

Abstract
Nimustine (ACNU) is a chloroethylating agent which was the most active chemotherapy agent used for patients with high‐grade gliomas until the introduction of temozolomide, which became the standard of care for patients with newly diagnosed glioblastomas in Japan. Since temozolomide was established as the standard first‐line therapy for glioblastoma multiforme (GBM), ACNU has been employed as a salvage chemotherapy agent for recurrent GBM in combination with other drugs. The acting molecular mechanism in ACNU has yet to be elucidated. ACNU is a cross‐linking agent which induces DNA double‐strand breaks (DSBs). The work described here was intended to clarify details in repair pathways which are active in the repair of DNA DSBs induced by ACNU. DSBs are repaired through the homologous recombination (HR) and non‐homologous end‐joining (NHEJ) pathways. Cultured mouse embryonic fibroblasts were used which have deficiencies in DNA DSB repair genes which are involved in HR repair (X‐ray repair cross‐complementing group 2 [XRCC2] and radiation sensitive mutant 54 [Rad54]), and in NHEJ repair (DNA ligase IV [Lig4]). Cellular sensitivity to ACNU treatment was evaluated with colony forming assays. The most effective molecular target which correlated with ACNU cell sensitivity was Lig4. In addition, it was found that Lig4 small‐interference RNA (siRNA) efficiently enhanced cell lethality which was induced by ACNU in human glioblastoma A172 cells. These findings suggest that the down‐regulation of Lig4 might provide a useful tool which can be used to increase cell sensitivity in response to ACNU chemotherapy. (Cancer Sci 2010)
High‐grade gliomas remain a therapeutic challenge. In the past, nitrosourea drugs such as nimustine (ACNU) in Japan and central Europe, or carmustine (BCNU) in the USA, were the standard drugs used in addition to radiation. This has changed since the introduction of temozolomide (TMZ) was shown to provide benefits with lower levels of toxicity.( 1 ) However, a recent meta‐analysis has suggested the possibility of a significant survival gain for the use of ACNU for newly diagnosed high‐grade gliomas.( 2 ) The therapeutic value weights of previously used drugs when compared with TMZ and with each other is still unknown. However, the widespread use of TMZ in patients with newly diagnosed disease, primarily glioblastomas, resulted in a reevaluation of nitrosoureas for tumor progression or for tumor recurrences.
ACNU and BCNU are both chloroethylating agents. Following cellular exposure to a chloroethylating agent, a chloroethyl group is transferred to the O 6 ‐position on guanine (G) residues in DNA, and this O 6 ‐chloroethylG is repaired by the activity of O 6 ‐methylG‐DNA methyltransferase (MGMT).( 3 ) Elevated levels of MGMT have been blamed for much of the observed resistance to chloroethylating agents,( 4 ) but it has also been suggested that repair of O 6 ‐chloroethylG by MGMT is not the sole mechanism responsible for resistance to chloroethylating agents.( 5 ) Kaina et al. ( 6 ) reported that about 5% of all solid tumors assayed in their laboratory completely lacked MGMT. Among gliomas the frequency of tumors lacking MGMT is 17–30%.( 7 , 8 ) Therefore, if only MGMT is targeted, improvements in drug efficacy are likely to be limited. A new target which can improve chemotherapy results obtained with chloroethylating agents is now being sought.
DNA double‐strand breaks (DSBs) are generated in wild‐type cells and in other cell culture systems in response to treatments with chloroethylating agents.( 9 ) Since DSBs are likely to be the final event leading to cell death, it would thus be expected that cells which are defective in DSB repair should be more sensitive to chloroethylating agents. DSBs are repaired through the homologous recombination (HR) and non‐homologous end‐joining (NHEJ) pathways.( 10 ) In human cells, the proteins involved in HR include members of the MRN complex (consisting of meiotic recombination 11 [MRE11]/radiation sensitive mutant 50 [Rad50]/Nijmegen breakage syndrome 1 [NBS1]), Rad51, the Rad51 paralogs (Rad51B, Rad51C, Rad51D, X‐ray repair cross‐complementing group 2 [XRCC2], and XRCC3), Rad54, and Rad54B.( 10 ) Proteins involved in the NHEJ pathway include Ku70/80, DNA‐dependent protein kinase catalytic subunit (DNA‐PKcs), DNA ligase IV (Lig4), XRCC4, and Artemis.( 10 )
The work described here was designed to lead to a better understanding of the details involved in DSB repair pathways which respond to ACNU sensitivity. The activity of specific components of HR repair (XRCC2 and Rad54) and NHEJ repair (Lig4) leading to the repair of ACNU‐induced DNA damage were assessed using clonogenic survival assays. A panel of p53 tumor suppressor gene knockout mouse embryonic fibroblast cell lines (MEFs) was used which contained cells which were defective in specific components in these repair pathways (XRCC2, Rad54, and Lig4).
