

Abstract
There is an enormous gap between the antiproliferative and in vivo antitumor efficacy of gemcitabine in cell line-based models and its clinical efficacy. This may be due to insensitiveness of the precursor, cancer stem cell (CSC) compartment to cytotoxic agents. The hedgehog pathway is associated with CSC signaling and control. We used a direct xenograft model of pancreatic cancer and a two-stage approach was used to test the hypotheses that targeting CSC could increase the efficacy of gemcitabine. Tumors from a gemcitabine-sensitive xenograft were treated with gemcitabine first, and randomized, after tumor regression to continuing treatment with gemcitabine, a hedgehog inhibitor alone or in combination with gemcitabine. We tested markers described as associated with CSC such as CD24, CD44, ALDH, nestin, and the hedgehog pathway. After induction with gemcitabine, treated tumor showed an enrichment in CSC markers such as ALDH and CD24. Subsequently, a release from gemcitabine prompted a repopulation of proliferating cells and a decrease in such markers to equilibrate from pretreatment levels. Combined treatment with gemcitabine and cyclopamine induced tumor regression and decrease in CSC markers and hedgehog signaling. Cytoplasmic CD24 and ALDH were inversely and strongly associated with growth and were expressed in a minority of cells that we propose constitute the CSC compartment. Hedgehog inhibitors as part of a dual compartment therapeutic approach were able to further reduce tumor growth and decreased both static and dynamic markers of CSC. Direct tumor xenografts are a valid platform to test multicompartment therapeutic approaches in pancreatic cancer.
Introduction
Pancreatic ductal adenocarcinoma, which constitutes >90% of pancreatic cancers in humans, is one of the most devastating human malignancies. Despite extensive research during the past decades, the dismal prognosis has not markedly improved and is almost uniformly lethal (1), with an average overall 5-year survival of <5% (2). To date, surgical resection is the only available potentially curative therapeutic option, but due to the lack of early symptoms and reliable screening methods for early detection, the vast majority of patients are diagnosed with already metastatic disease, precluding curative surgical treatment and rendering an overall remaining life expectancy of only 6 months (3). Thus, the development of new, potent therapeutic options is highly desirable. Conventional chemotherapy remains the mainstay of pancreatic cancer management. However, even patients whose tumors initially are arrested or regress with therapy eventually experience tumor regrowth even while still receiving primary therapy. This may indicate repopulation of proliferating cells originated by a senescent, cytotoxic therapy-resistant precursor.
An advantage of using direct xenografting of human cancers is that there is no clonal selection and all cellular fractions existing in a tumor are transplanted. Prior work by other groups used direct xenograft models to identify, isolate, and characterize pancreatic cancer stem cells (CSC; ref. 4). These models may provide a unique platform to test therapeutic approaches directed toward CSC. We have hypothesized that conventional cytotoxic therapy aims primarily at the proliferating, differentiated cellular fraction. Many studies have been undertaken to study the properties of CSC, but few address ways to target and inhibit them as a necessary step to control cancer growth. A frequent caveat has been the process of differentiation that starts immediately after isolation and that will seemingly be overcome with this intact in vivo system.
Recently, aberrant activation of the hedgehog pathway has been found in the majority of human pancreatic cancers and other gastrointestinal tract malignancies (5, 6). Moderate growth inhibition of ∼50% to 60% in vivo was shown in preestablished s.c. pancreatic cancer xenografts in response to hedgehog inhibition with the small-molecule smoothened antagonist cyclopamine; the effects were more pronounced when cyclopamine therapy was initiated simultaneously with s.c. implantation of cancer cells (5). Evidence is mounting on the relevance of the hedgehog pathway in CSC signaling, which could be responsible for the above-described antitumor activity (7, 8).
In this study, we show that combining gemcitabine with a hedgehog inhibitor eradicates CSC and results in improved antitumor efficacy. To this end, a gemcitabine-sensitive tumor was treated with gemcitabine first to determine whether this would enrich the proportion of CSC; then, we tested whether treatment with a hedgehog inhibitor alone and in combination with gemcitabine was able to modify the proportion of CSC and increase antitumor efficacy. We aimed at developing markers that could be applied in a clinical setting using immunohistochemistry. For this purpose, we evaluated a series of markers in the tumors that have been described as associated with TSC such as CD24, CD44, ALDH, nestin, and hedgehog pathway components as GLI1 (4, 9–11), some of which were adapted for testing in fixed paraffin tissues.
