

Abstract
Metal surfaces in contact with water, surfactants and biopolymers experience attractive polarization owing to induced charges. This fundamental physical interaction complements stronger epitaxial and covalent surface interactions and remains difficult to measure experimentally. We present a first step to quantify polarization on even gold (Au) surfaces in contact with water and with aqueous solutions of peptides of different charge state (A3 and Flg-Na3) by molecular dynamics simulation in all-atomic resolution and a posteriori computation of the image potential. Attractive polarization scales with the magnitude of atomic charges and with the length of multi-poles in the aqueous phase such as the distance between cationic and anionic groups. The polarization energy per surface area is similar on aqueous Au {1 1 1} and Au {1 0 0} interfaces of approximately −50 mJ m−2 and decreases to −70 mJ m−2 in the presence of charged peptides. In molecular terms, the polarization energy corresponds to −2.3 and −0.1 kJ mol−1 for water in the first and second molecular layers on the metal surface, and to between −40 and 0 kJ mol−1 for individual amino acids in the peptides depending on the charge state, multi-pole length and proximity to the surface. The net contribution of polarization to peptide adsorption on the metal surface is determined by the balance between polarization by the peptide and loss of polarization by replaced surface-bound water. On metal surfaces with significant epitaxial attraction of peptides such as Au {1 1 1}, polarization contributes only 10–20% to total adsorption related to similar polarity of water and of amino acids. On metal surfaces with weak epitaxial attraction of peptides such as Au {1 0 0}, polarization is a major contribution to adsorption, especially for charged peptides (−80 kJ mol−1 for peptide Flg-Na3). A remaining water interlayer between the metal surface and the peptide then reduces losses in polarization energy by replaced surface-bound water. Computed polarization energies are sensitive to the precise location of the image plane (within tenths of Angstroms near the jellium edge). The computational method can be extended to complex nanometre and micrometer-size surface topologies.
Keywords: metal nanostructures, induced charges, image potential, molecular dynamics, biomaterials, proteins
1. Introduction
The interaction of proteins, peptides and surfactants with metal surfaces is of great promise for the assembly of metal nanostructures with potential applications in nanoelectronics, sensors, medical technology and bioinspired materials (e.g. [1–12]). Peptides with high affinity to Au and Pd surfaces have been identified by phage display techniques [2,3,5,6,8,10] and specific non-covalent binding can be explained by complementarity of the molecular structure of the peptide to epitaxial face-centred cubic (FCC) lattice sites on the metal surface [12]. Accordingly, the binding strength of the peptide correlates with the surface energy of the metal and with competitive epitaxial binding preferences of sp2 and sp3 hybridized groups in comparison to solvent molecules on a given metal surface [12–14]. This concept of molecular epitaxy also explains the influence of curvature and roughness of metal nanoparticles on differential peptide adsorption [15]. Alternatively, covalently assisted binding is possible through R–S–Aun bonds of Cys-containing peptides and thiols to Au surfaces.
A yet much less considered contribution to the binding strength is polarization of the metal surface through the typically polar and often ionic environment created by solvents, surfactants, peptides or DNA. It has long been known that electrons and other point charges experience an attractive image potential near metal surfaces [16–30] and numerous reports explain the effect of point charges and single molecules on metal surfaces in vacuum and in the gas phase [16–21,24–28]. Yet few studies deal with polarization in solution or in the solid state [17,22,23,29,30], and approximations such as hard sphere electrolytes as well as the focus on interfacial capacity have not provided quantitative thermodynamic insight related to the competitive adsorption of organic and biological matter onto metal nanostructures in aqueous solution. The lack of quantitative knowledge of attraction mediated by induced charges is also related to the difficulty to measure such effects as separate contributions to adsorption experimentally and, thus, shortage of experimental data. At the same time, the impact of polarization on the self-assembly of surfactants and biomolecules on metal surfaces is undeniable, especially on metal surfaces of irregular geometry. The aim of our contribution is a first quantitative evaluation of the effects of induced charges on metal surfaces using a physically justified computational approach for systems up to millions of atoms on nanosecond timescales. We will discuss the impact of the image potential as an additional contribution to epitaxial and covalent binding on even metal surfaces with water, peptides of different charge state and amino acids.
Attractive polarization occurs in response to exterior charges in the vicinity of a grounded metal surface. Electrons enter or exit through the ground, or free valence electrons redistribute at the metal surface without grounding, to maintain the interior of the metal free of electric fields (figure 1). The induced, complementary electric field can be thought of as mediated by image charges [16,19]. According to a classical continuum model for distances z − zSf >2.5 Å from the top atomic layer of the metal surface, the attractive image potential Eim felt by a charged species q is proportional to 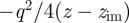 [16,27,28]. The image plane zim is located close to the jellium edge
[16,27,28]. The image plane zim is located close to the jellium edge 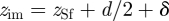 with a small system-dependent offset δ ([16,27,28]; figure 1a). At distances z − zSf <2.5 Å, the classical continuum model overestimates the attraction and alternative models such as discrete classical models perform better [27,28].
with a small system-dependent offset δ ([16,27,28]; figure 1a). At distances z − zSf <2.5 Å, the classical continuum model overestimates the attraction and alternative models such as discrete classical models perform better [27,28].
Figure 1.

Concept of induced charge and image potential on an Au {1 1 1} surface as example (a) in vacuum and (b) in contact with a peptide solution. (a) In vacuum, a point charge +q causes opposite charges to enter through the ground and locate at the surface of the metal (pink negative charges) to generate an electrical field opposite to that created by +q. The image potential can be calculated assuming a classical image charge −q [16,19]. Positions of the jellium edge (broken line) and of the image plane (solid line) are shown. (b) In contact with a peptide solution, we find a dense collection of multi-poles which is overall charge-neutral but typically of non-zero dipole moment on time average. The electrons in the metal rearrange similarly at the surface to counterbalance the local electric field at the solution interface (grounding is not required). The image potential can be computed on the assumption of a mirror image of the collection of all atomic charges (shaded in pink). δV and δI represent system-dependent small offsets between the image plane and the jellium edge in vacuum and in the condensed phase. zSol and zSf represent the average position of the first atomic layer of the solution and of the metal at the interface. The arithmetic mean (‘interface edge’) characterizes the position of the image plane which maps the first atomic layer of the solution onto the first atomic layer of the metal.
To analyse the contribution of induced charges to adsorption of water and peptides on metal surfaces, we use example systems which have been characterized in experiment [5,8,10] and by molecular dynamics (MD) simulation under neglect of polarization [12,31]. Using these previously reported equilibrium MD conformations over a period of several nanoseconds, we determined polarization energies a posteriori for a range of positions of the image plane. This decoupling of MD and image potential is only a first approximation as induced charges and the associated image potential remain difficult to integrate into the MD algorithm (see §2). Nevertheless, we discuss probable changes in molecular conformation and interface structure owing to the added image potential and show that the a posteriori calculation yields polarization energies of quantitative to semi-quantitative value.
To our knowledge, the contribution of image charges to adsorption in such all-atomic models has not been computed before and we begin with a derivation of the governing equations (§2). Then we employ the previously described model systems (§3) of approximately 3 × 3 × 5 nm3 dimension that consist of even Au {1 1 1} and Au {1 0 0} surfaces in contact with 1000 explicit water molecules and dissolved single peptide molecules (figure 2) to compute the image potential [12]. Metal interfaces with pure water, a solution of a non-charged and a solution of a charged biomolecule in solution were analysed using the charge-neutral dodecapeptide A3 and the threefold negatively charged octapeptide Flg-Na3 as examples. The MD simulation relies on the biomolecular force field CVFF [32] extended for FCC metals [33] and provides access to length scales of multiple nanometres, timescales of multiple nanoseconds and realistic peptide concentrations in chemical detail. Strong epitaxial adsorption and a high degree of direct surface contact was reported for both peptides on Au {1 1 1} surfaces [8,10,12] and weak epitaxial adsorption and a low degree of direct surface contact on Au {1 0 0} surfaces [12]. The analysis of the image potential on the basis of MD simulation benefits from physically justified atomic charges in biomolecular force fields which agree with experimentally determined dipole moments [32].
