cells within the CNS is also not rare in the human population. Considering just the example of T. gondii, the CDC estimates that ~30% of the world population (~20% in the US and ~40% in western Europe) is chronically infected by this parasite (Hofhuis et al. 2010; Jones and Dubey 2010). Thus ~30% of the world’s population has a lifelong influx of IFNγ-producing T cells in their CNS without overt signs of neurodegeneration. Even in the absence of CNS infection, T cells routinely enter and survey the healthy CNS. The apparent function is not only aimed at pathogen detection. T cells also play critical cytoprotective roles in maintaining neuronal function during injury and stress (Stalder et al. 1998; Byram et al. 2004; Derecki et al. 2010; Schwartz and Kipnis 2007; Schwartz and Cohen 2000). Thus, defining mechanisms regulating T cell influx is important both to prevent unwanted neurotoxic inflammation as well as to promote neuroprotective inflammatory responses. Multiple chemokines contribute to the regulated entry of T cells into the CNS (Bromley et al. 2008; Müller et al. 2010). In the current study, we examine the role of CCL21.
Why focus on the lymphoid chemokine CCL21? CCR7 is the high affinity receptor for CCL21 and CCL19 (Förster et al. 2008; Lalor and Segal 2010). In the absence of CCR7 expression, T. gondii infection cannot be controlled and leads to early lethality following infection (Noor et al. 2010). Restoration of CCR7 expression solely in T cells is sufficient to restore immune mediated control of the parasite and survival of the infected host. CCR7 expressing T cells are found within the CSF and lesions of individuals with active multiple sclerosis (reviewed in Förster et al. 2008; Lalor and Segal 2010). Moreover, constitutive and induced CNS expression of CCL21 is hypothesized to facilitate T cell entry from the CSF and blood into the CNS parenchyma (Wilson et al. 2009, Wilson et al. 2010). Ischemic injury, infection or experimental autoimmune triggers all rapidly induce increased CCL21 expression within the CNS roughly co-incident or just prior to the appearance of T cells within the CNS (Biber et al. 2002; Aloisi and Pujol-Borrell 2006; de Jong et al. 2005; Lalor and Segal 2010). Finally, CCL21 immunoreactivity has been observed in T. gondii infected mice associated with migratory pathways from the points of T cell influx into the CNS (perivascular and meningeal spaces) to brain regions surrounding T. gondii cysts (Wilson et al. 2009). Thus, CCL21 is a candidate molecule for regulating T cell influx into and within the CNS.
Outside the CNS, endogenous and transgenic CCL21 expression is sufficient to lead to accumulation and organization of large numbers of T cells into lymph node-like structures in most tissues (Fan et al. 2000; Aloisi and Pujol-Borrell 2006; Lalor and Segal 2010). In addition, increased CCL21 expression within tissues is sufficient to pre-activate auto-reactive T cells and trigger autoimmunity by promoting homeostatic proliferation (Fan et al. 2000; Ploix et al. 2001). Under T cell deficient (lymphopenic) conditions due to genetic, chemical, pathogen or other causes, naïve T cells proliferate in an antigen-independent manner in response to free CCL21 within lymph nodes. In non-lymphopenic mice there is no free CCL21, and homeostatic proliferation does not occur (Ploix et al. 2001, Li et al. 2007). Consequently, CD4+ T cells fail to undergo homeostatic proliferation in CCL21-deficient mice even under lymphopenic conditions; but do undergo homeostatic proliferation in the presence of transgenic pancreatic over-expression of CCL21 (Ins-CCL21 mice) even in the absence of lymphopenia. Furthermore, with each round of homeostatic proliferation, CD4+ T cells down regulate naïve T cell markers and increase expression of molecules associated with amplifying T cell receptor signaling (Ploix et al. 2001). Therefore, it is not surprising that lymphopenia and homeostatic proliferation are associated with onset of autoimmune disease in animal models and human patients (Marella et al. 2008, Milner et al. 2008, Data et al. 2009). Consistent with these observations, CCL21 expression proceeds or is co-incident with T cell influx in spontaneous models of autoimmunity (Förster et al. 2008; Aloisi and Pujol-Borrell 2006; Lalor and Segal 2010).
Here we sought to test whether CCL21 within the CNS was sufficient to recruit and organize lymphocytes in the healthy or infected CNS. We now report the generation of a viable transgenic murine model of astrocytic driven CCL21 expression (GFAP-CCL21 mice). GFAP-CCL21 mice developed normally without overt behavioral abnormalities. CNS transgenic expression of CCL21 did not cause lymphocyte or granulocyte influx into the healthy CNS. However, CNS expression of CCL21 did support homeostatic CD4+ T cell proliferation in T cells isolated from lymph nodes draining the CNS. Thus, CNS-produced CCL21 has the potential to contribute to priming T cell activation within cervical lymph nodes. Furthermore, CNS expression of CCL21 augmented the migration of CD4+ T cells from perivascular spaces into the CNS parenchyma of T. gondii infected mice. These data contribute to the growing literature demonstrating that the immune privileged CNS can regulate T cell-mediated immunity.
Materials and Methods
Generation of Mice and infection with Toxoplasma gondii (T. gondii)
GFAP-CCL21 transgenic mice were generated by standard methodologies (Fan et al. 2000). In brief, the previously reported CCL21 transgene was cloned into glial fibrillary acidic protein (GFAP) expression cassette (into the single BamHI transgene insertion site) (Fan et al 2000, Stalder et al. 1998). Transgenic offspring (GFAP-CCL21) were identified by PCR analysis of genomic DNA. GFAP-CCL21 mice were backcrossed onto B10.D2 and BALB/c genetic backgrounds for at least 10 generations prior to the experiments reported in this study. Ins-CCL21 mice were bred and characterized as previously reported for Ins-CCL21 transgenic mice. T. gondii strain Prugniaud was maintained in vitro in human foreskin fibroblasts (HFF) grown in Dulbecco’s modified Eagle’s medium (DMEM) complete (90% DMEM, 10% fetal bovine serum, 1% penicillin/streptomycin) as previously described (Noor et al. 2010). After infecting HFF, parasites were grown in D10 medium (70% DMEM, 20% medium 199, 10% fetal bovine serum, 5% penicillin/streptomycin, 5% gentamicin). The parasites were purified by passing them through a 22.5-gauge needle, followed by passage through a 5.0-um nylon filter, and were centrifuged at 3,000 rpm for 10 min at 4°C. After the supernatant was removed, the parasites were resuspended in 1 ml sterile phosphate-buffered saline (PBS) and counted. Ten thousand parasites were intraperitoneally (i.p.) injected in 200 ul of PBS into nontransgenic, plt/plt and GFAP-CCL21 mice. For all studies involving T. gondii infections, BALB/c was the genetic background for all mice used. All experiments in this study involving mice were performed in compliance with IACUC approved protocols. All reagents, tissues and experiments involving T. gondii were conducted under Biosafety Level 2 (BSL2) conditions and were performed in compliance with approved Biological Use Authorized protocols. Unless otherwise stated, the data presented are representative of a minimum of three replicate experiments.
Northern blot, RT-PCR and qPCR
Expression of CCL21 mRNA in wild-type (WT) and transgenic GFAP-CCL21 mice was assessed by northern blot, RT-PCR and qPCR. Total RNAs were purified from mice CNS, pancreas, intestine, kidney and lymph nodes using Trizol reagent (Invitrogen, Carlsbad, CA). For northern blots, 2ug per lane of poly A+ RNA per lane was resolved by electrophoresis in a 1.5% agarose/1.2 M formaldehyde gel, transferred to nylon membrane and hybridized with 32P-radiolabeled probes (Melchior et al. 2010; Thrash et al. 2009). For reverse transcriptase (RT)-polymerase chain reaction (PCR), first strand cDNA was synthesized (Amersham Biosciences, NJ) and amplified by PCR using specific primers as described (Ploix et al. 2001; Schmid et al. 2009). Real-time PCR was performed using the iQ5 real-time PCR detection system (Bio-Rad) and primer sequences described in (Noor et al. 2010; Toulme et al. 2010). The relative number of CCL21 transcripts per hypoxanthine phosphoribosyl transferase (HPRT) transcripts was determined by using calibration standards for both HPRT and CCL21 as previously described (Toulme et al. 2010). Briefly, PCR fragments served as standards for calibration of quantitative PCR were purified from preparative gels with an extraction kit (QiAquick; Qiagen Germany). Fragment concentrations were assessed based on their respective molecular weights and their 260 nm absorbance. These standards were subsequently diluted to obtain a standard curve of 107, 106, 105, 104, 103, and 102 copies for qPCR analysis. To minimize experimental variations from one sample to another, the copy number of CCL21 per sample was normalized to the expression of the housekeeping gene, HPRT. It was verified that the copy number of HPRT transcripts was of the same order of magnitude in all samples being compared.