To examine if the resulting observations were applicable to glioma cells, targeted repair pathways were down‐regulated using small‐interference RNA (siRNA), and the sensitivity of human glioblastoma A172 cells to ACNU was measured in the presence of siRNAs. In order to determine if DSBs were formed in response to ACNU, the expression of γH2AX was monitored at different times following treatment with ACNU in cells deficient in specific repair pathway components, and in the corresponding parental cells. Hopefully, an understanding of the DNA repair mechanisms which have been identified as contributing to ACNU resistance in this study will lead to the development of new tools or methods which can be utilized to improve drug efficacy.
Materials and Methods
Cell lines. The cell lines used in these studies were the MEF cell lines XRCC2−/−p53−/− (XRCC2−/−); XRCC2+/+p53−/− (XRCC2+/+); Rad54−/−Lig4+/+p53−/− (Rad54−/−); Rad54+/+Lig4−/−p53−/− (Lig4−/−); Rad54−/−Lig4−/−p53−/− (Rad54−/−Lig4−/−); and Rad54+/+Lig4+/+p53−/− (Rad54+/+Lig4+/+). Human glioblastoma A172 cells were purchased from the American Type Culture Collection of Cell Cultures (Manassas, VA, USA). Cells were cultured in DMEM‐10 (Dulbecco’s modified Eagle’s medium containing 10% [v/v] fetal bovine serum, 20 mM 2‐[4‐(2‐hydroxyethyl)‐1‐piperazinyl] ethanesulfonic acid, 50 units/mL penicillin, 50 μg/mL streptomycin, and 50 μg/mL kanamycin). Cells were cultured at 37°C in a conventional humidified CO2 incubator.
Drugs and drug treatments. ACNU (Sigma‐Aldrich, St. Louis, MO, USA) was dissolved at a stock concentration of 10 mM in sterile H2O. ACNU stock solutions were stored at −20°C until used. Cisplatin (provided by Nihonkayaku, Tokyo, Japan) was dissolved at a stock concentration of 100 mM in dimethylsulfoxide. Cisplatin stock solutions were stored at −80°C until used. For experimental procedures, cells were grown in medium containing ACNU or cisplatin at various concentrations for 3 h, and then rinsed twice with phosphate‐buffered saline.
Colony forming assays. Cell survival was measured using a standard clonogenic survival assay as previously described.( 11 )
Flow cytometry. To determine whether DSBs were formed in response to ACNU exposures, and how many DSBs were formed, the overall levels of phosphorylated H2AX (γH2AX) were measured with flow cytometry as previously described.( 11 )
siRNA transfection. siRNA for human XRCC2 (product name: si GENOME SMART pool M‐004361‐01‐0005) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). siRNA (50 nM) for human Lig4, XRCC2, or for non‐specific negative controls was transfected into human glioblastoma A172 cells as previously described.( 12 ) The cells were then trypsinized for colony forming assays and proteins were extracted for western blot analysis.
Western blot analysis. Total cellular protein amounts were quantified with a Bio‐Rad protein assay kit (Bio‐Rad Labs, Richmond, CA, USA). Aliquots of proteins (20 μg) were subjected to western blot analyses. After electrophoresis on 10% polyacrylamide gels containing 0.1% SDS, the proteins were transferred electrophoretically onto Poly Screen PVDF membranes (Dupont/Biotechnology Systems, NEN Research Products, Boston, MA, USA). The membranes were then incubated with mouse monoclonal anti‐Lig4 antibody (M02; Abnova, Taipei, Taiwan), mouse monoclonal anti‐XRCC2 antibody (2H4; Novus Biologicals, Littleton, CO, USA), and goat polyclonal anti‐actin antibody (I‐19; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Horseradish peroxidase‐conjugated anti‐mouse IgG antibody (Zymed Laboratories, San Francisco, CA, USA) was used for Lig4 and XRCC2. Horseradish peroxidase‐conjugated anti‐goat IgG antibody (Zymed Laboratories) was used for actin. For visualization of the bands, the ECL Plus Western Blotting Detection System (GE Healthcare UK, Buckinghamshire, England) was used according to the manufacturer’s protocol. The amounts of the proteins in the samples were quantified by scanning profiles using the Scion imaging program (Scion, Frederick, MD, USA).