Figure 2.

Models of the peptides in the simulation. (a) Peptide A3 was identified from a phage display library and is charge-neutral. (b) Peptide Flg-Na3 is a common binding motif to Au {1 1 1} and Pd {1 1 1} surfaces and contains 3 Na+ ions to compensate three negative charges on the Asp side chains. Peptide models were employed in zwitterionic form and the charge state of the side chains corresponds to pH = 7.
Potential alternative methods to determine the influence of induced charges include hybrid quantum mechanics/molecular mechanics (QM/MM) and full electronic structure calculations such as density functional theory (DFT). However, QM/MM methods could introduce a significant dependence of atomic charges on basis sets or pseudopotentials, substantial disagreement with experimental dipole moments [34] and uncertainty in the image potential. Electronic structure (DFT) calculations would be computationally a million times more expensive and limit the system size to peptide fragments, few water molecules, and reduce equilibration times to picoseconds to probe the image potential. Therefore, these methods were not further pursued here. Previous DFT calculations on subsystems [13,35–37], however, were detrimental to verify the absence of significant covalent bonding between peptides (excluding Cys) and metal surfaces in aqueous solution. The force field models employed here neglect residual amounts of charge transfer and covalent bonding across the metal–biological interface and thus remain a good approximation. A limitation of all methods is also the difficulty to define an exact position of the image plane (see §2.1). A typical uncertainty of ± 0.1 Å often introduces more uncertainty in the image potential than other details. Yet the classical continuum method [27,28] is a simple and good approximation to estimate polarization on metal interfaces as the distance between the top atomic layer of the metal surface and the first layer of atoms in solution is in the order of 2.5 Å, including some shorter individual distances down to 2.0 Å of less significant statistical weight (table 1).
Table 1.
Properties of Au {1 1 1} and {1 0 0} surfaces in contact with water and peptide solutions, including the average position of the jellium edge zJel and of the first layer of solution atoms zSol relative to the first layer of metal atoms zSf, the polarization energy EPol per surface area, per water molecule and per amino acid of the peptides in contact with the metal surface assuming an image plane located at the jellium edge.
| surface | zJel − zSf (Å) | zSol − zSfa (Å) | EPol per surface areaa (mJ m−2) | EPol per H2O (kJ mol−1) | EPol per amino acid (kJ mol−1) |
|---|---|---|---|---|---|
| Au {1 1 1} | 1.1773 | 2.535 ± 0.005 | −55 ± 5 | −2.38 (1st layer); −0.13 (2nd layer) | −40 to ± 0 (avg. −8.4) |
| Au {1 0 0} | 1.0196 | 2.299 ± 0.005 | −60 ± 10 | −2.18 (1st layer); −0.14 (2nd layer) | −29 to ± 0 (avg. n.a.) |
aAverage over all systems (water, A3, Flg-Na3).
The outline of the paper is as follows. In §2, we discuss the location of the image plane and introduce novel computational algorithms to compute the image potential of metal–biomolecular interfaces in solution. In §3, we explain details of the models, computation and analysis. Section 4 is dedicated to the results and discussion, including the total polarization energy per surface area of the metal, the contribution to the polarization energy by water, peptides, amino acids and the net contribution of polarization to peptide adsorption on the metal surface. The paper concludes with a summary in §5.
2. Computation of the image potential
2.1. Position of the image plane
The computation of the image potential, which we call equally polarization energy here, depends on the position of the image plane. In vacuum, the image plane 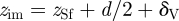 is typically located near or slightly above the jellium edge (figure 1a) which corresponds to a plane located half a lattice spacing d/2 above the top atomic layer of a given metal surface [19,26–29]. Surface state energies from photoemission and inverse photoemission measurement indicate metal-dependent locations of the image plane relative to the jellium edge δV between −0.13 Å (inwards) and +0.26 Å (outwards; [27]). Low values of δV are associated with higher total electron density of the metal, proportional to higher atomic number, for example δV(Au) < δV(Ag) < δV(Cu) [19,27]. The image plane can additionally be shifted inwards (i) owing to movement of charges, forcing a response of the electron gas and partial loss of the image potential and (ii) through the approach of charged entities closer than 2.5 Å to the surface atomic plane of the metal which causes image saturation and nonlinearity and would otherwise result in overestimates of the image potential [27,28]. On the contrary, the application of external electric fields and charge transfer at electrode surfaces can shift the image plane outwards up to δV = +0.8 Å [19,29].
is typically located near or slightly above the jellium edge (figure 1a) which corresponds to a plane located half a lattice spacing d/2 above the top atomic layer of a given metal surface [19,26–29]. Surface state energies from photoemission and inverse photoemission measurement indicate metal-dependent locations of the image plane relative to the jellium edge δV between −0.13 Å (inwards) and +0.26 Å (outwards; [27]). Low values of δV are associated with higher total electron density of the metal, proportional to higher atomic number, for example δV(Au) < δV(Ag) < δV(Cu) [19,27]. The image plane can additionally be shifted inwards (i) owing to movement of charges, forcing a response of the electron gas and partial loss of the image potential and (ii) through the approach of charged entities closer than 2.5 Å to the surface atomic plane of the metal which causes image saturation and nonlinearity and would otherwise result in overestimates of the image potential [27,28]. On the contrary, the application of external electric fields and charge transfer at electrode surfaces can shift the image plane outwards up to δV = +0.8 Å [19,29].
In contrast to vacuum, the presence of a condensed phase brings charged species very close to the surface (figure 1b) so that the electron density of the metal is pushed inwards and nonlinear effects are more important. An inward shift of the mirror plane δI − δV up to −0.5 Å was reported for mercury–water interfaces in theoretical models [29,30]. In consideration of experimental data [27], a zero shift or a small negative shift δI = 0 ± 0.2 Å of the image plane relative to the jellium edge on Au and Pd surfaces appears best justified [27]. The mirror image of the first atomic layer of water molecules and organic solutes is then anticipated on or slightly below the surface atomic plane of the metal similar as shown in figure 3b, with some tendency toward figure 3c. For a chosen metal interface, the position of the image plane remains fixed at this position in the absence of applied electric fields and nanoscopic flow.
Figure 3.

Illustration of different positions of the image plane zim on the Au {1 1 1} surface in contact with peptide A3 and water. Corresponding mirror images are shaded in pink. The image plane is shown (a) above the interface edge, (b) at the interface edge (approx. 0.1 Å above the jellium edge), (c) near the first layer of metal atoms, and (d) far below the interface edge, equal to zero image potential as employed in equation (2.6).
2.2. Image potential in solution
To our knowledge, numerical expressions for the image potential in all-atomic models containing solute molecules, explicit solvent and ions have not been developed. Therefore, this section contains a brief summary of the image potential of single point charges in vacuum and proceeds with a derivation of the image potential for a collection of charged molecules such as water, peptides and ions in contact with metal surfaces.