In situ hybridization and histologic analysis of inflammatory infiltrates
In situ hybridization was performed on free-floating cryosections sections as previously described (Thrash et al 2008, Melchior et al. 2010). Briefly, coronal sections were hybridized at 55°C for 16 hours with a 33P-labeled riboprobe (107 cpm/ml). GFAP positive cells were visualized using biotinylated antibodies directed against GFAP and standard strep-avidin, horseradish peroxidase methodology. Sections were mounted onto Fisherbrand superfrost/plus slides and dehydrated with ethanol and chloroform. Slides were exposed for 3 days to Kodak X-AR film and dipped in Ilford K-5 emulsion. After 3 weeks, slides were developed with Kodak D19 developer, fixed and counterstained with Mayer’s hematoxylin. For histologic analysis of inflammatory infiltrates, brain sections were labeled with hematoxylin, eosin, and antibodies against CD4, CD8 and VCAM-1 as described (Ploix et al. 2001, Noor et al. 2010). The proportion of CD4+ and CD8+ T cells in parenchymal and non-parenchymal sites were quantified in histologic images from three biologic replicates per condition reported.
Microglia, macrophage and lymphocyte isolation from adult murine brain
Microglia, macrophages and lymphocytes were isolated as previously described from the CNS of mice following intracardiac perfusion with PBS and halothane-mediated euthanasia (Carson et al. 1998; Carson et al. 1999). In brief, following euthanasia, the brains of the mice were rapidly removed and mechanically dissociated. The cell suspension was separated on a discontinuous 1.03/1.088 Percoll density gradient and microglia/macrophages were collected from the interface as well as from the 1.03 Percoll fraction. Microglia and CNS-infiltrating immune cells were identified by flow cytometry using APC-conjugated antibodies against pan-CD45 and phycoerythrin (PE)-conjugated antibodies against CD11b (labeling microglia and macrophages), PE- and PerCP antibodies conjugated to CD3 antibodies (labeling T cells), PE-, FITC, PerCP-conjugated anti-CD4 and anti-CD8 (labeling T cells) (BD Biosciences). FcR block (BD biosciences) was used to minimize any non-specific labeling of cells via Fc receptors. A BD FACs Calibur or Canto was used for flow cytometric quantification of immunoreactivity. Flow cytometric data were subsequently analyzed with FlowJo software (Tree Star Inc., Palo Alto, CA).
CFSE labeling and T cell transfer
T cells were isolated from murine peripheral lymph nodes as previously described (Ploix et al. 2001). The resulting cell populations comprised more than 95% Thy-1.2+ CD62Lhi, CD44lo cells as determined by flow cytometry. For CFSE labeling, T cells were resuspended at 1–5×107/ml and incubated for 10 minutes at 37°C with 10 μM carboxyfluorescein diacetate succinimidyl ester (CFSE) (Invitrogen). Cells were then washed 3 times with 10 ml ice cold PBS. An aliquot from the labeled population was stored for later flow cytometric analysis to define the parameters of the non-dividing cell population. Ten million T cells were injected intravenously into irradiated (600 rad) recipient mice (8–12 weeks old). Proliferation of CFSE-positive CD4+ T cells was analyzed in T cells recovered from cervical and pancreatic lymph nodes using a FACSCalibur and subsequent analysis of data with FlowJo software (Tree Star Inc., Palo Alto, CA). Five days post-transfer into recipient mice, T cells were harvested from cervical and pancreatic lymph nodes and CD4+ T cells were analyzed for CFSE fluorescence. All panels within the same figure were derived from the same batch of CFSE labeled T cells injected at the same time into different recipient mice. The fluorescence intensity of the undividing CD4+ T cell population was always determined for each batch of CFSE labeled CD4+T cells by analyzing the level of fluorescence observed in an aliquot of uninjected CFSE-labeled CD4+T cells stored at 4°C and analyzed at the same time as the CFSE-labeled cells recovered from recipient mice.
Quantification of T. gondii burden by real-time PCR
Parasite burden was measured by amplifying the T. gondii B1 gene by real-time PCR, using SYBR® GreenERTM SuperMix for iCyclerR (InvitrogenTM, Carlsbad, CA, USA) with a MgCl2 concentration adjusted to 3.5μM in a 30μl reaction volume, 2μg of total template DNA from each organ, and 1.50μl of 0.5μM primer (IDT; Forward primer 5′-TCCCCTCTGCTGGCGAAAAGT-3′ and Reverse primer 5′-AGCGTTCGTGGTCAACTATCGATTG-3′) and water for a final volume of 30μl. The gene was amplified in an iCyclerR RT-PCR machine (Bio-Rad Laboratories, Hercules, CA, USA) using a 10min initial denaturation at 95 °C followed by 50 cycles that consisted of 15s denaturation at 95°C, 30s annealing at 60°C, and 30s extension at 72°C. The melt curve was generated to check for primer-dimers and threshold values (Ct) were acquired and analyzed using the Bio-Rad iQ5 2.0 standard Edition Optical System Software v 2.0.148.060623.
Results
Several studies have reported increased expression of CCL21 within the CNS in response to multiple inflammatory insults, including infection by pathogens and triggers of CNS autoimmune disease (Lalor and Segal 2010). Within the CNS, CCL21 is hypothesized to regulate neuroinflammation both by recruiting CD4+ T cells into the CNS via CCR7, the high affinity receptor for CCL21 and CCL19 as well as by activating CNS-resident microglia via CXCR3, the low affinity receptor for CCL21. Here we sought to test whether CCL21 in the absence of other pathogenic stimuli was sufficient to recruit T lymphocytes into the CNS and to increase microglial expression of molecules associated with antigen-presentation to CD4+ T cells.
Expression of the CCL21 transgene is restricted to the CNS of GFAP-CCL21 transgenic mice
To test a similar question, a previous study generated transgenic mouse lines with CCL21 expression driven by the oligodendrocyte myelin basic protein (MBP) promoter (Chen et al. 2002). In these mice, transgenic oligodendrocyte-driven CCL21 expression resulted in neutrophil influx into the CNS during early post-natal development accompanied by lethality within 4 weeks of life. The MBP transgene promoter is active early in development and drives robust transgene expression. Therefore, it is uncertain how much of the observed lethal phenotype was a consequence of production of supraphysiologic levels of CCL21, CXCR3-mediated activation of microglia, and/or oligodendrocyte dysfunction and dysmyelination (Chen et al. 2002). Based on these data, we chose not to use oligodendrocyte promoters to generate a transgenic model of CNS CCL21 expression.
Both neurons and astrocytes are reported to increase CCL21 expression by at least 5–10 fold over control levels in response to CNS injury and infection (Gomez-Nicola et al. 2010, Lalor and Segal 2010, Wilson et al. 2009, Noor et al. 2010). Therefore, we chose to drive CCL21 expression in astrocytes using the well-characterized GFAP expression cassette and the CCL21 transgene reported in the generation of the Ins-CCL21 transgenic mouse line (Stalder et al. 1998, Fan et al 2000, Ploix et al. 2001). By northern blot analysis, we could detect expression of CCL21 mRNA in the brain and spinal cords of GFAP-CCL21 transgenic mice at levels much higher than observed in non-transgenic wild-type siblings (figure 1A). Aberrant transgene expression in the intestine by enteric glia and in the kidney has been reported in some transgenic animals generated with the GFAP expression cassette used in our studies. Therefore, we also analyzed CCL21 transgene expression in the intestine, kidney as well as in the CNS, pancreas and lymph nodes by RT-PCR analysis (40 cycles) (figure 1B). CCL21 expression could only be detected in the CNS (lane 1, figure 1B) and lymph nodes (lane 5, figure 1B) of GFAP-CCL21 transgenic mice. To illustrate that lymph node expression of CCL21 in the GFAP-CCL21 mouse line was due to the endogenous CCL21 genes, GFAP-CCL21 mice were bred to plt/plt deletion mutant mice lacking the endogenous CCL21-ser and CCL19-ser genes. CCL21 expression was only detected in the CNS (lane 6, figure 1B) and not in the pancreas, intestine, kidney or lymph nodes (lane 7–10) of mice homozygous for the plt deletion (plt/plt) but carrying the GFAP-CCL21 transgene. For comparison and as a positive control, endogenous CCL21 expression in the lymph nodes of non-transgenic wild-type is also shown (lane 11, figure 1B).
Figure 1. Transgenic expression of CCL21 is restricted to the CNS.

Panel A depicts northern blot analysis of CCL21 and cyclophillin (loading control) expression in the brains (BR) and spinal cords (SC) of wild-type non transgenic and GFAP-CCL21 transgenic adult mice. Panel B depicts RT-PCR (40 cycles) detection of CCL21 expression in cDNA templates prepared from the brain, pancreas, intestine, kidney and lymph nodes of mice expressing the GFAP-CCL21 transgene, CCL21/CCL19 deficient (plt/plt) mice expressing the GFAP-CCL21 transgene and non-transgenic wild-type mice. Panel C depicts qPCR analysis of CCL21 expression as a function of HPRT expression in the brains of wild-type nontransgenic and GFAP-CCL21 transgenic mice.