Statistical analysis. Statistical analysis was performed using the Student’s t‐test.
Results
Repair genes responding to ACNU‐induced DNA damage. In this study, in order to understand the relative contributions of the HR and NHEJ repair pathways for ACNU‐induced DNA damage, cellular responses to ACNU were examined using clonogenic survival assays after a 3 h exposure to ACNU. In these studies, XRCC2 defective cells (Fig. 1a), and Rad54 and/or Lig4 defective cells were used (Fig. 1b). The sensitivity of each cell line was assessed from its D 30 value, i.e. from the ACNU dose which reduced cell survival to 30% (Table 1). Each D 30 value was calculated from the cell survival data shown in Figure 1(a,b). All repair defective cells were more sensitive to ACNU than the corresponding proficient cells (Fig. 1a,b). Also, Rad54−/−Lig4−/− cells were more sensitive to ACNU than were Rad54−/− cells or Lig4−/− cells (Fig. 1b).
Figure 1.
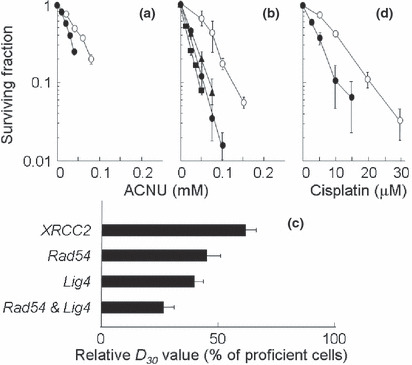
Sensitivity to nimustine (ACNU) or cisplatin. (a–c) ACNU, (d) cisplatin. (a) ○, XRCC2+/+ cells; •, XRCC2−/− cells. (b,d) ○, Rad54+/+Lig4+/+ cells; , Rad54−/− cells; , Rad54−/−Lig4−/− cells; •, Lig4−/− cells. (c) Relative D 30 value (% compared to proficient cells). Each point represents the mean of three independent experiments; bars indicate the SD. Lig4, DNA ligase IV; Rad54, radiation sensitive mutant 54; XRCC2, X‐ray repair cross‐complementing group 2.
Table 1.
Sensitivity of each cell line to ACNU assessed by D 30 value
| Genes | D 30 (μM) | |
|---|---|---|
| Proficient cells | Deficient cells | |
| XRCC2 | 57.7 | 34.5 |
| Rad54 | 83 | 37 |
| Lig4 | 32.6 | |
| Rad54 & Lig4 | 21.6 | |
Each D 30 value (i.e. the dose that reduces cell survival to 30%) was calculated from results of the cell survival data shown in Figure 1(a,b). ACNU, nimustine; Lig4, DNA ligase IV; Rad54, radiation sensitive mutant 54; XRCC2, X‐ray repair cross‐complementing group 2.
In order to accurately compare ACNU sensitivities in the repair defective cell lines, the relative D 30 values were normalized using the D 30 value of the corresponding proficient cell lines. The relative D 30 values listed sequentially in the order in which they increase (reflecting decreasing sensitivities to ACNU) are: Rad54−/−Lig4−/− cells < Lig4−/− cells < Rad54−/− cells < XRCC2−/− cells (Fig. 1c). These differences were statistically significant except for the difference between Lig4−/− cells and Rad54−/− cells. However in cell lines which were defective in a single repair gene, the relative D 30 value of the Lig4 defective cells was the smallest after treatment with ACNU reflecting their high sensitivity to ACNU. When Lig4−/− cells were treated with cisplatin which is as effective a cross‐linking agent as ACNU, similar hypersensitivities were observed in these cells (Fig. 1d).