Let us consider first a single point charge (figure 1a). For simplicity, we assume the vertical distance between the original charge q and the image charge −q along the z-axis to be z, equal to twice the distance of the original charge from the image plane z − zim = z/2 (figure 1a). According to Coulomb's law, the attractive force on the original charge owing to the image charge equals 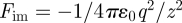 . The image potential corresponds to the energy gain Eim of moving the original charge from an infinite distance ∞ to the specified distance z from the image charge −q along the z-axis:
. The image potential corresponds to the energy gain Eim of moving the original charge from an infinite distance ∞ to the specified distance z from the image charge −q along the z-axis:
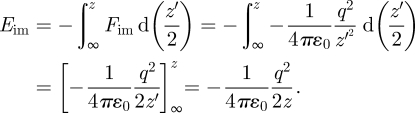 |
2.1 |
The integration variable is hereby d(z′/2), not dz′, owing to the symmetry between original and image: when the original charge moves downward by −d(z′/2), the image moves upwards by d(z′/2) so that their distance decreases by −dz′. Therefore, the denominator 2z in equation (2.1) equals twice the vertical distance z between original charge and image charge. Equation (2.1) describes the image potential of a single ion at a sufficiently large distance z/2 from the image plane on a metal surface [16,19,27,28].
The situation is similar for multi-polar, overall neutral molecules such as water H(+0.41e)–O(−0.82e)–H(+0.41e), peptides and condensed aqueous interfaces (figure 1b; [30]). Then, the ‘imprint’ of the opposite charge distribution of the dense liquid phase across the image plane creates the image potential. The evaluation of this potential includes all pairwise interactions between original charges and image charges, under additional consideration of periodic images of image charges. Original charges qi and image charges qj can then differ in absolute value, in x and y coordinates, and deviate from the ± z/2 symmetry across the image plane along the z-axis. Therefore, the expression for the image potential between a pair of a charged atom i and another image charge j changes to:
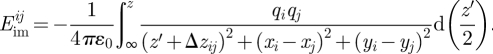 |
2.2 |
Equation (2.2) allows different charges, a possible horizontal offset 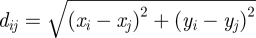 , and a non-zero asymmetry in z direction Δzij = −zi − zj as part of the vertical distance zij = zi − zj = 2zi − zi − zj = z′ + Δzij. The positive coordinate zi and the negative coordinate zj can be freely chosen and still conform with the previous definition of the z coordinate zi = z/2 and with the integration variable d(z′/2). Evaluation of the integral in equation (2.2) yields the image potential:
, and a non-zero asymmetry in z direction Δzij = −zi − zj as part of the vertical distance zij = zi − zj = 2zi − zi − zj = z′ + Δzij. The positive coordinate zi and the negative coordinate zj can be freely chosen and still conform with the previous definition of the z coordinate zi = z/2 and with the integration variable d(z′/2). Evaluation of the integral in equation (2.2) yields the image potential:
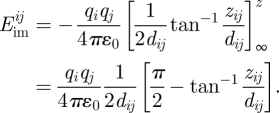 |
2.3 |
Equation (2.3) is the exact expression for the image potential between one pair of a charged atom i and an image charge j. The image potential depends on the vertical distance zij, regardless of whether it is symmetrically or asymmetrically intersected by the image plane. For the overall charge neutral system, every asymmetric pair zi /zj is accompanied by another asymmetric pair of opposite orientation. The position of the image plane is solely dictated by the positions of the image charges. For dij → 0, it can be shown that equation (2.3) converges into equation (2.1).
The image potential for the full system is given by a summation over all pairwise Coulomb interactions across the image plane as shown in equation (2.3), including the interactions between N original charges i and corresponding N image charges j as well as the interactions between original charges and periodic images of image charges in the xy plane under the condition of overall charge neutrality 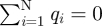 :
:
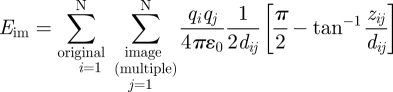 |
2.4 |
Equation (2.4) is the exact expression for the full image potential with respect to an image plane determined by the coordinates of the given image charges j, located at half the separation zij between original charges and their corresponding image charges (i = j).
For a simplified algorithmic implementation using common Ewald and particle–particle–particle mesh (PPPM) methods instead of the less convenient tan−1 function in equation (2.4), it is instructive to compare the integrands of the exact formula 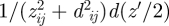 for the image potential to a hypothetical, extended version of the image potential of single ions
for the image potential to a hypothetical, extended version of the image potential of single ions 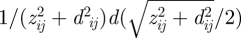 including a horizontal offset d ≠ 0 in the integration variable. The latter expression yields a larger absolute value of the integral owing to the augmented integration variable and corresponds to half the summation of Coulomb interactions as in equation (2.1). We thus obtain an upper bound of the absolute value of the (negative) polarization energy as:
including a horizontal offset d ≠ 0 in the integration variable. The latter expression yields a larger absolute value of the integral owing to the augmented integration variable and corresponds to half the summation of Coulomb interactions as in equation (2.1). We thus obtain an upper bound of the absolute value of the (negative) polarization energy as:
 |
2.5 |
This formula can be extended into a Coulomb summation over all interactions in the total pool of (2N) original charges and image charges for the location of the image plane (IP) (i) at the desired distance zim and (ii) at a distance −∞ far below for which Eim (equation (2.4)) equals zero. The benefit is the applicability of common Ewald and PPPM schemes and division by two:
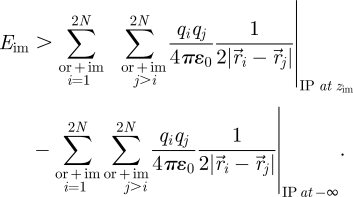 |
2.6 |
The first term in equation (2.6) includes original–original, original–image and image–image interactions and the second term subtracts original–original and image–image interactions, leading to the desired estimate of the polarization energy. We note that Eim is negative so that equation (2.6) yields a lower value than the exact formula equation (2.4). A position of the image plane at zim−300 Å in the second term can be regarded sufficient to reduce the image potential to zero. Typical dipole lengths of approximately 2 Å (in the presence of counter ions more than 10 Å) are then much smaller than the minimum distance of the mirror image of 600 Å.
In this section, we have derived two methods to compute the image potential at metal–aqueous interfaces in atomistic detail (equations (2.4) and (2.6)). Consideration of positions of the image plane slightly away from the jellium edge (figure 3a–c) helps understand the nature of the image potential and the sensitivity of computed results with regard to the exact position of the image plane. A reference position far below the metal surface (figure 3d) is required when using approximate Ewald and PPPM procedures (equation (2.6)).
3. Computational details
3.1. Molecular models and force field
Molecular models and the force field have been previously described [12]. We employed six model systems composed of {1 1 1} and {1 0 0} surfaces of Au in contact with water, an aqueous solution of peptide A3 and an aqueous solution of peptide Flg-Na3 in all atomic resolutions. Vertical metal slabs of at least 1.2 nm thickness with a surface area of 28.84 × 29.97 Å2 for Au {1 1 1} surfaces and a surface area of 28.5474 × 28.5474 Å2 for Au {1 0 0} surfaces were built using multiples of the metal unit cell according to X-ray data [33]. Models of the peptides were prepared according to the sequence of amino acids (figure 2) in different configurations (extended, helical, random coil) and extensively equilibrated by molecular dynamics simulation on the Au {1 1 1} and {1 0 0} surfaces in the presence of 1000 explicit water molecules at a density of the liquid phase of 1000 kg m−3. The corresponding peptide concentration was 56 mM, and the effective concentration was lower owing to isolation of one peptide molecule per simulation box and limited peptide–peptide interactions through periodic boundary conditions. We employed the graphical interfaces of Materials Studio [38] and Hyperchem [39] to prepare initial model configurations.
For molecular dynamics simulations, the CVFF [32]–METAL [33] force field (no cross no morse version) was employed which includes accurate Lennard-Jones parameters for FCC metals. The potential energy consists of the sum of the bond, angle, torsion, out of plane, Coulomb and van der Waals energies. Computed metal–water interface tensions [33] and the strength of metal–peptide interactions in aqueous solution were found to be in very good agreement with experimental data and ab initio studies [12]. The possibility of quantitative thermodynamic consistency between biomolecular force fields and validated extensions for inorganic components using standard combination rules has also been previously demonstrated [40]. Nevertheless, further validation of the accuracy and limitations of Lennard-Jones interactions between the metal and other molecules will be desirable.