In generating a CCL21 transgenic model, we sought to identify transgenic mouse lines expressing levels of CCL21 in the CNS roughly comparable to that detected in response to infection by a naturally occurring murine pathogen. In non-transgenic mice, low levels of CCL21 could be detected in the healthy nontransgenic adult murine CNS by northern blot analysis (figure 1A) and by quantitative PCR (qPCR) (figure 1C). Using qPCR, we determined that CCL21 expression within the CNS of GFAP-CCL21 mice was ~8-fold higher than that detected in the healthy non-transgenic CNS (figure 1C). For comparison, CCL21 expression in the brains of non-transgenic wild-type mice infected with Toxoplasma gondii is ~5- fold higher than in the brains of uninfected wild-type mice (Noor et al. 2010). Therefore, CCL21 levels in the CNS of GFAP-CCL21 mice are higher than that observed during a successful immune response against a natural murine CNS pathogen, but likely in the physiologic range of expression.
Several studies have reported that CNS neurons express low levels of CCL21 (Lalor and Segal 2010). Using in situ hybridization analysis (figure 2), we could not detect CCL21 expression in the healthy young (figure 2A) or aged (figure 2C) non-transgenic wild-type brain (figure 2A). CCL21 expression was readily detected in all brain regions of GFAP-CCL21 mice at all ages examined, but only in GFAP+ astrocytes (figure 2B, 2D and data not shown). It is likely that our in situ hybridization analysis was not sufficiently sensitive to detect neuronal expression in healthy wild-type CNS. Although not all astrocytes expressed detectable levels of CCL21, there were no overt differences in morphology among CCL21+ and CCL21- cells. A similar heterogeneous, but GFAP-restricted pattern of expression has been observed with multiple GFAP-driven transgenes (Stalder et al. 1998). Although CCL21 levels in the CNS of GFAP-CCL21 were higher than those of T. gondii infected wild-type mice, we did not observe overt signs of astrogliosis, microgliosis or hypercellularity characteristic of a CNS inflammatory responses and/or leukocyte infiltration in brain sections from unmanipulated GFAP-CCL21 mice (figure 2 and data not shown). Furthermore, no overt defects in spatial organization of the brain could be detected in routine histologic examination of brain sections from unmanipulated adult GFAP-CCL21. Finally no overt signs of aberrant behavior were observed and the lifespan of GFAP-CCL21 transgenic mice was comparable to non-transgenic siblings maintained in the same animal colony (>24 months).
Figure 2. Transgenic expression of CCL21 is localized to astrocytes.

In situ hybridization analysis of CCL21 expression in the CNS of unmanipulated adult wild-type (panels A and C) and GFAP-CCL21 (panel B and D), 3 month (panels A and B) and 12 month (panels C and D) mice. Astrocytes are visualized in brown using antibodies directed against GFAP, nuclei are visualized in blue with Mayer’s hematoxylin and CCL21 is visualized by presence of black grains in the film emulsion coating the tissue section using 33P labeled CCL21 anti-sense riboprobes. Arrows identify examples of CCL21 expressing astrocytes.
Astrocyte-driven expression of CCL21 does not lead to microglial activation or lymphocyte influx into the CNS
Transgenic overexpression of CCL21 in the pancreas is by itself sufficient to recruit T cells and antigen-presenting cells into uninfected healthy tissues (Fan et al. 2000, Ploix et al. 2001, Chen et al. 2002). In contrast to the effects of overexpressing CCL21 in non-immune privileged sites such as the pancreas, we could not detect lymphocyte, neutrophil or myeloid influx into the healthy CNS of GFAP-CCL21 mice. By histologic analysis, we did not detect hypercellularity characteristic of leukocyte influx or the presence of CD3+ cells in regions with high levels of transgenic CCL21 expression (figure 2 and data not shown). To confirm these observations, we also assayed leukocyte accumulation in the CNS by flow cytometric analysis of brain cell suspensions prepared from healthy adult nontransgenic wild-type and GFAP-CCL21 transgenic mice (figure 3). In brain cell suspensions, microglia can be distinguished from all other mature blood derived leukocytes by their lower level of CD45 expression. Thus, CNS-resident microglia are defined as CD45lo, CD11b+ cells, CNS-infiltrating macrophages as CD45hi, CD11b+, while lymphocytes are defined as CD45hi but CD11b negative. In the healthy CNS of both GFAP-CCL21 (figures 3A and 3b) and wild-type mice (figure 3B), only a few CD45hi cells could be detected. The numbers of these CD45hi cells ranged from 0.5–2% of total CD45 cells (low and high) isolated from the brains of wild-type and GFAP-CCL21 mice and did not significantly differ between non-transgenic and non-transgenic mice. The few CD45hi cells detected expressed the myeloid marker, CD11b. Thus, these flow cytometric data confirm our histologic data. No lymphocytes (CD45hi, CD3+, CD11bnegative) could be detected in the CNS of adult GFAP-CCL21 at any age examined from 2 months to 24 month old (figures 3A, 3B, and data not shown).
Figure 3. Astrocyte driven expression of CCL21 does not lead to microglial activation or lymphocyte influx into the CNS.

Panel A depicts flow cytometric analysis of brain cell suspensions (gated on live cells) from 2 month old GFAP-CCL21 mice labeled with antibodies for CD11b (y-axis) and CD45 (x-axis). In panels B–E, flow cytometric analyses of brain cell suspensions from unmanipulated adult non-transgenic wild type mouse are depicted in light grey filled histograms and from unmanipulated age matched GFAP-CCL21 mice in unfilled black histograms. Panel B depicts the relative numbers of CD45+ cells. Histograms (panels B–E) are gated on live CD45+ cells (upper right and lower right quadrants of panel A, that include CD11b+ and CD11b-cells). Histograms (panels C–E) are gated on CD45lo cells (see figure 6A, for example of gate) and depict the relative expression levels of MHC class II (panel C), B7.2 (panel D) and CD40 (panel E) by CD45lo microglia.
High concentrations of CCL21 can trigger pro-inflammatory activation of microglia via the CXCR3 receptor (Lalor and Segal 2010). Approximately 25% of microglia express CXCR3 as determined by flow cytometric analysis of brain cell suspensions of healthy uninfected wild-type mice (Nance and Wilson, ms in prep). Therefore, microglia could potentially respond to transgenically expressed CCL21. Microglial activation can be detected by several measures. In vivo, increased expression of CD45 and MHC class II are sensitive measures of general microglial activation, while CD40 and B7.2 are measures of pro-inflammatory activation (Carson et al.1998). No difference in microglial expression of these markers was detected in brain cell suspensions prepared from healthy wild-type and GFAP-CCL21. These data suggest that either the levels of CCL21 expressed in the GFAP-CCL21 model are insufficient to trigger pro-inflammatory microglial activation via the CXCR3 receptor or insufficient to overcome the inhibitory actions of neuronally expressed microglial regulatory molecules (such as CD22, CD200 and fractalkine) (reviewed in Carson et al. 2006).
Astrocyte driven CCL21 expression is both bioactive and bioavailable
Transgenic expression of CCL21 in many non-immune privileged sites is by itself sufficient to recruit lymphocytes and trigger formation of organized ectopic lymphoid structures (Fan et al. 2000). Other tissues such as the skin are refractory to lymphoid formation even in the presence of transgenic CCL21 expression (Chen et al. 2002). Therefore, the absence of lymphocytes and activated microglia in our GFAP-CCL21 transgenic model could indicate that astrocyte driven transgene expression does not produce bioactive or bioavailable CCL21. Alternatively, our data may indicate the CNS, like the skin is refractory to formation of lymphoid structures that serve to facilitate lymphocyte retention and activation during chronic inflammation.
To determine whether GFAP-CCL21 transgenic mice produced CCL21 that was both bioavailable and bioactive, we tested whether astrocyte driven CCL21 expression could support homeostatic proliferation of CD4+ T cells. Under normal conditions, the numbers of T cells present in secondary lymphoid organs are determined by thymic production and export (reviewed in Ploix et al. 2001). However, under lymphopenic conditions (T cell-deficient conditions) that can result from pathogen encounter, chemotherapy or sub-lethal irradiation, T cells undergo antigen-independent homeostatic proliferation in secondary lymphoid organs. T cells “recognize” lymphopenia by availability and competition for specific chemokines and cytokines. For naïve CD4+ T cells, homeostatic proliferation is dependent on the levels of available CCL21 and thus homeostatic CD4+ T cell proliferation is a sensitive assay of CCL21 bioavailability and bioactivity (figure 3) (Ploix et al. 2001, Li et al. 2007).
