

Tautomycetin (TMC) is a polyketide metabolite produced by Streptomyces sp. CK4412 and Streptomyces griseochromogenes.[1] This intriguing molecule was previously shown to possess activated T-cell specific immunosuppressive activity with a novel mode of pharmacological action, both in vivo and in vitro.[2] More recent studies have also revealed promising anticancer activity in a variety of models.[3–5]
The biosynthesis of complex polyketide compounds in bacteria often occurs by an assembly-line type mechanism that is catalyzed by type I modular polyketide synthases (PKSs). In a key final step the characteristic macrolactone scaffold is generated for the macrolide antibiotics erythromycin and pikromycin.[6] This termination process is catalyzed by a thioesterase (TE) domain located at the carboxy-terminus of the final PKS elongation module. The activity of this domain results in cleavage of the acyl chain from the adjacent ACP followed (typically) by macrocyclization.[7, 8] The macrolactone is often further modified to yield the final bioactive compound.[9]
The structure of TMC is highly unusual;[10] one of the few known examples of a polyketide natural product that bears a terminal alkene (Figure 1). The TMC biosynthetic gene cluster (tmc) has recently been characterized, revealing two putative type I PKSs (TmcA and TmcB), along with 16 additional gene products presumably involved in chain construction, tailoring and regulation (Figure 2).[11, 12]
Figure 1.

TMC, produced by Streptomyces sp. CK4412.
Figure 2.

Proposed Steps in the TMC biosynthetic pathway. Abbreviations: KS – ketosynthase, AT – acyltransferase, ACP – acyl carrier protein, DH – dehydratase, KR – ketoreductase, ER – enoylreductase, TE – thioesterase. Domains predicted to be inactive are marked with an “X”. Domains that are active but not utilized are indicated with an “*”. The ordering of post-PKS tailoring reactions is unknown.
Based on pathway annotation and biosynthetic principles, we hypothesized that the TMC TE catalyzes termination of chain assembly through generation of the free acid, as it contains the highly conserved sequence GxSxG and GdH motifs, as well as the Ser-His-Asp catalytic triad characteristic of the α,β-hydrolase class of serine hydrolases. Installation of the terminal olefin is presumed to occur through decarboxylative dehydration during the post-PKS maturation of the polyketide to form the final TMC product. DNA sequence analysis of open reading frames downstream of tmc reveals several potential candidate enzymes for catalyzing this transformation: two putative decarboxylases and a dehydratase.[11, 12] These biosynthetic steps remain unclear, motivating us to elucidate the biochemical details of this process.
Based on our efforts to understand chain termination and to decipher terminal alkene formation in TMC biosynthesis, we report here the cloning, biochemical characterization, and a 2.0-Å crystal structure of the TE domain for this pathway; the first high-resolution structural analysis of a linear chain terminating TE.
The TMC TE was amplified from cosmid pTMC2290 and inserted into the vector pMCSG7 (Supporting Information).[11] To assess enzyme function, two short-chain enantiomerically pure TMC substrate mimics were synthesized in two steps (Scheme 1). [13] [14] We first evaluated the hydrolysis of model substrates 4 and 5 by the TMC TE. Following overnight incubation and LC/MS analysis, we found that hydrolysis of the (R) isomer 4 occurred > 350 fold more efficiently compared to (S) isomer 5 (Figure 3). This was surprising, as previously characterized TEs from macrolactone-forming PKSs exhibit a high degree of substrate and stereochemical tolerance.[15, 16] However, this selectivity is consistent with the predicted stereochemistry of the β-hydroxyl group (eliminated during decarboxylative dehydration) based on sequence analysis of the preceding module 9 KR domain (Figure S2).[17] Mutation of the TMC TE active site Ser132 to Ala completely abrogated hydrolysis of substrate (not shown) and confirmed its key role in catalysis.
Scheme 1.
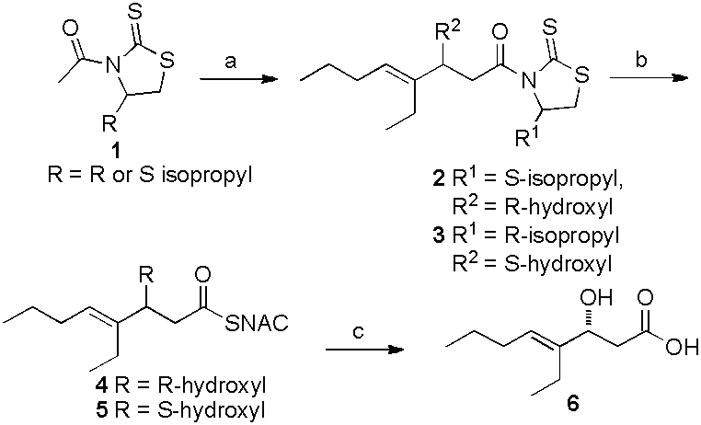
Synthesis of N-acetylcysteamine (NAC) short chain TMC analogues. Reagents and conditions: a) trans-2-ethyl-2-hexenal, tin triflate, N-ethylpiperidine, CH2Cl2, −78 °C, 70%; b) NAC, imidazole (3 equiv), CH2Cl2, 85%; c) LiOH, H2O2, 10% aq. THF, 90%.
Figure 3.