3.2. Molecular dynamics simulation
Equilibrium configurations of the Au–water and Au–water–peptide systems were obtained using various peptide start conformations on the surface by energy minimization (1000 steps) and subsequent molecular dynamics simulation in the NVT ensemble as previously described [12]. A spherical cutoff at 1.2 nm was employed for van der Waals interactions and Ewald summation for Coulomb interactions using high accuracy of 10−6 kcal mol−1. The total simulation time exceeded 5 ns for the metal–peptide–water systems (1 ns sufficient for metal–water interfaces) with a time step of 1 fs and temperature control by the Andersen thermostat at 298.15 K. Five hundred snapshots from the equilibrium trajectory were chosen for each system to analyse polarization energies. For visualizations of representative structures and further details, see [12].
3.3. Analysis
The first step in the analysis involved the definition of the image plane. The average z coordinate of the superficial layer of Au atoms zSf was obtained as an average over all Au atoms in the first atomic layer over 500 snapshots with negligible uncertainty (±0.002 Å). Notably, however, individual atoms of the FCC surface differed in z coordinate nearly up to 1 Å in the course of molecular dynamics. The average z coordinate of the first atomic layer of the liquid surface zSol was determined as an average over the 150 nearest atoms above the {1 1 1} Au surface and over the 141 nearest atoms above the {1 0 0} Au surface in each snapshot as an average over 500 snapshots, corresponding to the same number of atoms per surface area. Using the values of zSol and zSf (table 1), we defined a default position of the image plane zim = 0 Å at the interface edge (zSol + zSf)/2 (figure 3). The statistical uncertainty of ±0.005 Å was negligible although the perceived uncertainty on the basis of visual inspection was ± 0.1 Å. The position of the image plane was then varied in 17 steps from zim = +0.3 Å to zim = −300 Å to evaluate the image potential for each position.
The second step in the analysis involved the generation of image charges and the computation of the total polarization energy per surface area (figure 4). For each snapshot of each system, a copy of all atoms in the solution phase, including peptide, counter ions and water, was reflected on the image plane as shown in figure 3 with opposite atomic charges. Periodicity in the z direction was eliminated and the polarization energy was computed using equation (2.4) as an average over all 500 snapshots, using the coordinates and point charges of the original atoms and of the image atoms. For this purpose, original atoms were translated into the box without dissecting charge-neutral molecules. We employed a cell-based cutoff for Coulomb interactions between original charges i and image charges j, including five layers of cells in the x and in the y direction for the image charges j to take into account sufficient periodic replicas (a total of 112 = 121 cells in the xy plane for the image charges). Approximate results using the Ewald method according to equation (2.6) were also obtained (see the electronic supplementary material).
Figure 4.

Polarization energy per surface area as a function of the position of the image plane (a) on the Au {1 1 1} surface and (b) on the Au {1 0 0} surface in contact with water and peptide solutions. The position of the image plane is near the jellium edge. Other positions serve to illustrate the nature of charge-induced polarization. Differences in polarization energy between a peptide solution and pure water indicate the net contribution of induced charges to adsorption (filled square, A3 (neutral peptide); filled circle, Flg-Na3 (charged peptide); filled triangle, water).
The third step in the analysis involved the breakdown of polarization energies per water molecule in the first and second molecular layers on the Au {1 1 1} and {1 0 0} surfaces in contact with water (figure 5). The density profile of water oxygen atoms was recorded as an average over all 500 snapshots of the Au–water interfaces and partitioned into first, second and residual water layers on the basis of visual inspection (figure 5). The partition coincides with significant changes in the O density along the z coordinate. Given the dimensions of the simulation box, the first molecular layer of water on the {1 1 1} surface contained on average 99 water molecules (8.7 Å2 per molecule) and the second layer 75 water molecules. The first molecular layer of water on the {1 0 0} surface contained on average 98 water molecules (8.3 Å2 per molecule) and the second layer 71 water molecules. The polarization energy per water molecule was then computed by running the summation in equation (2.4) selectively for the original atoms i of the water molecules in the respective molecular layer and over the images j of all original atoms and their periodic replicas in five layers of cells as an average over 500 snapshots, similar as described for the entire system.
Figure 5.

Density profile of water in contact with the metal surfaces, showing the distribution of water oxygen atoms along the z-axis. (a) On the Au {1 1 1} surface and (b) on the Au {1 0 0} surface. The average polarization energy per mol water molecules in the first two molecular layers is indicated, assuming that the image plane is located at the jellium edge.
The fourth step in the analysis involved the breakdown of polarization energies per peptide molecule and per constituting amino acid (figure 6). For this purpose, the summation in equation (2.4) was carried out selectively for the original atoms i of a given amino acid residue, as well as counter ions, and over the images j of all original atoms and their periodic replicas in five layers of cells as an average over 500 snapshots, similar as described for the entire system. The polarization energy per peptide corresponds to the sum of the polarization energy of all constituting amino acid residues including counter ions.
Figure 6.

Contributions of constituting amino acids to the polarization energy of (a) peptide A3 and (b) peptide Flg-Na3, including three sodium ions, in aqueous solution on Au {1 1 1} and {1 0 0} surfaces. The image plane was located at the jellium edge (black bar, Au {1 1 1} grey bar, Au {1 0 0}).
As a last step, the net contribution of polarization to peptide adsorption was determined as the difference between the polarization energy of the adsorbed peptides on the metal surface in solution compared with that of pure water adsorbed on the metal surface, using the difference in total polarization energy per surface area. The net contribution of polarization to peptide adsorption was also independently estimated from the balance of the polarization energy of the peptide and loss of polarization energy of the corresponding amount of replaced surface-bound water. The latter approximation neglects reorientation of water molecules in contact with the peptide compared with their orientation in the absence of peptide.
4. Results and discussion
On the basis of the molecular dynamics model, we attempt a first quantitative evaluation of the contribution of induced charges to the adsorption of water and biomolecules in solution on metal surfaces. Such polarization remains difficult to quantify in experiment and no laboratory data have been available. We analyse total polarization energies per surface area for water and peptide solutions on Au {1 1 1} and {1 0 0} surfaces, average contributions by water molecules, peptides, constituting amino acids, as well as the net contribution of polarization to peptide adsorption on the metal surfaces. We find that the net contribution of polarization to adsorption is an order of magnitude smaller than epitaxial contributions on {1 1 1} surfaces or covalent bonds. In contrast, polarization can be the major contribution to adsorption of highly charged peptides on epitaxially unattractive {1 0 0} metal surfaces (table 2).
Table 2.
Net contribution of polarization to peptide adsorption on even Au surfaces 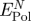 in comparison to the net contribution of epitaxial interaction
in comparison to the net contribution of epitaxial interaction 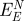 . The main contributions are the polarization energy per peptide
. The main contributions are the polarization energy per peptide 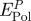 and the loss in polarization energy by replaced surface-bound water
and the loss in polarization energy by replaced surface-bound water 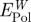 .
.
| surface | peptide | net attraction by polarizationa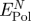 (kJ mol−1) (kJ mol−1) |
net attraction by epitaxyb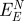 (kJ mol−1) (kJ mol−1) |
contributions to net attraction by polarization |
||
|---|---|---|---|---|---|---|
peptide 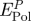 (kJ mol−1) (kJ mol−1) |
replaced surface-bound waterc |
|||||
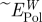 (kJ mol−1) (kJ mol−1) |
no. | |||||
| Au {1 1 1} | A3 | −28 ± 4 [−6 ± 16] | −260 ± 20 | −80 | +74 | 31 |
| Flg-Na3 | −12 ± 4 [−10 ± 16] | −260 ± 20 | −82 | +72 | 30 | |
| Au {1 0 0} | A3 | −16 ± 12 [+10 ± 16] | −38 ± 20 | −23 | +33 | 15 |
| Flg-Na3 | −80 ± 20 [−69 ± 8] | 0 ± 20 | −77 | +9 | 4 | |
aExact values from the difference in polarization energy of the peptide solution relative to a pure aqueous interface for an image plane located at the jellium edge (figure 4). The values in square brackets are simplified additive estimates 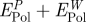 .