Both the number of CD4+ T cells proliferating and the number of rounds of proliferation completed can be quantified using flow cytometric analysis of CFSE-labeled CD4+ T cells (Ploix et al. 2001). The level of CFSE fluorescence per CD4+ T cell is reduced by ½ with each round of proliferation. For example, when naïve (antigen inexperienced) CD4+ T cells are transferred into non-lymphopenic wild-type recipients (with a normal complement of CD4+ T cells and thus no free endogenous CCL21), nearly all of the transferred cells fail to proliferate. The absence of proliferation is visualized by the presence of a single uniform population of CFSEhi CD4+ T cells harvested 5 days post-transfer (figure 4A). By contrast, CD4+ T cells undergo multiple rounds of proliferation when transferred into lymphopenic wild-type mice, resulting in detection of multiple peaks of decreasing CFSE fluorescence (figure 4B). The data depicted in figures 4A and 4B represent CD4+ T cells isolated and pooled from cervical lymph nodes draining the CNS and the mesenteric and pancreatic lymph nodes draining the pancreas. However, in wild-type mice no difference is observed in the relative degree of homeostatic proliferation in T cells isolated from cervical versus mesenteric/pancreatic lymph nodes (Ploix et al. 2001 and data not shown). To determine the relative bioavailability of CCL21 in non-transgenic and CCL21 transgenic mice, a single batch of naïve CFSE labeled CD4+ T cells was used for transfer into non-lymphopenic and lymphopenic wild-type, Ins-CCL21 and GFAP-CCL21 mice (figure 4). CD4+ T cells were harvested from the lymph nodes draining the pancreas when using Ins-CCL21 recipient mice and from cervical lymph nodes when using GFAP-CCL21 recipient mice.
Figure 4. Astrocyte-driven expression of CCL21 promotes homeostatic proliferation of adoptively transferred naïve CD4+ T cells in non-lymphopenic recipients.

Flow cytometric analysis of CFSE labeled naïve CD4+ T cells 5-days post-adoptive transfer into non-transgenic mice (panels A and B), into Ins-CCL21 transgenic mice (panels C and D) and into GFAP-CCL21 mice (panels E and F). Panels A, C and E represent non-lymphopenic recipients. Panels B, D and F represent sublethally irradiated (600 rads) lymphopenic recipients. All recipient mice (panels A–F) received CFSE CD4+ T cells labeled in the same experiment.
We have previously demonstrated that transgenically produced CCL21 is sufficient to promote homeostatic proliferation of CD4+ T cells in the lymph nodes draining the pancreas even in non-lymphopenic Ins-CCL21 recipient mice (Ploix et al. 2001 and figure 4C). The observed proliferation was greater than that observed in lymphopenic non-transgenic recipients and is a consequence of the higher levels of CCL21 being produced in the Ins-CCL21 transgenic mice (Ploix et al. 2001 and figure 4B). Inducing lymphopenia in Ins-CCL21 by sublethal irradiation did not lead to increased homeostatic proliferation of transferred CD4+ T cells. Therefore, CCL21 in the pancreatic lymph nodes was not rate limiting for homostatic proliferation in the presence of transgenic overexpression of CCL21 (figure 4D, Ploix et al. 2001). Strikingly, transgenic overexpression of CCL21 by CNS astrocytes was also sufficient to promote CD4+ T cell proliferation in non-lymphopenic GFAP-CCL21 recipients (figure 4E). The degree of homeostatic proliferation in non-lymphopenic GFAP-CCL21 mice was also greater than that observed in lymphopenic wild-type mice, although the degree of proliferation was not as great as that observed in Ins-CCL21. As observed with Ins-CCL21 recipient mice, homeostatic proliferation of CFSE labeled CD4+ T cells harvested from lymphopenic GFAP-CCL21 recipients was similar to those harvested from non-lymphopenic GFAP-CCL21 recipients (figures 4E and 4F). Taken together these data indicate that transgenic overexpression of CCL21 from within the CNS is both bioavailable and bioactive for T cells in lymph nodes outside the CNS.
We also bred the GFAP-CCL21 onto the plt/plt genetic background to determine if CNS-produced CCL21 was by itself sufficient to support homeostatic CD4+ T cell proliferation (figure 5). Very little proliferation is observed when naïve CFSE labeled CD4+ T cells are transferred into lymphopenic plt/plt mice (which are CCL19/CCL21 deficient) (figure 5A). By contrast, homeostatic proliferation of adoptively transferred CD4+ T cells is restored in lymphopenic plt/plt mice expressing transgenic CCL21 solely in the CNS (figure 5B). We have previously reported that transgenic CCL21 expressed solely in the pancreas of plt/plt mice was similarly sufficient to support homeostatic CD4+ T cell proliferation (Ploix et al. 2001). Together these data illustrate that similar to CCL21 produced in non-immune privileged tissues, CNS-derived CCL21 is bioavailable to lymph node CD4+ T cells. However, these data also suggest that the CNS itself is refractory for the formation of lymphoid-like structures observed in the pancreas of healthy Ins-CCL21 mice or that insufficient numbers of CD4+ T cells could breach the blood brain barrier of healthy GFAP-CCL21 mice.
Figure 5. Astrocyte driven expression of CCL21 is sufficient to restore homeostatic CD4+ T cell proliferation in CCL19/CCL21 deficient mice.

Flow cytometric analysis of CFSE labeled naïve CD4+ T cells 5-days post-adoptive transfer into sublethally irradiated CCL19/CCL21 deficient mice (plt/plt) that do not carry the GFAP-CCL21 transgene (panel A) or that do carry the GFAP-CCL21 transgene (panel B). All recipient mice (panels A–B) received CFSE T cells labeled in the same experiment.
CNS-derived CCL21 is required for efficient CD4+ T cell migration from perivascular sites into the CNS parenchyma
In the absence of injury and/or pathogens, CD4+ T cells may not have sufficient access to the CNS. We therefore tested the dependence and consequence of CCL21 deficiency and overexpression following T. gondii infection. We chose this model for three reasons. First, mice are a natural host for T. gondii (Dzierszinski and Hunter 2008; Wilson et al. 2009). Following initial infection, the CNS is the site of chronic life-long T. gondii infection. Second, IFNγ producing T cells are required to limit infection and drive T. gondii tachyzoites into latent cyst forming bradyzoites during the early phase of infection. Chronic influx of T cells into the CNS for the lifetime of the infected host is required to limit emergence of active cytotoxic tachyzoites from latent cysts (Dzierszinski and Hunter 2008; Wilson et al. 2009). Third, CCL21 immunoreactivity is induced following infection and is associated with T cell migratory pathways leading from perivascular and meningeal sites of T cell influx to regions surrounding cysts (Wilson et al. 2009, Noor et al. 2010).
T. gondii infection causes an influx of CD45hi, CD11b+ macrophages and CD45hi CD11b-negative lymphocytes evident by 14 days post-infection in wild-type adult mice (figure 6A). CXCR3 expression is detected by flow cytometric analysis on approximately 30–50% of CNS-resident microglia and macrophages in infected mice (Nance and Wilson, ms in prep). Thus both CNS-resident microglia and macrophages have the potential to respond to CCL21 via low affinity interactions with this receptor. Figure 6B depicts the CD45 levels of only CD11b+ cells isolated from uninfected and infected CNS. Infection is sufficient to cause a 2-fold increase in the levels of CD45 expressed by microglia (compare light grey unfilled histogram with filled grey histogram; figure 6B). However, CD45 histograms of CD11b+ cells isolated from infected wild-type mice (filled grey histogram) and infected GFAP-CCL21 mice (unfilled black histogram) are coincident. These data indicate that there is no difference in the influx of CD45hi macrophages into the brains of infected wild-type versus infected GFAP-CCL21 mice. Furthermore, while macrophages display higher levels of pro-inflammatory molecules than microglia in brains of infected mice, there are no differences in the activation states between wild-type and GFAP-CCL21 mice (figure 6B–F). Specifically, there are no differences in the levels of CD45 (figure 6B), MHC class II (figures 6C and 6E) or B7.2 (figures 6D and 6F) expressed by microglia (figures 6C and 6D) and macrophages (figures 6E and 6F) from infected brains.
Figure 6. Astrocyte-driven expression of CCL21 does not alter microglial activation in T. gondii infected brains.

Panel A depicts flow cytometric analysis of brain cell suspensions isolated from non-transgenic wild-type mice 14 days post-infection with T. gondii. Boxed regions depict gates used to identify CD11b+, CD45lo microglia (MG), CD11b+, CD45hi macrophages (MP) and CD11b-, CD45hi lymphocytes (lymph). Panel B depicts the relative degree of CD45 expression by CD11b+ cells (both CD45lo and CD45hi) in brain cell suspensions from uninfected wild-type (light grey unfilled histogram), from T. gondii-infected wild-type (grey filled histogram) and from T. gondii-infected GFAP-CCL21 (black unfilled histogram) mice. Panels C–D depict expression of MHC class II (panel C) and B7.2 (panel D) by CD45lo microglia isolated from uninfected wild-type (light grey unfilled histogram), infected wild-type (filled grey histogram) and infected GFAP-CCL21 (dark line unfilled histogram) mice. Panels E–F depict expression of MHC class II (panel E) and B7.2 (panel F) by CD45lo microglia isolated from infected wild-type (light grey unfilled histogram), by CD45hi macrophages isolated from infected wild-type (filled grey histogram) and by CD45hi macrophages isolated from infected GFAP-CCL21 (black unfilled histogram) mice.