Hydrolysis by the TMC TE. A) 4 or 5, no enzyme; B) 4 with TE; C) 5 with TE; D) pikromycin hexaketide, no enzyme; E) pikromycin hexaketide with TE. All substrates were at 5 mM and incubated overnight with 1 μM enzyme before analysis by LC/MS. All masses were as expected and for traces B and C the retention time of the acid matched that of standard 6 (Figure S1). Comparison of the shape and size of the active site tunnel in TMC TE, relative to the homologous Pik and DEBS TEs, reveals why the enzyme forms a linear hydrolysis product as opposed to a macrolactone. The bulky side chains of Tyr161, Phe163 and Leu205 constrict the “exit” side of the substrate tunnel, leaving only enough space for an extended acyl chain (Figure 4C).
Steady state kinetic analysis of the TMC TE was performed with compound 4 using a discontinuous coupled fluorescence-based assay.[19] The TE was found to have a catalytic efficiency of 22 ± 2 m−1 s−1, with a kcat of 2.2 ± 0.2 min−1 and a Km of 1.7 ± 0.3 mM, based on the non-linear regression fit to the Michaelis-Menten equation (R2=0.85) (Figure S3). While relatively slow against 4, it is well within the spectrum of previously described kinetic parameters for the pikromycin and erythromycin TEs using model diketide substrates.[20] Interestingly, the data fit equally well to the allosteric kinetic model (R2=0.88), yielding similar kinetic constants as the previous fit (Figure S3). Moreover, the Hill coefficient calculated from this fit (1.7) is consistent with possible enzyme cooperativity. If true, this finding, to the best of our knowlege, represents the first report of a thioesterase to demonstrate allosterism; however, these results should be interpreted with care given that the recombinant TE domain is removed from its native polypeptide context for this study. No hydrolysis of the corresponding S isomer 5 was observed at these concentrations over this time course.
Next, we examined the ability of the TMC TE to catalyze intramolecular cyclization of the linear pikromycin hexaketide intermediate to form the 12-membered ring macrolactone 10-deoxymethynolide.[18] This substrate possesses the (R)-β-hydroxyl stereochemistry that is preferred by the TMC TE. Although cyclization of the hexaketide did not occur, partial hydrolysis to the linear carboxylic acid was observed after overnight incubation with the enzyme (Figure 3). This hydrolysis reveals some substrate flexibility of the TMC TE as its native substrate (final chain elongation intermediate, Figure 2) lacks an α-methyl group, and the distal portions of the two compounds differ considerably.
To understand further the basis for its unique selectivity, we pursued structural analysis of the TMC TE protein, which is a dimeric member of the α/β-hydrolase family bearing a fold similar to other type I PKS TEs.[15, 19] The protein structure consists of two discrete motifs, an α/β-hydrolase core capped by an α-helix lid domain. The α/β-hydrolase core fold is a seven–stranded predominantly parallel β-sheet surrounded by five α-helices (Figure 4A). Canonical α/β-hydrolase domains include a helix between β6 and β7 which is lacking in the macrolactone forming Pik and DEBS TEs. This helix (α4) is present in TMC TE and is proposed to impact processing of the product, directing hydrolysis as opposed to cyclization (see below). The lid domain consists of four α-helices, two (αL1 and αL2) from the N-terminal thirty-three residues, and two (αL3 and αL4) from a forty-five residue region between β5 and β6. As with Pik and DEBS TEs, the dimer interface is mediated exclusively through the two N-terminal helices (αL1 and αL2) that form the top of the lid domain (Figure 4B).
Figure 4.