Materials and Methods
Drugs
Gemcitabine (Eli Lilly) was obtained from commercially available sources. Cyclopamine was a kind gift from Anirban Maitra.
In vivo Growth Inhibition Studies
Six-week-old female athymic nude mice (Harlan) were used. The research protocol was approved by the Johns Hopkins University Animal Care and Use Committee and animals were maintained in accordance to guidelines of the American Association of Laboratory Animal Care. The xenografts were generated according to methodology published elsewhere (12). Briefly, surgical nondiagnostic specimens of patients operated at the Johns Hopkins Hospital were reimplanted s.c. to 1 to 2 mice for each patient (this is the first passage of the human tumor on the mouse or F1 generation). Tumors were let to grow to a size of 1.5 cm3 at which point were harvested, divided, and transplanted to another 5 mice (F2 generation). After a second growth passage, tumors were excised and propagated to cohorts of ≥30 mice, which constituted the treatment cohort (F3 generation). Tumors from this cohort were allowed to grow until reaching ∼200 mm3, at which time they were evenly distributed by size in the following two treatment groups: (a) control (n = 10 tumors) and (b) gemcitabine 100 mg/kg twice weekly i.p. (n = 50 tumors). Tumors from this treatment stage were treated for 25 days. After this, the control tumors and 10 gemcitabine-treated tumors were excised and aliquoted for analysis (three portions were analyzed fresh for flow cytometry, flash frozen, and embedded in paraffin). The remaining 40 gemcitabine-treated tumors were evenly redistributed in the following four groups (with n = 10 tumors each): (a) control, (b) gemcitabine 100 mg/kg twice weekly i.p., (c) cyclopamine 25 mg/kg twice a day by oral gavage, and (d) gemcitabine plus cyclopamine at the above doses. Treatment was given for 28 days. Mice were monitored daily for signs of toxicity and were weighed two times per week. Tumor size was evaluated two times per week by caliper measurements using the following formula: tumor volume = [length × width2] / 2. Relative tumor growth inhibition was calculated by relative tumor growth of treated mice divided by relative tumor growth of control mice since the initiation of therapy (T/C).
Gene Expression Analysis by Reverse Transcription-PCR
Total RNA was extracted from tumors using the RNeasy Mini Kit (Qiagen). cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad) following the manufacturer's instructions. Relative quantification of ALDH, human nestin, and mouse nestin and housekeeping ubiquitin C mRNA was achieved using an iCycler iQ real-time PCR detection system (Bio-Rad) using ABI Taqman probes. Accumulation of the specific PCR products was detected as an increase in fluorescence that was plotted against cycle number to determine the CT values. Relative expression of the mRNA analyzed was estimated using the formula: relative expression = 2−ΔCT, where ΔCT = CT (mRNA) - CT (ubiquitin C). Samples were analyzed twice in quadruplicate in a blinded manner.
Immunohistochemical Analysis
Sections (5 μm) were used for CD44, CD24, and ALDH (Cell Signaling Technology) and staining was done using citrate-steam recovery followed by Catalyzed Signal Amplification (DAKO). For CD24, both membrane and cytoplasmic staining were done separately. Samples were analyzed by expert pathologists (A.S., F.L-R., and A.M.) in a blinded manner.
Results and Discussion
Gemcitabine-Induced Debulking and Enrichment for Putative CSC
Compared with the control arm that grew rapidly forcing to stop the experiment after 3.5 weeks, gemcitabine induced tumor growth inhibition in the Panc185 xenograft model (Fig. 1). This resulted in a 6-fold increase in cytoplasmic CD24 (cCD24) in both the proportion of positive cells and the intensity of staining (Fig. 2A). The resulting cCD24 indexes were 11 ± 1 and 69 ± 17 in control and gemcitabine-treated tumors, respectively. Control tumors were 5-fold larger than those treated indicating an intact CSC population. Membrane CD24 and CD44 did not significantly change between groups. Human nestin did not significantly change, but mouse nesting decreased in gemcitabine-treated tumors. ALDH testing by reverse transcription-PCR had a wide dynamic range, and a 4.9-fold increase was documented in treated tumors (Fig. 2B). ALDH by immunohistochemistry gave even more dramatic differences, as was only detected in ∼1% to 2% of cells in gemcitabine-treated tumors, with homogeneously negative staining in control tumors. These results suggest that a debulking approach is efficient in significantly enriching the population of precursor cells by decreasing the proliferating, chemotherapy-sensitive population. We then evaluated whether this strategy would allow for testing CSC-directed therapies.