Following infection, lymphocytes infiltrate the CNS in a two-step procedure. First, the cells extravasate from the blood into the perivascular or extraparenchymal (submeningeal and ventricular) spaces. Second, extravasated immune cells may either accumulate in these extraparenchymal sites or infiltrate the parenchyma. The regulation of this latter step is actively debated but is likely multifactoral (for review see Wilson et al. 2010). Histologic examination of brains during the early phase of infection (14 days post-infection) revealed CCL21-dependent accumulation of inflammatory infiltrates at perivascular and meningeal sites (figure 7). Inflammatory infiltrates could be detected under the meninges of all strains of mice (figures 7A–C). However, accumulation of inflammatory infiltrates was much greater and much more extensive in CCL19/CCL21 deficient mice than in non-transgenic wild-type mice. Similarly, accumulation of inflammatory infiltrates at perivascular sites within the brain parenchyma were readily detected in infected wild-type and plt/plt mice (figure 7D–E); but, perivascular infiltrates were much greater in infected plt/plt mice. Conversely, perivascular accumulation of inflammatory infiltrates in the parenchyma of GFAP-CCL21 was rare and difficult to detect (figure 7F).
Figure 7. CCL21 is required for efficient migration of CD4+ T cells into T. gondii-infected brain.

Hematoxylin and eosin histology of meningeal (panels A–C) and cortical parenchymal (panels D–F) brain sections from wild-type (panels A, D, G), CCL19/21 deficient (plt/plt, panels B, E, H) and GFAP-CCL21 (panels C, F, I) mice 14 days post-infection. Left downward pointing arrows indicate meningeal inflammatory infiltrates (panels A–C). Right downward pointing arrows indicate perivascular regions (panels D, F, E). In panels G–I, CD4+ T cells are visualized in red with PE-conjugated anti-CD4 antibodies CD8+ cells are visualized in green with FITC-conjugated anti-CD8 antibodies. Upward pointing arrows indicate CD4+ T cells in perivscular spaces, downward pointing arrows indicated CD4+ T cells in parenchymal spaces. Quantitation of the ratio of CD4+ Tcells/CD8+ T cells is depicted in panels J (ratio in extraparenchymal spaces) and K (ratio within parenchyma). In both panels, error bars represent mean +/− SEM. Significant difference from wild-type was determined using a 2-tailed Student’s T test: *p<0.05.
Both CD4+ and CD8+ T cells can respond to CCL21 via high affinity interactions with CCR7 (reviewed in Ploix et al. 2001). In addition, both CD4+ and CD8+ T cells infiltrate the brain in response to T. gondii infection (Grazinelli et al. 1992 and figure 7G–I). Strikingly, while CD8+ T cells infiltrated the parenchyma of all three strains of infected mice, CD4+ T cells preferentially accumulated at perivascular and extraparenchymal sites in plt/plt mice. Specifically, the ratio of CD4+ to CD8+ T cells in extraparenchymal sites was more than 2-fold greater in plt/plt mice than in either wild-type or GFAP-CCL21 (figure 7J). Conversely, the ratio of CD4+ to CD8+ T cells within the parenchyma was reduced more than 2-fold in plt/plt mice as compared to both wild-type or GFAP-CCL21 (figure 7K). Lastly, confirming our flow cytometric data, no inflammatory infiltrates, CD4+ or CD8+ T cells could be detected by histologic analysis in any of the three strains mice in the absence of infection (figure 3 and data not shown). These data indicate that there is a dependency on CCL21 expression for CD4+, but not CD8+ T cell migration from perivascular to intraparenchymal sites, even in the presence of a strong antigenic signal presented in an inflammatory context.
Toxoplasma cannot be cleared from the brains of infected rodents or humans. Instead, the intracellular parasite is driven into and maintained in a latent bradyzoite state by a stable but chronic lymphocyte influx into the infected brain. We therefore examined the CCL21-dependence of inflammatory infiltrate accumulation at the stable, chronic phase of infection (figures 8–9). At 6 weeks post-infection, inflammatory infiltrates were detected in all three strains of mice by hematoxylin and eosin histology (figure 8). Notably, the magnitude of inflammatory infiltrate accumulation at extra-parenchymal sites (submenigneal, ventricular and perivascular) in the brains of plt/plt was greater than that observed in wild-type and GFAP-CCL21 mice (figure 8).
Figure 8. Requirement for CCL21 continues in chronic T. gondii-infected brain.

Hematoxylin and eosin histology of meningeal (panels A–C), ventricular (panels D–F) and cortical parenchymal (panels G–I) brain sections from wild-type (panels A, D, G), CCL19/21 deficient (plt/plt, panels B, E, H) and GFAP-CCL21 (panels C, F, I) mice 6 weeks post-infection. Left downward pointing arrows indicate meningeal inflammatory infiltrates (panels A–C). Right downward pointing arrows indicate ventricular/subventricular regions (panels D–E). Right upward pointing arrows indicate perivascular regions (panels G–I).
Figure 9. CCL21 is not required for CD4+ T cell extravasation in chronically infected brains.
Panel A depicts the total number of CD45+ cells isolated from the CNS of wild-type, CCL19/CCL21 deficient (plt/plt) and GFAP-CCL21 mice isolated 6 weeks following T. gondii infection. The absolute numbers of CD45lo microglia (panel B), CD45hi macrophages (panel C), CD4+ T cells (panel D) and C8+ T cells (panel E) isolated from infected mice were determined using flow cytometric analysis of brain cell suspensions. Panel F depicts the burden of parasite DNA per mg brain weight. In all panels, the error bars represent mean +/− SEM. Significant difference from wild-type was determined using a 2-tailed Student’s T test: *p<0.05, **p<0.01.
To determine if the greater accumulation of inflammatory infiltrates in extraparenchymal sites in chronically infected plt/plt mice reflected a greater total influx of inflammatory cells, we quantified inflammatory infiltrates isolated from mice 6 weeks post-infection by flow cytometry (figure 9). We found no statistical difference between total cells, CD45lo microglia, CD45hi macrophages nor CD8+ T cells isolated from the brains of infected wild-type, plt/plt and GFAP-CCL21 mice (figure 9A–C, E). By contrast, 2-fold more CD4+ T cells were isolated from the brains of both plt/plt and GFAP-CCL21 mice than from wild-type mice (figure 9D). In the absence of CCR7 or CCL19/CCL21, T cells are inefficient at limiting and controlling T. gondii infection in peripheral tissues and brain resulting in significantly higher parasite burdens (~3-fold in plt/plt versus wild-type mice) (Noor et al 2010 and figure 9F). Thus, the higher levels of CD4+ T cells in plt/plt mice may reflect more robust stimulation of the immune response provided by the higher parasite burden. However, these data also indicate that in the presence of strong antigenic stimulus such as T. gondii, extravasation of activated myeloid cells and lymphocytes from the blood into extraparenchymal spaces is CCL21-independent.