Lig4 activity in the repair of ACNU‐induced DSBs. To determine whether DSBs are formed in response to exposures to ACNU, and how many DSBs are formed, overall levels of γH2AX were measured with flow cytometry. γH2AX is formed in response to the presence of DSBs( 10 ) in Lig4−/− cells and in the corresponding proficient cells, and the level of γH2AX is different at different times following treatment with ACNU. In proficient cells, γH2AX levels did not vary much from their initial levels at 12 and 24 h after ACNU treatment. However, in Lig4−/− cells, γH2AX levels increased more than 4‐fold at 12 h and 6‐fold at 24 h after ACNU treatment when compared to their initial levels. At 12 and 24 h after treatment with ACNU, there were significant differences in γH2AX levels between the proficient and deficient Lig4 cell lines (Fig. 2).
Figure 2.
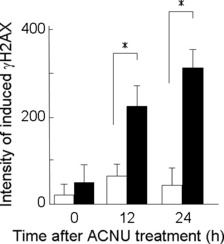
Phosphorylation of H2AX after nimustine (ACNU) treatment with 60 μM ACNU in proficient cells (open columns) or in Lig4−/− cells (closed columns). Columns show the mean of three independent experiments; bars indicate the SD. An asterisk (*) indicates that the difference is statistically significant (P < 0.05).
Effect of silenced Lig4 activity on A172 glioblastoma cell sensitivity to ACNU. A172 cells were transfected with Lig4 or XRCC2 siRNA for 48 h, and the expression of these proteins was observed using western blots (Fig. 3a). The level of Lig4 proteins decreased to 68% as compared with negative control. The level of XRCC2 proteins decreased to 62% as compared with negative control. Lig4 siRNA leads to a slightly lower expression (95%) of the XRCC2 protein when compared with the negative control. X‐ray repair cross‐complementing group 2 (XRCC2) siRNA leads to a slightly decreased expression (95%) of the Lig4 protein when compared with the negative control.
Figure 3.

siRNA silencing of DNA ligase IV (Lig4) or X‐ray repair cross‐complementing group 2 (XRCC2) in glioblastoma A172 cells. (a) Expression analysis of Lig4 and XRCC2 in cells transfected with each type of siRNA using western blots. Actin was used as a loading control, and the relative ratios of protein were normalized using actin levels. (b) Effect of siRNA silencing of Lig4 or XRCC2 on sensitivity to ACNU. ○, negative control siRNA; , XRCC2 siRNA; •, Lig4 siRNA. Each point represents the mean of three independent experiments; bars indicate the SD.
After an ACNU treatment at each concentration, Lig4 or XRCC2 silencing caused a remarkable reduction in colony formation ability in each case when compared to the negative control cells transfected with siRNA (Fig. 3b). However, in comparing Lig4 siRNA with XRCC2 siRNA, the Lig4 siRNA resulted in a significantly more effective sensitization towards ACNU than did XRCC2 siRNA (Fig. 3b).
Discussion
The data in this paper provide the first evidence that both HR and NHEJ play a critical role in the repair of ACNU‐mediated DNA damage, and observations of the relative D 30 values support this idea. The parental cells from which the Rad54−/− and Lig4−/− cells had the same genetic background as the defective cells, but the relative D 30 values of the Rad54−/− cells and the Lig4−/− cells were 44.6% and 39.5% of the parental cells, respectively (Fig. 1c). In addition, the sensitivity of Rad54−/−Lig4−/− cells to ACNU was higher than that of cells defective in either gene alone, which indicates that the HR and NHEJ double knockout cells displayed an additive effect from these two knockouts (Fig. 1b,c). These results clearly show the importance of both HR and NHEJ in the repair of ACNU‐induced DNA damage. Results for XRCC2 (Fig. 1a) are in agreement with previous studies which revealed that XRCC2 knockout MEFs showed a hypersensitivity to other DNA interstrand cross‐linking (ICL) agents such as fotemustine, cisplatin, and mitomycin C.( 13 ) Although it was reported that DT40 cells appear to possess a significantly higher HR repair efficiency than any mammalian cell line, and that NHEJ plays only a minor role in ICL repair,( 14 ) the data here support the idea that not only HR, but also NHEJ both play a major role in protecting cells against ACNU‐induced DNA lesions. It is assumed that DSBs induced not only by ACNU, but also by cisplatin, are repaired by Lig4 (Fig. 1d). DNA–DNA crosslinks might result in the production of DSBs during the replication process, and these DSBs could be repaired by Lig4.