Figure 1.
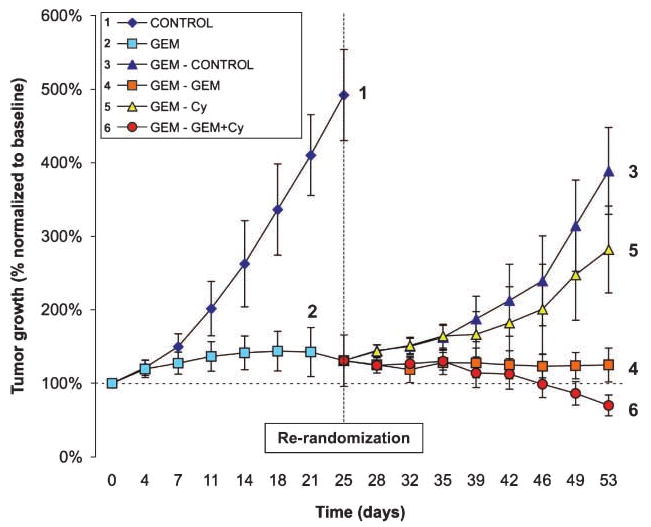
After 4 wk of induction with gemcitabine, the cohort 2 showed a plateau in effect. After re-randomization, the group released from gemcitabine (3) rapidly regrew, as did the group receiving cyclopamine (5). The group that continued gemcitabine (4) remained in an activity plateau. Only the combination therapy group (6) showed incremental effect, and these tumors homogeneously regressed to 50% of the initial size after 4 wk. Bars, SD.
Figure 2.
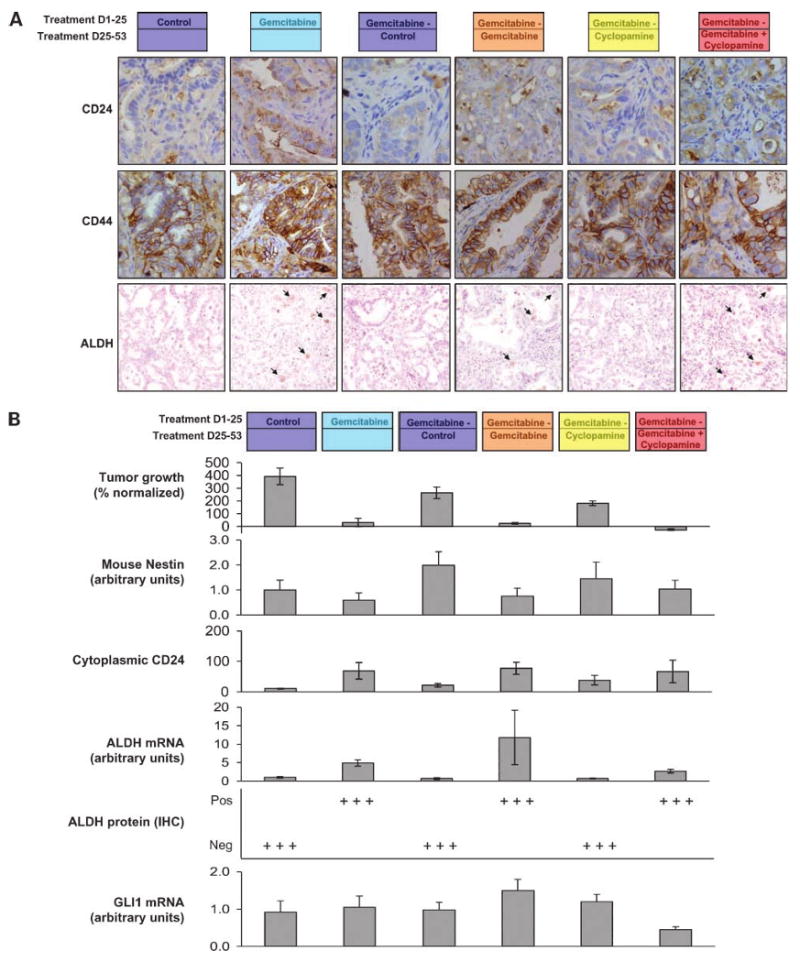
A, immunohistochemical assessment of CD24, CD44, and ALDH by immunohistochemistry. CD44 presented a flat staining that was not different between groups. However, both CD24 and ALDH had a pattern consisting in inversely intense staining with tumor size, with maximal staining in gemcitabine-treated and gemcitabine + cyclopamine-treated groups. B, tumor growth, mouse nestin expression, cytoplasmic CD24, ALDH by reverse transcription-PCR and immunohistochemistry, and GLI1 mRNA expression. ALDH by immunohistochemistry was particularly differentiating as the low-level expression followed an all-or-nothing pattern. ALDH was only positive in 2% of the cells in the gemcitabine-treated groups. In the group where gemcitabine is ceased, the TSC markers (both CD24 and ALDH) decreased back to the levels of the untreated group, suggesting a repopulation of proliferating cells and a dilution of the TSC-enriching effect.