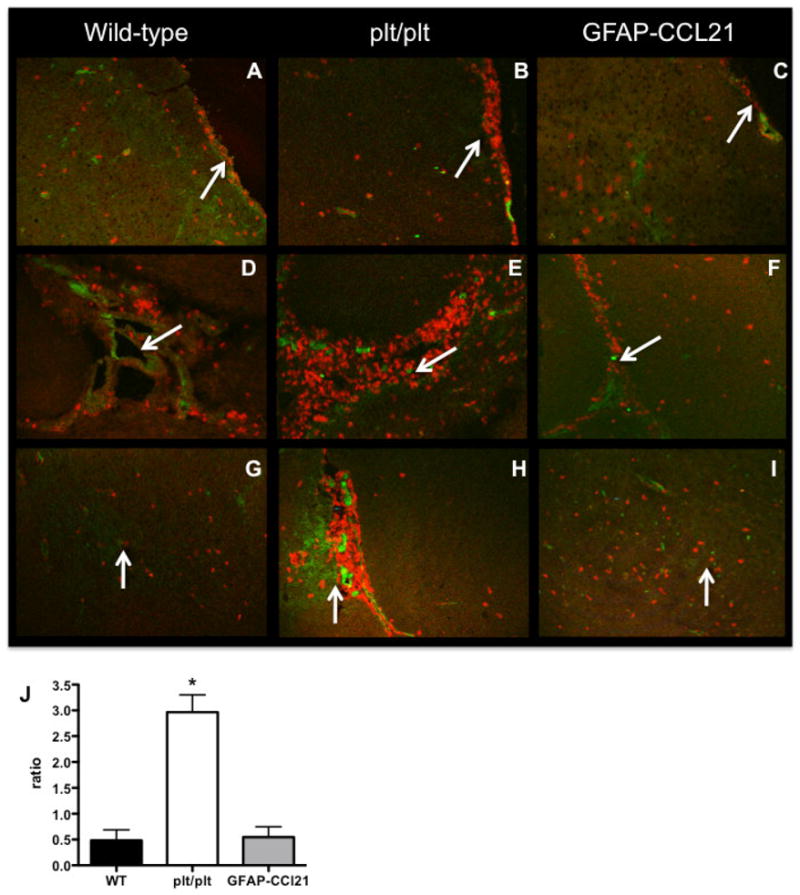
These flow cytometric data also reveal that the greater accumulation of inflammatory infiltrates in plt/plt mice is not a simple consequence of higher numbers of CNS-infiltrating cells (figure 8 and 9). Although similar numbers of CD4+ T cells were present in infected plt/plt and GFAP-CCL21 mice, they were not similarly distributed within the chronically infected brain (figure 10). While at least some CD4+ T cells were found in the parenchyma of all chronically infected mice, the proportion of CD4+ T cells in extraparenchymal (submenigeal and ventricular, figure 10A–F) versus parenchymal sites was greatest in plt/plt mice (figure 10G–I). The few CD4+ T cells detected within the parenchymal of plt/plt mice were often detected in close proximity to VCAM-1 expression that is indicative of activated vascular endothelium and sites of lymphocyte influx. Furthermore, the ratio of CD4+ T cells in extraparenchymal sites versus intraparenchymal sites was ~6-fold higher in plt/plt mice than in either wild-type or GFAP-CCL21 mice at 6 weeks post-infection (figure 10J).
Figure 10. CD4+ T cells preferentially accumulate in non-parenchymal sites of chronically infected brains in the absence of CCL21.

Immunofluorescence of CD4+ T cells (red) and VCAM-1 (green) at meningeal (panels A–C), ventricular (panels D–F) and parenchymal (panels G–I) brain regions from wild-type (panels A, D, G), CCL19/21 deficient (plt/plt, panels B, E, H) and GFAP-CCL21 (panels C, F, I) mice 6 weeks post-infection. Arrows indicate CD4+ T cells in the chronically infected brain. Panel J represents the quantitation of the ratio of CD4+ T cells in extraparenchymal sites/CD4+ T cells within parenchymal sites. Error bars represent mean +/− SEM. Significant difference from wild-type was determined using a 2-tailed Student’s T test: *p<0.05.
Despite the high numbers of CD4+ T cells present in the CNS of GFAP-CCL21 mice, no organization of T cells and/or inflammatory infiltrates into neo lymphoid structures could be detected. Instead, CD4+ T cells were found dispersed throughout the infected CNS during chronic inflammation. Thus, the CNS remains refractory to CCL21-driven organization of chronic inflammatory infiltrates even in the presence of constitutive CCL21 expression, chronic influx of CD4+ T cells and ongoing pathogenic stimulus. Lastly, CD4+ T cell influx into the parenchyma of infected GFAP-CCL21 was much greater than in either wild-type or plt/plt mice and was detected in all brain regions. These data may in part explain the highly variable parasite burden detected in GFAP-CCL21 mice at 6 weeks post-infection that did not correlate with CD4+ T cell or inflammatory infiltrate numbers (figure 9). Specifically, we speculate that in GFAP-CCL21 mice expression of the CCL21 transgene throughout the brain and spinal cord recruits lymphocytes into the parenchyma of all brain regions (infected and non-infected). Therefore GFAP-CCL21 mice may be less efficient at localizing CD4+ T cell responses to the sites of brain infection than wild-type mice.
Discussion
CCL21 is a potent chemoattractant and functional regulator of CD4+ T cells (Fan et al. 2000, Ploix et al. 2001, Forster et al. 2008, Lalor and Segal, 2010). For example, in non-immune privileged organs, CCL21 plays at least three distinct roles regulating CD4+ T cell function. First, CCL21 promotes homeostatic proliferation of CD4+ but not CD8+ T cells (Ploix et al. 2001). Second, CCL21 recruits CD4+ T cells and other immune cells involved in adaptive (lymphocyte-mediated) immunity to the sites of expression (Fan et al. 2000, Chen et al. 2002). Third, CCL21 expression is sufficient to organize chronic inflammatory infiltrates into lymphoid-like structures (Ploix et al. 2001, Chen et al. 2002). In the CNS, CCL21 expression is induced by a broad array of injury, infectious and autoimmune triggers (Lalor and Segal 2010). The consequences of CCL21 expression within the CNS are ill-defined. Here we report a novel and viable transgenic murine model to examine the consequences of CCL21 expression within the CNS.
Outside the CNS, induction of CCL21 within a tissue such as the pancreas promotes antigen-independent homeostatic proliferation of CD4+ T cells within the lymph nodes draining the tissue (Ploix et al. 2001). CCL21-driven homeostatic proliferation also increases CD4+ T cell expression of molecules required for extravasation (LFA-1 and VLA-1) as well as amplifying CD4+ T cell responses to their target antigens. For example, in experimental autoimmune models of diabetes, transfer of auto-reactive CD4+ T cells causes insulitis but does not cause autoimmune destruction of insulin producing islets (reviewed in Lo, 2002, Ploix et al. 2001). However, autoimmune destruction of insulin producing islets does occur when the same autoreactive CD4+ T cells are transferred into lymphopenic recipients or recipient mice overexpressing CCL21 in islets. Furthermore, in experimental and spontaneous models of autoimmunity, CCL21 expression is also observed in the target tissue during the preclinical phases of disease (Forster et al. 2008, Lalor and Segal 2010).
The role of homeostatic proliferation in amplifying T cell responses is not just relevant to experimental animal models. Lymphopenia and concurrent homeostatic T cell proliferation is a risk factor for subsequent autoimmunity and underlies the severe autoimmunity observed in genetically lymphopenic individuals with Omenn’s disease (Marella et al. 2008, Milner et al. 2008, Data et al. 2009). Homeostatic proliferation is even implicated in driving autoimmune disease in individuals without lymphopenia. For example, naïve CD4+ T cells in individuals with autoimmune disorders such as multiple sclerosis and rheumatoid arthritis display features consistent with higher levels of ongoing homeostatic proliferation (decreased numbers of T cell receptor DNA excision circles) than healthy individuals (Gleeson et al. 1995, Hug et al. 2003, Duszczyszyn et al. 2006, Datta et al. 2009). In these latter cases, it has been hypothesized that homeostatic proliferation may be promoted by chemokine/cytokine dysregulation.
In the GFAP-CCL21 model reported here, we demonstrate that CCL21 produced within the CNS is bioavailable. CD4+ T cells do not undergo homeostatic proliferation in non-lymphopenic recipients lacking free CCL21 or in CCL19/CCL21 deficient recipients. However, CNS-expressed CCL21 not only was sufficient to support CD4+ T cell proliferation within the cervical lymph nodes of CCL19/21 deficient mice (plt/plt), it was sufficient to support homeostatic proliferation in non-lymphopenic wild-type mice. CCL21 can activate its high affinity receptor, CCR7 in either soluble or extracellular matrix bound forms: the former triggering chemokinesis and the latter triggering adhesion (Schumann et al. 2010). As yet, we have not identified where and in what form CFSE labeled CD4+ T cells encounter transgenically expressed CCL21. Soluble CCL21 is a small molecule and thus able to passively drain to cervical lymph nodes via Virchow-Robbins spaces as demonstrated for a wide variety of molecules expressed or placed in the brain (reviewed in Carson et al. 2006, Wilson et al. 2010). Because we detected CFSE labeled CD4+ T cells in cervical lymph nodes and not in the CNS, we favor the hypothesis that adoptively transferred CD4+ T cells encounter soluble CCL21 in the cervical lymph nodes draining the uninfected CNS. However, we cannot exclude the possibility that as part of routine surveillance of the uninfected healthy CNS, adoptively transferred T cells entered the CNS of transgenic GFAP-CCL21 mice in numbers below the limits of our detection and subsequently migrated to and proliferated within the cervical lymph nodes. In either situation, these data provide proof of principle that induction of CCL21 within the CNS by neurons or glia can promote CD4+ T cell homeostatic proliferation and thus lower the threshold for CNS autoimmunity.