.
bFrom Heinz et al. [12]. Using the more accurate CHARMM-METAL force field, lower epitaxial adsorption energies of −160 kJ mol−1 (A3) and −80 kJ mol−1 (Flg-Na3) on {1 1 1} surfaces were reported [15].
cEstimates from an average number count of replaced surface-bound water molecules.
Visualizations of the metal–peptide–water systems on the basis of molecular dynamics simulation in atomic detail can be found in Heinz et al. [12]. Peptides A3 and Flg-Na3 are in direct contact and strongly bound to {1 1 1} surfaces in aqueous solution, and weakly bound to {1 0 0} surfaces with less direct contact and at least a partial water interlayer between the metal surface and the peptide.
4.1. Total polarization energy per surface area
A first impression of the magnitude of charge-induced polarization can be formed on the basis of the polarization energy per surface area (figure 4). The image plane is positioned stationary near the jellium edge between the first layer of metal atoms zSf and the first layer of solution atoms zSol (table 1), although some ambiguity about the exact position of the image plane causes uncertainty in image potential (§2.1). It is thus instructive to follow the magnitude of the image potential across the range of possible positions of the image plane, which also provides further insight into the nature of charge-induced polarization and can play a role in the calculation of the image potential (equation (2.6)). The polarization energy on Au {1 1 1} and on Au {1 0 0} surfaces is significant near the jellium edge and increases to zero when the image plane would shift towards the metal (figure 4).
On the Au {1 1 1} surface, the polarization energy per surface area with reference to the jellium edge amounts to between −50 and −55 mJ m−2 for water, a solution of the charged peptide Flg-Na3, and a solution of the neutral peptide A3 (figure 4a). When the position of the image plane is hypothetically varied, similar polarization energies are observed for all three systems across the range of positions, related to comparable polarity of the peptides and of water. Differences in polarization energy of the metal–peptide solution interfaces in comparison to metal–water interfaces indicate the net contribution of polarization to peptide adsorption (table 2, §4.4). A hypothetical outward shift of the image plane towards the first layer of solution atoms would decrease the polarization energy by multiples, and a hypothetical shift toward the first atomic layer of Au (at zim = −1.27 Å) would increase the polarization energy to −15 mJ m−2.
On the Au {1 0 0} surface, the polarization energy per surface area with reference to the jellium edge is between −50 and −70 mJ m−2 for water, a solution of peptide A3, and a solution of peptide Flg-Na3 (figure 4b). The polarization energy is then mainly determined by water molecules in direct contact with the surface and enhanced by the polarity of ionic groups in the peptides. Significant polarization is particularly seen for Flg-Na3, consistent with a strong net contribution of polarization to adsorption on the Au {1 0 0} surface (table 2, §4.4).
At hypothetical inward positions of the image plane zim between −5 and −10 Å, the polarization energy per surface area exhibits tail contributions different from zero (figure 4). These non-zero contributions result from net dipole moments in the simulation box in spite of overall charge neutrality. The smallest tail contribution for water (−5 mJ m−2) on the Au {1 1 1} and Au {1 0 0} surfaces is associated with molecular orientation of water molecules through hydrogen bonds and a short dipole length. The intermediate tail contribution for peptide Flg-Na3 (−7 mJ m−2) on the Au {1 1 1} surface (figure 4a) arises from a larger dipole length between carboxylate anions and individual Na+ cations [12]. A low tail contribution for peptide A3 (−10 mJ m−2) on the Au {1 1 1} surface (figure 4a) is associated with the large distance of approximately 30 Å between the positively charged N terminal and the negatively charged C-terminal end. On the Au {1 0 0} surface, a smaller tail contribution for peptide A3 (−6 mJ m−2) is found related to surface detachment of the peptide [12]. A low tail contribution for peptide Flg-Na3 (−14 mJ m−2) is caused by several large distances more than 10 Å between anionic groups and Na+ cations related to separation of the peptide from the surface by a water interlayer. The difference in tail contributions for a given system relative to pure water also points toward net contributions of polarization to peptide adsorption.
The polarization energy per surface area in the order of −50 mJ m−2 is significant on an absolute scale, however, it amounts only to 3 per cent of the surface tension γSV = 1540 mJ m−2 of the Au {1 1 1} surface on a relative scale [33]. The attractive nature of the image potential tends to lower the metal–aqueous interface tension γSL. The interface tension suggested by the Young equation γSL = γSV − γLV cosθ, in which contact angles of polar and non-polar liquids on clean noble metal surfaces are θ = 0° [41], may thus be further reduced by 50 mJ m−2. A trend in this direction was indicated by simulation [33].
Polarization energies per surface area were also computed using the approximate method equation (2.6) using Ewald techniques (see the electronic supplementary material, figure S1). Results are similar for the image plane positioned near the jellium edge, lower for an outward (solution) shift of the image plane owing to the modification of the integrand, and higher for an inward (surface) shift of the image plane. However, an arbitrary assumption of zero dipole moment on the system in the Ewald summation scheme leads to a loss of tail contributions and renders quantitative conclusions impossible [38].
4.2. Contribution by water
The density profile of water molecules above the Au {1 1 1} and {1 0 0} surfaces, represented by the water oxygen atoms, indicates the formation of two distinct molecular layers, followed by a rather continuous distribution of water molecules further away from the surface (figure 5). The polarization energy per surface area is −50 mJ m−2 with respect to the jellium edge (figure 4), and translates into an average attraction of −2.4 kJ mol−1 surface-bound water molecule on the Au {1 1 1} surface and of −2.2 kJ mol−1 surface-bound water on the Au {1 0 0} surface. Water molecules in the second molecular layer are attracted only by –0.13 kJ mol−1 on the Au {1 1 1} surface and by −0.14 kJ mol−1 on the Au {1 0 0} surface on average. Water molecules further away from the metal surface make negligible contributions (figure 5 and table 1). The associated increase in molecular mobility with increasing distance from the surface is driven both by loss in molecular epitaxy and in polarization, and consistent with an increase in dielectric constant from 6 to 32 to 78.5 in the first water layer, the second water layer, and bulk water on a metal surface reported by measurements and calculations [17].
The averaged total attraction per water molecule in the first molecular layer on the Au surface ranges between −8 and −12 kJ mol−1 in classical MD simulation [33]. Experimental results or data from other computational methods on the adsorption of bulk water on Au surfaces were not available, however, a similar attraction of −10 kJ mol−1 of single water molecules to Au {1 1 1} surfaces in vacuum was reported by DFT calculations [35]. The contribution of polarization of −2.3 kJ mol−1 to total water adsorption of approximately −10 kJ mol−1 is therefore in the order of 20 per cent. This does not induce significant changes in molecular structure of the metal–water interface compared with MD models which neglect polarization. A test using an additional surface attraction of 20 per cent in molecular dynamics models under neglect of polarization slightly shortens the distance of water molecules from the surface and increases the maximum-to-minimum ratio in the density profile (figure 5). However, considerable differences in molecular orientation (layering) at the metal–water interface were observed when the surface attraction of the metal was increased in excess of 100 per cent [33].
4.3. Contribution by peptides and amino acids
The polarization energy per peptide molecule 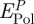 varies as a function of the sequence of the peptide and of the type of metal surface between −20 and −80 kJ mol−1 peptide (table 2). The composition of these contributions is shown in more detail for individual amino acids as an average over 500 snapshots (figure 6).
varies as a function of the sequence of the peptide and of the type of metal surface between −20 and −80 kJ mol−1 peptide (table 2). The composition of these contributions is shown in more detail for individual amino acids as an average over 500 snapshots (figure 6).