Assessment of an Anti-CSC Treatment Modality
The 40 remaining gemcitabine-treated tumors were then evenly allocated in four groups and were either observed or treated with gemcitabine, cyclopamine, or the combination of both drugs for another 4 weeks. As expected, on discontinuation of therapy, tumors regrew at an incremental pace that peaked 2 weeks after gemcitabine discontinuation. Long-term gemcitabine did not increment its effect and tumors remained stable. Cyclopamine as a single agent had minimal activity, with slower growth initially but ultimately comparable with the untreated group. However, the combination of both induced progressive tumor regression, with an average final tumor volume that was 30% smaller than on day 0 and 50% of that on day 25. The tumors in the combination arm were ∼45% smaller than in the chronic gemcitabine arm.
There was again a strong inverse correlation between tumor growth and CSC markers. The cCD24 indices were 21 ± 6 and 36 ± 14 in the untreated and cyclopamine-treated groups (that incremented 300% and 200% their volumes in this period) and 77 ± 16 and 67 ± 24 in the gemcitabine and combination groups that did not grow or regress, respectively. The intermediate cCD24 staining index and rate of growth in the cyclopamine single-agent arm further support the primary hypothesis, as can be interpreted as a restrained repopulation due to CSC-specific signaling inhibition. Overall, there was a negative correlation between cCD24 index and tumor growth that reached statistical significance (P < 0.01). Again, ALDH, at both gene expression and protein levels by immunohistochemistry, showed increments in the sustained gemcitabine group compared with the group where treatment was discontinued. Human nestin was lowest in the combination arm but with a narrow dynamic range; mouse nestin was directly correlated with tumor growth and inversely correlated to putative CSC markers. Overall, assuming that cCD24 and ALDH are CSC markers, the observation that gemcitabine treatment increments 5-fold their expression and that stopping gemcitabine reverses this enrichment gives support to the primary hypothesis that gemcitabine targets the proliferating and not the CSC population.
Interestingly, in the combination arm, there was a lower expression of ALDH by reverse transcription-PCR compared with the gemcitabine group, and this was the only group where an inhibition of human GLI1 expression was documented. The delay in growth arrest in the combination arm coincides with the estimates for a slowly proliferating cancer type as pancreatic cancer and suggests a half-life of 2 weeks for the transforming cells. It also coincides with the repopulating slope documented in the group “released” from gemcitabine therapy. Whereas the proportion of putative CSC seemed equivalent in the gemcitabine versus the combination arm, it needs to be stressed that the average tumor volume was roughly half in the combination arm. As these readings are normalized by cell count or RNA content in the immunohistochemistry and reverse transcription-PCR analyses, respectively, this indicates a parallel reduction in both compartments' cell mass. This observation, together with the 2.5-fold decrease in GLI1 signaling in the combined therapy arm compared with cytotoxic treatment alone, supports the primary hypothesis that a dual approach meaningfully targets the different cell subpopulations.