Outside the CNS, expression of CCL21 in the absence of infection is sufficient to recruit CD4+ T cells to the site of expression and trigger the formation of ectopic non-inflammatory lymphoid structures (Ploix et al. 2001, Chen et al. 2002). Despite the demonstration that CNS-produced CCL21 was bioavailable in GFAP-CCL21 mice, lymphocytes could not be detected in the uninfected perfused brain by histologic or flow cytometric measures. It is possible that in the absence of infection, CD4+ T cells could not gain access to the CNS. Therefore, we also examined the recruitment and organization of CD4+ T cells in the brains of mice with chronic T. gondii infections.
Outside the CNS, expression of CCL21 is not required to recruit lymphocytes to the sites of infection or autoimmune triggers (Forrester et al. 2008, Ploix et al. 2009). Thus it was not surprising that lymphocytes could be recruited to extra-parenchymal sites within the brains of CCL21/CCL19 deficient mice in response to T. gondii infection. CCL21 expression during chronic inflammation is associated with the organization of inflammatory infiltrates into lymph node-like structures (Ploix et al. 2001, Chen et al. 2002, Shomer et al. 2003). Therefore, it was also not unexpected that in the absence of CCL21/CCL19 these inflammatory infiltrates would fail to organize into lymph node structures within the brain parenchyma. However, within the CNS, it is notable that chronic inflammation is not associated with neo-lymphoid formation, even in T cell mediated disorders such as MS; rather, lymphoid-like structures form outside of the brain parenchyma in meningeal sites (Aloisi and Pujol-Borrell 2006; Columba-Cabezas et al. 2003). From the literature to date, it is unclear if this is due to a deficit in CCL21 production or the inability of the CNS to support lymphoid formation within the parenchyma. Our studies support the latter possibility. Despite the presence of ~3-fold higher numbers of CD4+ T cells in the brain parenchyma of infected GFAP-CCL21 mice than in wild-type mice, CD4+ T cells did not form organized lymphoid-like structures. Instead they remained dispersed throughout the CNS. Our studies did however reveal a role for CCR7 ligands in regulating T cell migration within the CNS. In CCL19/CCL21 deficient mice, most CD4+ T cells failed to enter the infected CNS parenchyma, while in GFAP-CCL21 mice nearly all CD4+ T cells entered the infected CNS parenchyma and migrated widely throughout the CNS.
CCL21 can also activate cells through its low affinity receptor, CXCR3. CXCR3 is expressed by subsets of activated CNS-resident microglia, CNS-infiltrating macrophages and astrocytes (Biber et al. 2002; de Jong et al. 2005, Van Weering et al. 2010). We did not observe any differences in the phenotypes of microglia and macrophages in the presence or absence of infection between wild-type and GFAP-CCL21. We also did not observe changes in astrocyte morphology or GFAP expression indicative of gliosis. Future studies will explore whether this is a consequence of inhibition provided by the intact CNS or insufficient production of CCL21. Independent of which of these two explanations applies to our glial observations, our data indicate that CNS expressed CCL21 not only can regulate immune responses occurring outside the CNS, it can differentially target adaptive (lymphocytic) versus innate (glial and myeloid) immune responses.
Acknowledgments
These studies were supported in part by grants to MJC from NIH (NS045735), the Dana Foundation, CRCC and the UCR Division of Biomedical Sciences, a grant to DL from NIH (AI63426) and grants to EHW from CRCC, the UCR Division of Biomedical Sciences and the UCR academic senate. CP was a fellow of the Christopher Reeve Paralysis Foundation. IL Campbell generously provided the GFAP expression cassette used in these studies.
Footnotes
All authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aloisi Francesca, Pujol-Borrell Ricardo. Lymphoid neogenesis in chronic inflammatory diseases. Nature Reviews Immunology. 2006 March;6(3):205–217. doi: 10.1038/nri1786. [DOI] [PubMed] [Google Scholar]
- Baxter Alan G. The origin and application of experimental autoimmune encephalomyelitis. Nature Reviews Immunology. 2007 November;7(11):904–912. doi: 10.1038/nri2190. [DOI] [PubMed] [Google Scholar]
- Biber K, Rappert A, Kettenmann H, Brouwer N, Copray SCVM, Boddeke HWGM. Neuronal SLC (CCL21) expression: implications for the neuron-microglial signaling system. Ernst Schering Research Foundation Workshop. 2002;(39):45–60. doi: 10.1007/978-3-662-05073-6_4. [DOI] [PubMed] [Google Scholar]
- Bromley Shannon K, Mempel Thorsten R, Luster Andrew D. Orchestrating the orchestrators: chemokines in control of T cell traffic. Nature Immunology. 2008 September;9(9):970–980. doi: 10.1038/ni.f.213. [DOI] [PubMed] [Google Scholar]
- Byram Susanna C, Carson Monica J, DeBoy Cynthia A, Serpe Craig J, Sanders Virginia M, Jones Kathryn J. CD4-positive T cell-mediated neuroprotection requires dual compartment antigen presentation. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2004 May 5;24(18):4333–4339. doi: 10.1523/JNEUROSCI.5276-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998 January;22(1):72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Carson MJ, et al. Disproportionate recruitment of CD8+ T cells into the central nervous system by professional antigen-presenting cells. The American Journal of Pathology. 1999 February;154(2):481–494. doi: 10.1016/S0002-9440(10)65294-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson Monica J, Doose Jonathan M, Melchior Benoit, Schmid Christoph D, Ploix Corinne C. CNS immune privilege: hiding in plain sight. Immunological Reviews. 2006 October;213:48–65. doi: 10.1111/j.1600-065X.2006.00441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Shu-Cheng, Leach Michael W, Chen Yuetian, Cai Xiao-Yan, Sullivan Lee, Wiekowski Maria, Dovey-Hartman BJ, Zlotnik Albert, Lira Sergio A. Central nervous system inflammation and neurological disease in transgenic mice expressing the CC chemokine CCL21 in oligodendrocytes. Journal of Immunology (Baltimore, Md: 1950) 2002 February 1;168(3):1009–1017. doi: 10.4049/jimmunol.168.3.1009. [DOI] [PubMed] [Google Scholar]
- Columba-Cabezas Sandra, Serafini Barbara, Ambrosini Elena, Aloisi Francesca. Lymphoid chemokines CCL19 and CCL21 are expressed in the central nervous system during experimental autoimmune encephalomyelitis: implications for the maintenance of chronic neuroinflammation. Brain Pathology (Zurich, Switzerland) 2003 January;13(1):38–51. doi: 10.1111/j.1750-3639.2003.tb00005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S, Sarvetnick N. Lymphocyte proliferation in immune-mediated diseases. Trends in Immunology. 2009;30:430–438. doi: 10.1016/j.it.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Davis DS, Carson MJ. When the tail can’t wag the dog: the implications of CNS-intrinsic initiation of neuroinflammation. ASN NEURO. 2009;1(2):e00008. doi: 10.1042/AN20090024. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derecki NC, Privman E, Kipnis J. Rett syndrome and other autism spectrum disorders--brain diseases of immune malfunction? Molecular Psychiatry. 2010 April;15(4):355–363. doi: 10.1038/mp.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duszczyszyn DA, Beck JD, Antel J, Bar-Or A, Lapierre Y, Gadag V, Haegert DG. Altered naïve CD4 and CD8 T cell hoeostasis in patients with relapsing-remitting multiple sclerosis: thymic versus peripheral (non-thymic) mehanisms. Clin Exp Immunol. 2006;143:305–313. doi: 10.1111/j.1365-2249.2005.02990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzierszinski FS, Hunter CA. Advances in the use of genetically engineered parasites to study immunity to Toxoplasma gondii. Parasite Immunology. 2008 April;30(4):235–244. doi: 10.1111/j.1365–3024.2007.01016.x. [DOI] [PubMed] [Google Scholar]
- Fan L, Reilly CR, Luo Y, Dorf ME, Lo D. Cutting edge: ectopic expression of the chemokine TCA4/SLC is sufficient to trigger lymphoid neogenesis. Journal of Immunology (Baltimore, Md: 1950) 2000 April 15;164(8):3955–3959. doi: 10.4049/jimmunol.164.8.3955. [DOI] [PubMed] [Google Scholar]
- Förster Reinhold, Davalos-Misslitz Ana Clara, Rot Antal. CCR7 and its ligands: balancing immunity and tolerance. Nature Reviews Immunology. 2008 May;8(5):362–371. doi: 10.1038/nri2297. [DOI] [PubMed] [Google Scholar]