On the Au {1 1 1} surface in aqueous solution, peptides A3 and Flg-Na3 experience strong non-covalent epitaxial interactions and approximately 80 per cent of the residues are in direct contact during molecular dynamics. Polarization energies of −80 and −82 kJ mol−1 peptide, respectively, are accompanied by a loss of polarization energy of approximately 30 replaced surface-bound water molecules and even without consideration of such losses considerably smaller than epitaxial binding energies which exceed −200 kJ mol−1 (table 2; [8,10,12]). The composition of the polarization energy per peptide by individual amino acids varies in a wide range. In the peptide A3 on the {1 1 1} surface, the positively charged N-terminal 1Ala residue exhibits the highest contribution to polarization of −40 kJ mol−1, followed by the negatively charged C-terminal 12Phe (figure 6a). All other polar residues such as Tyr and Ser contribute less than −10 kJ mol−1 to polarization and contributions less than −5 kJ mol−1 are made by lowly polar Gly, Ala, Met and Pro residues. Therefore, the total attraction by induced charges of peptide A3 is primarily caused by the long distance between the charged N- and C-terminal ends and the resulting large dipole moment. In the peptide Flg-Na3 on the Au {1 1 1} surface, the polarization energy is primarily mediated by residues with a net charge as well (figure 6b). A large contribution of −30 kJ mol−1 is seen for 3Lys in combination with neighbouring Asp residues which exhibit positive contributions. The observation of positive contributions to polarization underlines that contributions from groups with a net charge must be considered jointly with corresponding groups of opposite charge as an overall charge neutral unit. Significant contributions lower than –10 kJ mol−1 are also made by the bipolar 8Lys residue and by sodium counter ions in combination with neighbouring Asp residues. The neutral 1Asp N-terminus and 2Tyr make only minor contributions.
On the Au {1 0 0} surface in aqueous solution, peptides A3 and Flg-Na3 are not attracted by epitaxy and at least a partial water interlayer is found between the metal surface and the peptides [12]. The polarization energy of −23 and −77 kJ mol−1 peptide, respectively, is then significant in relation to the small net attraction (table 2). In the peptide A3 on the Au {1 0 0} surface, the comparatively small polarization energy of −23 kJ mol−1 arises predominantly from the dipole between the positively charged N-terminal 1Ala residue and the negatively charged C-terminal 12Phe residue. Other amino acids make virtually no contribution (figure 6a). The loss of surface contact compared with the Au {1 1 1} surface and the absence of electrically charged amino acids other than N-terminal and C-terminal ends leads to loss in polarization. In the peptide Flg-Na3 on the Au {1 0 0} surface, the total polarization energy of −77 kJ mol−1 includes large contributions of −20 kJ mol−1 by negatively charged Asp residues in combination with sodium counter ions (figure 6b). Charged groups in the peptide and counter ions are spatially further separated (more than 10 Å) in comparison to the average peptide structure on the Au {1 1 1} surface, leading to a similarly high polarization energy and significant tail contributions at image plane positions less than −5 Å (figure 4b). Minor contributions to polarization are made by the electrically neutral residues 1Asp, 2Tyr and 8Lys.
As polarization energies were excluded in the iterative MD simulation (§3), the additional attraction by polarization modifies reported peptide conformations on the metal surface [12]: amino acid residues of lowest polarization energy are probably found closer to the metal surface than under neglect of polarization. On the {1 1 1} surface with peptides strongly bound by molecular epitaxy and in direct surface contact, some additional polarization does not appreciably affect the structure of the interface. Equally, peptide detachment from the surface is unfavourable as it would result in loss of epitaxial binding strength and in polarization energy. On the {1 0 0} surface with peptides weakly bound to the surface and water molecules located between the surface and the peptide, polarization probably alters the reported interface structure. Attractive polarization brings highly polar amino acid residues closer to the surface and decreases the number of water molecules in the interlayer between the peptide and the surface. In the peptide A3 on the {1 0 0} surface, tighter surface contact of the polar end groups may occur and decrease the polarization energy, while additionally released surface-bound water molecules partially offset this gain in polarization energy (table 2). In the peptide Flg-Na3 on the Au {1 0 0} surface, tighter surface contact of several charged residues is also probable. However, closer surface contact does not enhance epitaxial binding and up to +60 kJ mol−1 polarization energy must be afforded to release 26 surface-bound water molecules. Therefore, the charged peptide Flg-Na3 may prefer adsorption states between direct contact with the metal surface of low net attraction and with a distance of about one water interlayer between the surface and the peptide of maximum attraction. Further detachment from the {1 0 0} surface is unfavourable for both A3 and Flg-Na3 peptides owing to ultimate loss of polarization energy.
In conclusion, we find that amino acids with net charges and counter ions, including N terminal and C terminal groups, contribute up to −40 kJ mol−1 to the polarization energy. Amino acids of low polarity contribute less than −5 kJ mol−1 to the polarization energy of peptides even when in direct epitaxial contact with the surface. The presence of covalent and significant epitaxial attraction as on the Au {1 1 1} surface supersedes contributions by polarization, in the absence of such interactions as on the Au {1 0 0} surface polarization can control adsorption.
4.4. Net contribution to peptide adsorption
The net contribution of induced charges to peptide adsorption 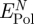 equals the difference between the polarization energy at the metal–peptide–water interface and at the neat metal–water interface (figure 4). It is thus smaller than the polarization energy by the peptide alone and we compare its magnitude to the previously reported net contribution of non-covalent epitaxial interaction to peptide adsorption
equals the difference between the polarization energy at the metal–peptide–water interface and at the neat metal–water interface (figure 4). It is thus smaller than the polarization energy by the peptide alone and we compare its magnitude to the previously reported net contribution of non-covalent epitaxial interaction to peptide adsorption 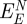 (table 2; [12]).
(table 2; [12]).
On the Au {1 1 1} surface, the net contribution of induced charges to peptide adsorption is about an order of magnitude less than the contribution of molecular epitaxy, and thus negligible in first approximation (table 2). The net contribution of polarization to peptide adsorption is only between −10 and −30 kJ mol−1 peptide compared with the total peptide adsorption energy in the order of −200 kJ mol−1, owing to the high degree of surface contact and epitaxial fit of the peptides A3 and Flg-Na3. The absolute contribution of polarization by the peptides of −80 kJ mol−1 peptide is reduced by the similar polarity of approximately 30 displaced water molecules to between −10 and −30 kJ mol−1 as a net contribution to adsorption. The similar polarity of peptide and solvent thus largely eliminates a competitive advantage in polarization of one component over the other, as evidenced by similar polarization energies per surface area and similar tail contributions (figure 4a). On the Au {1 1 1} surface, strong molecular epitaxy thus creates a high energy barrier for surface detachment of peptides regardless of induced charges.
On the Au {1 0 0} surface, the net contribution of induced charges to peptide adsorption plays a major role (table 2). The charge-neutral peptide A3 shows a net attraction of −16 kJ mol−1 owing to polarization and of −38 kJ mol−1 owing to weak epitaxial interaction. The significant relative contribution of polarization likely decreases the distance of the charged terminal groups (figure 6a) to the metal surface compared with MD simulation under neglect of polarization. More direct contact of other peptide residues with the metal surface, however, is unlikely owing to their small contribution to polarization and unfavourable replacement of more surface-bound water molecules (figure 6a). The highly charged, surface-detached peptide Flg-Na3 exhibits a net attraction owing to induced charges to the {1 0 0} surface of −80 kJ mol−1 which dominates over zero epitaxial attraction (table 2). This attraction draws the peptide closer to the metal surface than observed in MD simulation, although an increased degree of direct contact with the metal surface causes a penalty in polarization energy up to +60 kJ mol−1 by release of surface-bound water molecules and enforces further unfavourable epitaxial contact. Therefore, the peptide Flg-Na3 can be partially in direct contact with the metal surface yet the energetically favoured position is at a distance of a water layer away from the surface.