DSB repair pathways for ACNU‐induced DNA damage are summarized in Figure 4. Following cellular exposures to ACNU, O 6‐ G is chloroethylated and transformed into O 6 ‐chloroethylG. If this O 6 ‐chloroethylG is not repaired by MGMT, this adduct is unstable, and undergoes an intramolecular rearrangement leading to an intermediary N 1 ‐O 6 ‐ethanoG. The N 1 ‐O 6 ‐ethanoG adduct may react with cytosine in the complementary strand to yield a highly toxic DNA–DNA cross‐link between position 1 in the guanine and position 3 in the cytosine (1‐(3‐cytosinyl)‐2‐(1‐guanosinyl)‐ethane).( 15 ) The mechanism involved in the repair of this type of DNA ICL seems to involve a combination of Fanconi anemia (FA) proteins and nucleotide excision repair (NER) factors.( 16 ) It has been suggested that different FA proteins are involved in positive and negative regulation which determines which repair pathway is activated (HR or NHEJ).( 17 ) However, the details of this repair pathway remain to be elucidated. The XRCC2 protein plays a role in HR via its interaction with Rad51.( 18 ) Therefore, it may be argued that XRCC2 must be required for the Rad51 mediated strand invasion step to occur during HR. Without strand invasion of the homologous DNA, tolerance of the replication blocking lesion cannot occur. The Rad54 protein interacts with Rad51 directly during the HR process after the induction of DNA damage in mammalian cells.( 19 ) In the NHEJ pathway, after DSB formation, the Ku70/80 heterodimer binds to the damaged DNA ends. This facilitates the recruitment of the DNA‐PKcs to the DSB. The sequential binding of these proteins activates the phosphorylation function of the DNA‐PKcs which then phosphorylates itself, the Ku heterodimer, and other proteins involved in cell cycle regulation.( 20 ) It has been suggested that Ku70/80 might also function as an alignment factor which binds DSB ends, and can thus provide ready access for, and greatly stimulate the functioning of, the Lig4–XRCC4 complex. This can increase the efficiency and accuracy of NHEJ.( 21 ) The Lig4–XRCC4 complex then rejoins the juxtaposed DNA ends. In conclusion, both HR and NHEJ play an important role in the repair of ACNU‐mediated DNA damage. However, among the single repair gene defective cells, the D 30 value of the Lig4−/−cells was the smallest, so it appears that Lig4 activity could provide a new molecular target for ACNU chemotherapy.
Figure 4.
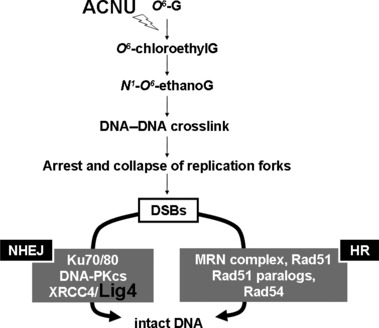
DNA double‐strand breaks (DSBs) repair pathways for nimustine (ACNU)‐induced DNA damage. DNA‐PKcs, DNA‐dependent protein kinase catalytic subunit; HR, homologous recombination; Lig4, DNA ligase IV; MRN, MRE11, Rad50, NBS1; NHEJ, non‐homologous end‐joining; Rad 51, radiation sensitive mutant 51; XRCC4, X‐ray repair cross‐complementing group 4.
The data presented here suggests that Lig4 can generate cellular resistance to ACNU exposure by repairing lesions which trigger the activation of DNA damage response cascades. H2AX, a histone protein, is rapidly phosphorylated at Ser139 when DSBs are introduced in mammalian cell DNA in response to damage and replication fork collapse.( 22 , 23 , 24 ) Many of the early components in the DNA damage response pathway co‐localize with γH2AX at sites of DNA breaks.( 25 , 26 ) Therefore, the detection and quantitation of γH2AX is a useful tool to monitor the induction of DNA damage response signaling pathways. In Lig4 proficient cells, ACNU‐induced DSBs showed small increases at 12 and 24 h after ACNU treatment. However, in Lig4−/− cells DSBs accumulated more than 4‐fold at 12 h and 6‐fold at 24 h after ACNU treatment when compared with their initial levels (Fig. 2). The data indicate that in Lig4−/− cells, more DSBs were produced from ICL processing and left unrepaired, whereas in the corresponding proficient cells, DSBs were repaired. This data is consistent with the enhanced sensitivity of Lig4−/− cells to ACNU when compared with the corresponding proficient cells (Fig. 1c). The formation of DSBs after ICL induction is a possible consequence of stalled replication forks during S phase.( 27 )
Lig4 could be a more effective target than XRCC2 during ACNU treatments for glioblastoma cells (Fig. 3b). It was reported that A172 cells exhibit very low levels of MGMT activity.( 28 ) This suggests that Lig4 down‐regulation could potentially be a useful strategy for augmenting the therapeutic effects of ACNU in treatments for glioblastoma cells.