- Frank Gregory M, Lepisto Andrew J, Freeman Michael L, Sheridan Brian S, Cherpes Thomas L, Hendricks Robert L. Early CD4(+) T cell help prevents partial CD8(+) T cell exhaustion and promotes maintenance of Herpes Simplex Virus 1 latency. Journal of Immunology (Baltimore, Md: 1950) 2010 January 1;184(1):277–286. doi: 10.4049/jimmunol.0902373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson PA, Toh BH, van Driel IR. Organ-specific autoimmunity induced by lymphopenia. Immunol Rev. 1996;149:97–125. doi: 10.1111/j.1600-065x.1996.tb00901.x. [DOI] [PubMed] [Google Scholar]
- Grazzinelli RT, Xu Y, Hieny S, Sher A. Simulataneous depletion of CD4+ and CD8+ T lymphocytes is required to reactivate chronic infection with Toxoplasma gondii. J Immunol. 1992;14:175–180. [PubMed] [Google Scholar]
- Hofhuis A, VAN Pelt W, VAN Duynhoven YTHP, Nijhuis CDM, Mollema L, VAN DER Klis FRM, Havelaar AH, Kortbeek LM. Decreased prevalence and age-specific risk factors for Toxoplasma gondii IgG antibodies in The Netherlands between 1995/1996 and 2006/2007. Epidemiology and Infection. 2010 May 24;:1–9. doi: 10.1017/S0950268810001044. [DOI] [PubMed] [Google Scholar]
- Hug A, Korporal M, Schröder I, Haas J, Glatz K, Storch-Hagenlocher B, Wildemann B. Thymic export function and T cell homeostasis in patients with relapsing remitting multiple sclerosis. J Immunol. 2006;171:432–437. doi: 10.4049/jimmunol.171.1.432. [DOI] [PubMed] [Google Scholar]
- Jones JL, Dubey JP. Waterborne toxoplasmosis--recent developments. Experimental Parasitology. 2010 January;124(1):10–25. doi: 10.1016/j.exppara.2009.03.013. [DOI] [PubMed] [Google Scholar]
- de Jong Eiko K, Dijkstra Ineke M, Hensens Marjolein, Brouwer Nieske, van Amerongen Machteld, Liem Robert SB, Boddeke Hendrikus WGM, Biber Knut. Vesicle-mediated transport and release of CCL21 in endangered neurons: a possible explanation for microglia activation remote from a primary lesion. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2005 August 17;25(33):7548–7557. doi: 10.1523/JNEUROSCI.1019-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara Taku, Ishikawa Fumio, Yasuda Takuwa, Aritomi Kentaro, Nakano Hideki, Tanaka Yuriko, Okada Yayoi, Lipp Martin, Kakiuchi Terutaka. CCR7 ligands are required for development of experimental autoimmune encephalomyelitis through generating IL-23-dependent Th17 cells. Journal of Immunology (Baltimore, Md: 1950) 2009 August 15;183(4):2513–2521. doi: 10.4049/jimmunol.0800729. [DOI] [PubMed] [Google Scholar]
- Lalor Stephen J, Segal Benjamin M. Lymphoid chemokines in the CNS. Journal of Neuroimmunology. 2010a July 27;224(1):56–61. doi: 10.1016/j.jneuroim.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert H, Barragan A. Modeling parasite dissemination: host cell subversion and immune evasion by Toxoplasma gondii. Cell Microbiol. 2010;12:292–300. doi: 10.1111/j.1462-5822.2009.01417.x. [DOI] [PubMed] [Google Scholar]
- Le Saout Cecile, Mennechet Sandie, Taylor Naomi, Hernandez Javier. Memory-like CD8+ and CD4+ T cells cooperate to break peripheral tolerance under lymphopenic conditions. Proceedings of the National Academy of Sciences of the United States of America. 2008 December 9;105(49):19414–19419. doi: 10.1073/pnas.0807743105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski GA, Lo D, Bloom FE. Interference with major histocompatibility complex class II-restricted antigen presentation in the brain by herpes simplex virus type 1: a possible mechanism of evasion of the immune response. Proceedings of the National Academy of Sciences of the United States of America. 1993 March 1;90(5):2005–2009. doi: 10.1073/pnas.90.5.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CR, Santoso S, Lo DD. Quantitative analysis of T cell homeostatic proliferation. Cell Immunol. 2007;250:40–54. doi: 10.1016/j.cellimm.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marella V, Poliani PL, Sobacchi C, Grassi F, Villa A. Of Omenn and mice. Trends Immunol. 2008;29:133–140. doi: 10.1016/j.it.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Melchior Benoit, Garcia Angie E, Hsiung Bor-Kai, Lo Katherine M, Doose Jonathan M, Thrash J Cameron, Stalder Anna K, Staufenbiel Matthias, Neumann Harald, Carson Monica J. Dual induction of TREM2 and tolerance-related transcript, Tmem176b, in amyloid transgenic mice: implications for vaccine-based therapies for Alzheimer’s disease. ASN Neuro. 2010;2(3):e00037. doi: 10.1042/AN20100010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner JD, Fasth A, Etzioni A. Autoimmunity in severe combined immunodeficiency (SCID): lessons from patients and experimental models. J Clin Immunol. 2008;28(s1):s29–s33. doi: 10.1007/s10875-007-9159-y. [DOI] [PubMed] [Google Scholar]
- Müller M, Carter S, Hofer MJ, Campbell IL. Review: The chemokine receptor CXCR3 and its ligands CXCL9, CXCL10 and CXCL11 in neuroimmunity--a tale of conflict and conundrum. Neuropathology and Applied Neurobiology. 2010 August;36(5):368–387. doi: 10.1111/j.1365–2990.2010.01089.x. [DOI] [PubMed] [Google Scholar]
- Noor Shahani, Habashy Andrew S, Nance J Philip, Clark Robin T, Nemati Kiav, Carson Monica J, Wilson Emma H. CCR7-dependent immunity during acute Toxoplasma gondii infection. Infection and Immunity. 2010 May;78(5):2257–2263. doi: 10.1128/IAI.01314-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploix C, Lo D, Carson MJ. A ligand for the chemokine receptor CCR7 can influence the homeostatic proliferation of CD4 T cells and progression of autoimmunity. Journal of Immunology (Baltimore, Md: 1950) 2001 December 15;167(12):6724–6730. doi: 10.4049/jimmunol.167.12.6724. [DOI] [PubMed] [Google Scholar]
- Corinne Ploix, Zuberi Riaz I, Liu Fu-Tong, Carson Monica J, Lo David D. Induction and effector phase of allergic lung inflammation is independent of CCL21/CCL19 and LT-beta. International Journal of Medical Sciences. 2009;6(2):85–92. doi: 10.7150/ijms.6.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid Christoph D, Melchior Benoit, Masek Kokoechat, Puntambekar Shweta S, Danielson Patria E, Lo David D, Sutcliffe J Gregor, Carson Monica J. Differential gene expression in LPS/IFNγamma activated microglia and macrophages: in vitro versus in vivo. Journal of Neurochemistry. 2009 May;109(Suppl 1):117–125. doi: 10.1111/j.1471–4159.2009.05984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann Kathrin, Lämmermann Tim, Bruckner Markus, Legler Daniel F, Polleux Julien, Spatz Joachim P, Schuler Gerold, et al. Immobilized chemokine fields and soluble chemokine gradients cooperatively shape migration patterns of dendritic cells. Immunity. 2010 May 28;32(5):703–713. doi: 10.1016/j.immuni.2010.04.017. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Cohen IR. Autoimmunity can benefit self-maintenance. Immunology Today. 2000 June;21(6):265–268. doi: 10.1016/s0167-5699(00)01633-9. [DOI] [PubMed] [Google Scholar]
- Schwartz Michal, Kipnis Jonathan. Model of acute injury to study neuroprotection. Methods in Molecular Biology (Clifton, NJ) 2007;399:41–53. doi: 10.1007/978-1-59745-504-6_4. [DOI] [PubMed] [Google Scholar]
- Stalder AK, Carson MJ, Pagenstecher A, Asensio VC, Kincaid C, Benedict M, Powell HC, Masliah E, Campbell IL. Late-onset chronic inflammatory encephalopathy in immune-competent and severe combined immune-deficient (SCID) mice with astrocyte-targeted expression of tumor necrosis factor. The American Journal of Pathology. 1998 September;153(3):767–783. doi: 10.1016/S0002-9440(10)65620-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrash J Cameron, Torbett Bruce E, Carson Monica J. Developmental regulation of TREM2 and DAP12 expression in the murine CNS: implications for Nasu-Hakola disease. Neurochemical Research. 2009 January;34(1):38–45. doi: 10.1007/s11064-008-9657-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toulme Estelle, Garcia Angie, Samways Damien, Egan Terrance M, Carson Monica J, Khakh Baljit S. P2X4 receptors in activated C8-B4 cells of cerebellar microglial origin. The Journal of General Physiology. 2010 April;135(4):333–353. doi: 10.1085/jgp.200910336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson Emma H, Harris Tajie H, Mrass Paulus, John Beena, Tait Elia D, Wu Gregory F, Pepper Marion, et al. Behavior of parasite-specific effector CD8+ T cells in the brain and visualization of a kinesis-associated system of reticular fibers. Immunity. 2009 February 20;30(2):300–311. doi: 10.1016/j.immuni.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest. 2010;120:1368–1379. doi: 10.1172/JCI41911. [DOI] [PMC free article] [PubMed] [Google Scholar]