Therefore, the net contribution of polarization to peptide adsorption is minor on metal surfaces with strong epitaxial attraction and significant, or dominant for highly charged peptides, on metal surfaces with weak epitaxial attraction.
5. Conclusion
The interaction of metal surfaces with biomolecules and water encompasses interfacial polarization owing to induced charges in addition to non-covalent epitaxial and covalent bonding interactions. Polarization by induced charges remains difficult to measure so that a computational approach was developed and applied as a first step to evaluate the nature and magnitude of charge-induced polarization. Using all-atomic models in classical molecular dynamics simulation and the concept of image charges in a a posteriori computation of the image potential, we determined the influence of induced charges on even Au surfaces on the interfacial structure and adsorption of water, peptides, and corresponding single amino acids. The polarization energy per surface area amounts to between −50 and −70 mJ m−2 for aqueous and biomolecular interfaces. Molecular contributions by surface-bound water molecules (−2.3 kJ mol−1) and peptide residues (0 to −40 kJ mol−1 per amino acid) are comparable on average owing to similar polarity. Charged amino acids with a large time-averaged distance between cationic and anionic groups are most strongly attracted to the metal surface owing to polarization. On metal surfaces with favourable epitaxial interactions and direct surface contact of peptides such as Au {1 1 1}, the net contribution of polarization to peptide adsorption is small. On metal surfaces with weak epitaxial attraction and a remaining water interlayer between the surface and the peptide such as Au {1 0 0}, the net contribution of induced charges to peptide adsorption is significant or even dominant. For the highly charged octapeptide Flg-Na3 on an Au {1 0 0} surface, for example, adsorption is entirely driven by polarization up to −80 kJ mol−1 peptide. Contributions of polarization to biomolecular adsorption decrease with increasing distance of the peptide from the surface and can range several nanometres for charged peptides compared with only about 1 nm for non-covalent epitaxial interactions.
This investigation is a first step toward quantitative understanding of polarization on metal nanostructures. When the polarity of biomolecules would differ more substantially from the solvent such as in ethers or hydrocarbons as opposed to water, adsorption onto metal surfaces would be more strongly influenced by induced charges. In addition to even metal surfaces investigated here, the geometry of edges and curved surfaces of nanorods and nanoparticles probably affects the adsorption of organic surfactants and peptides by different strength of induced electric fields which could be further investigated using the proposed model. The integration of polarization forces into molecular dynamics and QM/MM algorithms would also be desirable.
Acknowledgements
We are grateful for support by the Air Force Research Laboratory, the Ohio Department of Development, Sika Technology AG, the University of Akron, and the allocation of computer resources by the Ohio Supercomputing Center.
References
- 1.Brown S. 2001. Protein-mediated particle assembly. Nano Lett. 1, 391–394 10.1021/nl0155173 (doi:10.1021/nl0155173) [DOI] [Google Scholar]
- 2.Fu X., Wang Y., Huang L. X., Sha Y. L., Gui L. L., Lai L. H., Tang Y. Q. 2003. Assemblies of metal nanoparticles and self-assembled peptide fibrils—formation of double helical and single-chain arrays of metal nanoparticles. Adv. Mater. 15, 902–906 10.1002/adma.200304624 (doi:10.1002/adma.200304624) [DOI] [Google Scholar]
- 3.Sarikaya M., Tamerler C., Jen A. K.-Y., Schulten K., Baneyx F. 2003. Molecular biomimetics: nanotechnology through biology. Nat. Mater. 2, 577–585 10.1038/nmat964 (doi:10.1038/nmat964) [DOI] [PubMed] [Google Scholar]
- 4.Sarikaya M., Tamerler C., Schwartz D. T., Baneyx F. 2004. Materials assembly and formation using engineered polypeptides. Annu. Rev. Mater. Res. 34, 373–408 10.1146/annurev.matsci.34.040203.121025 (doi:10.1146/annurev.matsci.34.040203.121025) [DOI] [Google Scholar]
- 5.Slocik J. M., Wright D. W. 2003. Biomimetic mineralization of noble metal nanoclusters. Biomacromolecules 4, 1135–1141 10.1021/bm034003q (doi:10.1021/bm034003q) [DOI] [PubMed] [Google Scholar]
- 6.Levy R., Thanh N. T. K., Doty R. C., Hussain I., Nichols R. J., Schiffrin D. J., Brust M., Fernig D. G. 2004. Rational and combinatorial design of peptide capping ligands for gold nanoparticles. J. Am. Chem. Soc. 126, 10 076–10 084 10.1021/ja0487269 (doi:10.1021/ja0487269) [DOI] [PubMed] [Google Scholar]
- 7.Murphy C. J., Sau T. K., Gole A. M., Orendorff C. J., Gao J. X., Gou L. F., Hunyadi S. E., Li T. 2005. Anisotropic metal nanoparticles: synthesis, assembly, and optical applications. J. Phys. Chem. B 109, 13 857–13 870 10.1021/jp0516846 (doi:10.1021/jp0516846) [DOI] [PubMed] [Google Scholar]
- 8.Slocik J. M., Stone M. O., Naik R. R. 2005. Synthesis of gold nanoparticles using multifunctional peptides. Small 1, 1048–1052 10.1002/smll.200500172 (doi:10.1002/smll.200500172) [DOI] [PubMed] [Google Scholar]
- 9.Huang X., El-Sayed I. H., Qian W., El-Sayed M. A. 2006. Cancer cell imaging and photothermal therapy in the near-infrared region by using gold nanorods. J. Am. Chem. Soc. 128, 2115–2120 10.1021/ja057254a (doi:10.1021/ja057254a) [DOI] [PubMed] [Google Scholar]
- 10.Slocik J. M., Naik R. R. 2006. Biologically programmed synthesis of bimetallic nanostructures. Adv. Mater. 18, 1988–1992 10.1002/adma.200600327 (doi:10.1002/adma.200600327) [DOI] [Google Scholar]
- 11.Mandal H. S., Kratz H. B. 2007. Effect of the surface curvature on the secondary structure of peptides adsorbed on nanoparticles. J. Am. Chem. Soc. 129, 6356–6357 10.1021/ja0703372 (doi:10.1021/ja0703372) [DOI] [PubMed] [Google Scholar]
- 12.Heinz H., Farmer B. L., Pandey R. B., Slocik J. M., Patnaik S. S., Pachter R., Naik R. R. 2009. Nature of molecular interactions of peptides with gold, palladium, and Pd–Au bimetal surfaces in aqueous solution. J. Am. Chem. Soc. 131, 9704–9714 10.1021/ja900531f (doi:10.1021/ja900531f) [DOI] [PubMed] [Google Scholar]
- 13.Hong G., Heinz H., Naik R. R., Farmer B. L., Pachter R. 2009. Toward understanding amino acid adsorption at metallic interfaces: a density functional theory study. ACS Appl. Mat. Interf. 1, 388–392 10.1021/am800099z (doi:10.1021/am800099z) [DOI] [PubMed] [Google Scholar]