For ACNU chemotherapy, the focus was on DSBs which are induced by DNA–DNA crosslinks following ACNU treatment (Fig. 4), and it was demonstrated that the depression of Lig4 can enhance the sensitivity of glioblastoma cells to ACNU (Fig. 3b). From the results reported in this study, it appears that Lig4 activity could provide a new molecular target to sensitize cells to ACNU. Previously, we also found that Lig4 could be the molecular target for TMZ.( 12 ) In view of the work shown here, it appears likely that Lig4 contributes significantly towards the repair of ACNU‐induced DSBs, and that modulating Lig4 activity could enhance cellular sensitivity to chemotherapeutic agents such as ACNU as well as TMZ.
Acknowledgments
This work was supported by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan. This work was also funded in part by a grant from the Central Research Institute of the Electric Power Industry in Japan, and by a Grant for Exploratory Research for Space Utilization from the Japan Space Forum. The authors thank Dr G. Iliakis (University of Duisburg‐Essen Medical School, Essen, Germany) and Dr Y. Takagi (Fukuoka Dental College, Fukuoka, Japan) for support, and thank Dr F.W. Alt (Howard Hughes Medical Institute, The Children’s Hospital, Boston, MA, USA) for kindly providing Rad54−/− and/or Lig4−/− cells.
Disclosure Statement
The authors have no conflict of interest.
References
- 1. Stupp R, Mason WP, Van Den Bent MJ et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352: 987–96. [DOI] [PubMed] [Google Scholar]
- 2. Wolff JE, Berrak S, Koontz Webb SE et al. Nitrosourea efficacy in high‐grade glioma: a survival gain analysis summarizing 504 cohorts with 24193 patients. J Neurooncol 2008; 88: 57–63. [DOI] [PubMed] [Google Scholar]
- 3. Gonzaga PE, Potter PM, Niu TQ et al. Identification of the cross‐link between human O 6 ‐methylguanine‐DNA methyltransferase and chloroethylnitrosourea‐treated DNA. Cancer Res 1992; 52: 6052–8. [PubMed] [Google Scholar]
- 4. Kokkinakis DM, Bocangel DB, Schold SC, Moschel RC, Pegg AE. Thresholds of O 6 ‐alkylguanine‐DNA alkyltransferase which confer significant resistance of human glial tumor xenografts to treatment with 1,3‐bis(2‐chloroethyl)‐1‐nitrosourea or temozolomide. Clin Cancer Res 2001; 7: 421–8. [PubMed] [Google Scholar]
- 5. Bobola MS, Berger MS, Silber JR. Contribution of O 6 ‐methylguanine‐DNA methyltransferase to resistance to 1,3‐(2‐chloroethyl)‐1‐nitrosourea in human brain tumor derived cell lines. Mol Carcinog 1995; 13: 81–8. [DOI] [PubMed] [Google Scholar]
- 6. Kaina B, Christmann M, Naumann S, Roos WP. MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair (Amst) 2007; 6: 1079–99. [DOI] [PubMed] [Google Scholar]
- 7. Preuss I, Eberhagen I, Haas S et al. O 6 ‐methylguanine‐DNA methyltransferase activity in breast and brain tumors. Int J Cancer 1995; 61: 321–6. [DOI] [PubMed] [Google Scholar]
- 8. Lees NP, Harrison KL, Hill E, Hall CN, Povey AC, Margison GP. Heterogeneity of O 6 ‐alkylguanine‐DNA alkyltransferase activity in colorectal cancer: implications for treatment. Oncology 2002; 63: 393–7. [DOI] [PubMed] [Google Scholar]
- 9. Batista LF, Roos WP, Christmann M, Menck CF, Kaina B. Differential sensitivity of malignant glioma cells to methylating and chloroethylating anticancer drugs: p53 determines the switch by regulating xpc, ddb2, and DNA double‐strand breaks. Cancer Res 2007; 67: 11886–95. [DOI] [PubMed] [Google Scholar]