Global Marker Analysis
A global correlative analysis of all the individual tumors was conducted, assessing the hedgehog pathway components, putative CSC markers, and tumor growth. There was a high level of correlation within the hedgehog pathway, with SHH-PTCH, SHH-SMO, SHH-GLI1, and SMO-PTCH showing P < 0.01 by Pearson analysis and GLI1-PTCH showing borderline association (P = 0.03). Only SMO and GLI1 were not significantly associated (P = 0.14). GLI1 expression correlated with human but not mouse nestin (P < 0.01). In the analysis of the putative CSC markers, only cCD24 correlated with tumor volume (P < 0.01). Interestingly, PTCH and cytoplasmic CD24 were strongly associated (P < 0.01). This suggests that both cCD24 and PTCH levels are indicative of CSC presence.
For this work, we took advantage of the PancXenoBank, a collection of individual pancreatic cancer tumors obtained by directly implanting surgical specimens in immunodeficient mice (12). Generally, before entering clinical trials, new agents are tested against high-passage cell lines and typically a few xenografts established from these lines. It is unclear how representative those models are of the biology of pancreatic cancer, in view of the historic disconnect between preclinical and clinical results in this disease. We have shown that directly xenografted tumors retain the key features of the originator tumor, represent the heterogeneity of the disease, are easily amenable to treatment with different drugs, and offer an endless source to tumors for complex biological studies (13). In addition, in this study, we were able to conduct biological studies directed at elucidating the proportion of several compartments in the same tumor. Indeed, these direct xenografts have been used as a source of precursor cells for their biological characterization (4). Although obviously clinical specimens and clinical response data are more valuable, the two-step treatment process and the detailed biological and therapeutic assessment conducted in this work are not possible in the clinical setting at early developmental stages and would be difficult even at a validation/confirmation phase. We propose that this platform is useful for screening purposes and best drug and biomarker candidate selection that after can be tested in focused clinical studies.
Overall, these results suggest that gemcitabine is not efficacious in decreasing the CSC compartment, which precludes the cure of tumors even with long-term treatment. Cytoplasmic CD24 and ALDH were inversely and strongly associated with growth and were expressed in a minority of cells that we propose constitute the CSC compartment. Hedgehog inhibitors as part of a dual-compartment therapeutic approach were able to further reduce tumor growth and decreased both static and dynamic markers of CSC. Direct tumor xenografts are a valid platform to test multicompartment therapeutic approaches in pancreatic cancer.
Acknowledgments
Grant support: Sol Goldman Center for Pancreatic Cancer Research, Viragh Family Foundation, Lee family, and grants CA116554 and CA129963.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Carpelan-Holmstrom M, Nordling S, Pukkala E, et al. Does anyone survive pancreatic ductal adenocarcinoma? A nationwide study re-evaluating the data of the Finnish Cancer Registry. Gut. 2005;54:385–7. doi: 10.1136/gut.2004.047191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 3.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–57. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 4.Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 5.Thayer SP, di Magliano MP, Heiser PW, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–6. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berman DM, Karhadkar SS, Maitra A, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425:846–51. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 7.Parisi MJ, Lin H. The role of the hedgehog/patched signaling pathway in epithelial stem cell proliferation: from fly to human. Cell Res. 1998;8:15–21. doi: 10.1038/cr.1998.2. [DOI] [PubMed] [Google Scholar]
- 8.Peacock CD, Wang Q, Gesell GS, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci U S A. 2007;104:4048–53. doi: 10.1073/pnas.0611682104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang P, Wang CY, Gou SM, Wu HS, Liu T, Xiong JX. Isolation and biological analysis of tumor stem cells from pancreatic adenocarcinoma. World J Gastroenterol. 2008;14:3903–7. doi: 10.3748/wjg.14.3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–67. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008;27:6120–30. doi: 10.1038/onc.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rubio-Viqueira B, Jimeno A, Cusatis G, et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res. 2006;12:4652–61. doi: 10.1158/1078-0432.CCR-06-0113. [DOI] [PubMed] [Google Scholar]
- 13.Jimeno A, Rubio-Viqueira B, Amador ML, et al. Dual mitogen-activated protein kinase and epidermal growth factor receptor inhibition in biliary and pancreatic cancer. Mol Cancer Ther. 2007;6:1079–88. doi: 10.1158/1535-7163.MCT-06-0448. [DOI] [PubMed] [Google Scholar]