- 14.Pandey R. B., Heinz H., Feng J., Farmer B. L., Slocik J. M., Drummy L. R., Naik R. R. 2009. Adsorption of peptides (A3, Flg, Pd2, Pd4) on gold and palladium surfaces by a coarse-grained Monte Carlo simulation. Phys. Chem. Chem. Phys. 11, 1989–2001 10.1039/b816187a (doi:10.1039/b816187a) [DOI] [PubMed] [Google Scholar]
- 15.Feng J., Pandey R. B., Berry R. J., Slocik J. M., Farmer B. L., Naik R. R., Heinz H. Submitted. Adsorption of amino acids and surfactants on Au {111} surfaces in aqueous solution by molecular epitaxy. [Google Scholar]
- 16.Bardeen J. 1940. The image and van-der-Waals forces at a metallic surface. Phys. Rev. 58, 727–736 10.1103/PhysRev.58.727 (doi:10.1103/PhysRev.58.727) [DOI] [Google Scholar]
- 17.Bockris J. O. 'M., Devanathan M. A. V., Mueller K. 1963. On the structure of charged interfaces. Proc. R. Soc. Lond. A 274, 55–79 10.1098/rspa.1963.0114 (doi:10.1098/rspa.1963.0114) [DOI] [Google Scholar]
- 18.Lang N. D., Kohn W. 1970. Theory of metal surfaces: charge density and surface energy. Phys. Rev. B 1, 4555–4568 10.1103/PhysRevB.1.4555 (doi:10.1103/PhysRevB.1.4555) [DOI] [Google Scholar]
- 19.Lang N. D., Kohn W. 1973. Theory of metal surfaces: induced surface charge and image potential. Phys. Rev. B 7, 3541–3550 10.1103/PhysRevB.7.3541 (doi:10.1103/PhysRevB.7.3541) [DOI] [Google Scholar]
- 20.King F. W., Van Duyne R. P., Schatz G. C. 1978. Theory of Raman scattering by molecules adsorbed on electrode surfaces. J. Chem. Phys. 69, 4472–4481 10.1063/1.436436 (doi:10.1063/1.436436) [DOI] [Google Scholar]
- 21.McRae E. G. 1979. Electronic surface resonances of crystals. Rev. Mod. Phys. 51, 541–568 10.1103/RevModPhys.51.541 (doi:10.1103/RevModPhys.51.541) [DOI] [Google Scholar]
- 22.Badiali J. P., Rosinberg M. L., Goodisman J. 1983. Contribution of the metal to the differential capacity of an ideally polarisable electrode. J. Electroanal. Chem. 143, 73–88 10.1016/S0022-0728(83)80255-1 (doi:10.1016/S0022-0728(83)80255-1) [DOI] [Google Scholar]
- 23.Schmickler W., Henderson D. 1984. Approximate solution for the electronic density profile at the surface of jellium. Phys. Rev. B 30, 3081–3083 10.1103/PhysRevB.30.3081 (doi:10.1103/PhysRevB.30.3081) [DOI] [Google Scholar]
- 24.Schmickler W., Henderson D. 1984. The interphase between jellium and a hard sphere electrolyte. A model for the electric double layer. J. Chem. Phys. 80, 3381–3386 10.1063/1.447092 (doi:10.1063/1.447092) [DOI] [Google Scholar]
- 25.Weinert M., Hulbert S. L., Johnson P. D. 1985. Image planes and surface states. Phys. Rev. Lett. 55, 2055–2058 10.1103/PhysRevLett.55.2055 (doi:10.1103/PhysRevLett.55.2055) [DOI] [PubMed] [Google Scholar]
- 26.Serena P. A., Soler J. M., Garcia N. 1989. Image-plane position dependence on metal crystallographic face. Europhys. Lett. 8, 185–188 10.1209/0295-5075/8/2/013 (doi:10.1209/0295-5075/8/2/013) [DOI] [Google Scholar]
- 27.Smith N. V., Chen C. T., Weinert M. 1989. Distance of the image plane from metal surfaces. Phys. Rev. B 40, 7565–7573 10.1103/PhysRevB.40.7565 (doi:10.1103/PhysRevB.40.7565) [DOI] [PubMed] [Google Scholar]
- 28.Finnis M. W. 1991. The interaction of a point charge with an aluminium (111) surface. Surface Sci. 241, 61–72 10.1016/0039-6028(91)90212-B (doi:10.1016/0039-6028(91)90212-B) [DOI] [Google Scholar]
- 29.Schmickler W., Leiva E. 1995. The double layer at the interface between a simple metal and an aqueous solution. Mol. Phys. 86, 737–744 10.1080/00268979500102331 (doi:10.1080/00268979500102331) [DOI] [Google Scholar]
- 30.Guidelli R., Schmickler W. 2000. Recent developments in models for the interface between a metal and an aqueous solution. Electrochim. Acta 45, 2317–2338 10.1016/S0013-4686(00)00335-2 (doi:10.1016/S0013-4686(00)00335-2) [DOI] [Google Scholar]
- 31.Heinz H. 2010. Computational screening of biomolecular adsorption and self-assembly on nanoscale surfaces. J. Comp. Chem. 31, 1564–1568 10.1002/jcc.21421 (doi:10.1002/jcc.21421) [DOI] [PubMed] [Google Scholar]
- 32.Dauber-Osguthorpe P., Roberts V. A., Osguthorpe D. J., Wolff J., Genest M., Hagler A. T. 1988. Structure and energetics of ligand binding to proteins: Escherichia coli dihydrofolate reductase-trimethoprim, a drug–receptor system. Proteins: Struct. Funct. Genetics 4, 31–47 10.1002/prot.340040106 (doi:10.1002/prot.340040106) [DOI] [PubMed] [Google Scholar]
- 33.Heinz H., Vaia R. A., Farmer B. L., Naik R. R. 2008. Accurate Simulation of surfaces and interfaces of face-centered cubic metals using 12-6 and 9-6 Lennard-Jones potentials. J. Phys. Chem. C 112, 17 281–17 290 10.1021/jp801931d (doi:10.1021/jp801931d) [DOI] [Google Scholar]
- 34.Heinz H., Suter U. W. 2004. Atomic charges for classical simulations of polar systems. J. Phys. Chem. B 108, 18 341–18 352 10.1021/jp048142t (doi:10.1021/jp048142t) [DOI] [Google Scholar]
- 35.Schravendijk P., van der Vegt N., Delle Site L., Kremer K. 2005. Dual-scale modeling of benzene adsorption onto Ni(111) and Au(111) surfaces in explicit water. ChemPhysChem 6, 1866–1871 10.1002/cphc.200400591 (doi:10.1002/cphc.200400591) [DOI] [PubMed] [Google Scholar]
- 36.Ferretti A., Baldacchini C., Calzolari A., Di Felice R., Ruini A., Molinari E., Betti M. G. 2007. Mixing of electronic states in pentacene adsorption on copper. Phys. Rev. Lett. 99, 046802. 10.1103/PhysRevLett.99.046802 (doi:10.1103/PhysRevLett.99.046802) [DOI] [PubMed] [Google Scholar]
- 37.Calzolari A., Cicero G., Cavazzoni C., Di Felice R., Catellani A., Corni S. 2010. Hydroxyl-rich β-sheet adhesion to the gold surface in water by first-principle simulations. J. Am. Chem. Soc. 132, 4790–4795 10.1021/ja909823n (doi:10.1021/ja909823n) [DOI] [PubMed] [Google Scholar]
- 38.Materials Studio Visualizer and Discover Program, version 4.0 2006. Accelrys, Inc., San Diego, CA, USA. [Google Scholar]
- 39.Hyperchem Molecular Modeling Program, version 7.5 2006. HyperChem(TM), Hypercube, Inc., Gainesville, FL, USA [Google Scholar]
- 40.Heinz H., Koerner H., Vaia R. A., Anderson K. L., Farmer B. L. 2005. Force field for phyllosilicates and dynamics of octadecylammonium chains grafted to montmorillonite. Chem. Mater. 17, 5658–5669 10.1021/cm0509328 (doi:10.1021/cm0509328) [DOI] [Google Scholar]
- 41.Osman M. A., Keller B. A. 1996. Wettability of native silver surfaces. Appl. Surf. Sci. 99, 261–263 10.1016/0169-4332(96)00101-8 (doi:10.1016/0169-4332(96)00101-8) [DOI] [Google Scholar]