- 10. Ohnishi T, Mori E, Takahashi A. DNA double‐strand breaks: Their production, recognition, and repair in eukaryotes. Mutat Res 2009; 669: 8–12. [DOI] [PubMed] [Google Scholar]
- 11. Takahashi A, Matsumoto H, Nagayama K et al. Evidence for the involvement of double‐strand breaks in heat‐induced cell killing. Cancer Res 2004; 64: 8839–45. [DOI] [PubMed] [Google Scholar]
- 12. Kondo N, Takahashi A, Mori E et al. DNA ligase IV as a new molecular target for temozolomide. Biochem Biophys Res Commun 2009; 387: 656–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsaryk R, Fabian K, Thacker J, Kaina B. Xrcc2 deficiency sensitizes cells to apoptosis by MNNG and the alkylating anticancer drugs temozolomide, fotemustine and mafosfamide. Cancer Lett 2006; 239: 305–13. [DOI] [PubMed] [Google Scholar]
- 14. Nojima K, Hochegger H, Saberi A et al. Multiple repair pathways mediate tolerance to chemotherapeutic cross‐linking agents in vertebrate cells. Cancer Res 2005; 65: 11704–11. [DOI] [PubMed] [Google Scholar]
- 15. Margison GP, Santibañez‐Koref MF. O 6 ‐alkylguanine‐DNA alkyltransferase: role in carcinogenesis and chemotherapy. Bioessays 2002; 24: 255–66. [DOI] [PubMed] [Google Scholar]
- 16. McHugh PJ, Spanswick VJ, Hartley JA. Repair of DNA interstrand cross‐links: molecular mechanisms and clinical relevance. Lancet Oncol 2001; 2: 483–90. [DOI] [PubMed] [Google Scholar]
- 17. Donahue SL, Lundberg R, Saplis R, Campbell C. Deficient regulation of DNA double‐strand break repair in Fanconi anemia fibroblasts. J Biol Chem 2003; 278: 29487–95. [DOI] [PubMed] [Google Scholar]
- 18. Shim KS, Schmutte C, Tombline G, Heinen CD, Fishel R. hXRCC2 enhances ADP/ATP processing and strand exchange by hRAD51. J Biol Chem 2004; 279: 30385–94. [DOI] [PubMed] [Google Scholar]
- 19. Tan TL, Essers J, Citterio E et al. Mouse Rad54 affects DNA conformation and DNA‐damage‐induced Rad51 foci formation. Curr Biol 1999; 9: 325–8. [DOI] [PubMed] [Google Scholar]
- 20. Weterings E, Van Gent DC. The mechanism of non‐homologous end‐joining: a synopsis of synapsis. DNA Repair (Amst) 2004; 3: 1425–35. [DOI] [PubMed] [Google Scholar]
- 21. Thode S, Schafer A, Pfeiffer P, Vielmetter W. A novel pathway of DNA end‐to‐end joining. Cell 1990; 60: 921–8. [DOI] [PubMed] [Google Scholar]
- 22. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double‐stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998; 273: 5858–68. [DOI] [PubMed] [Google Scholar]
- 23. Takahashi A, Ohnishi T. Does γH2AX foci formation depend on the presence of DNA double strand breaks? Cancer Lett 2005; 229: 171–9. [DOI] [PubMed] [Google Scholar]
- 24. Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR‐dependent manner in response to replicational stress. J Biol Chem 2001; 276: 47759–62. [DOI] [PubMed] [Google Scholar]
- 25. Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 2000; 10: 886–95. [DOI] [PubMed] [Google Scholar]
- 26. Takahashi A, Yamakawa N, Kirita T et al. DNA damage recognition proteins localize along heavy ion induced tracks in the cell nucleus. J Radiat Res (Tokyo) 2008; 49: 645–52. [DOI] [PubMed] [Google Scholar]
- 27. Bessho T. Induction of DNA replication‐mediated double strand breaks by psoralen DNA interstrand cross‐links. J Biol Chem 2003; 278: 5250–4. [DOI] [PubMed] [Google Scholar]
- 28. Hermisson M, Klumpp A, Wick W et al. O 6‐methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J Neurochem 2006; 96: 766–76. [DOI] [PubMed] [Google Scholar]