Structure of TMC TE. A) Stereodiagram of TMC TE monomer rainbow colored from N-terminus (blue) to C-terminus (red). Active site triad is shown as spheres. Lid and α/β hydrolase core domains are indicated on the side. The pointer indicates the direction of entry into the substrate tunnel. B) TMC TE dimer interactions are mediated by the lid domains (yellow). The substrate tunnel (shown for one monomer) passes through the protein with the active site (spheres) in the center. Dimer axis is vertical in this view. Pointer is same as for A. C) Stereodiagram of active site looking from the entrance (along the pointer in part A). Pik TE (grey) is superimposed on TMC TE (blue) with TMC TE substrate channel (yellow surface). The catalytic triad and residues that constrict the active site relative to Pik TE are labeled. Equivalent residues are labeled with the TMC TE designation above the Pik TE designation. Helix α4 is present only in the TMC TE structure. Figure S7 shows comparison with the Pik TE substrate tunnel.
In accord with other type I TEs, the active site triad (Ser132, Asp159, and His255) is located on loops at the C-terminal edge of the core β-sheet after β4, β5, and β7. Ser132 is positioned in a classic nucleophilic elbow within the signature sequence Gly130-His131-Ser132-Xaa-Gly134. The oxyanion hole is formed by the backbone amide groups from Ser133 and Thr66. As observed previously with Pik TE and DEBS TE,[8, 15, 19] the active site is located at the center of an elongated substrate tunnel, which is open at both ends, spanning the width of the enzyme (Figure 4B). Compared to Pik and DEBS TEs, the TMC TE substrate tunnel is relatively narrow and constricted in the region bearing the polyketide intermediate during hydrolysis.
Despite these insights, a structural rationale for the observed chiral preference of TMC TE for (R)-β-OH is not readily apparent. We surmise that in the free-enzyme structure, the Ser133 side chain (adjacent to the catalytic Ser132) blocks the presumed oxyanion hole by hydrogen bonding with the backbone NH of Thr66. The Ser133 side chain Cα-Cβ bond must rotate in order for the substrate thioester oxygen to occupy the oxyanion hole. However, substrate modeling into the active site suggests that due to the restricted chamber dimensions, only the (R)-β-OH can be accommodated with the Ser133 side chain rotated to either of the available alternative rotamer positions.
Based on previous work that revealed a strategy for converting the rat FAS type II TE from a hydrolase to an acyltransferase by double mutation of the active site Ser 132 to Cys and His 255 to Arg, we were motivated to determine whether the corresponding mutations in the TMC TE would provide both an acyl-enzyme intermediate for use in further structural studies, as well as a valuable acyltransferase for generating new acyl-CoA species.[20]
The double mutant was generated and overnight incubation with either thioester 4 or 5 resulted in formation of an acyl-enzyme intermediate by low-resolution MS (Figure S4), with no evidence of hydrolysis of the thioesters to the carboxylic acids (not shown). High-resolution FTICR MS analysis revealed that the intact protein was labeled in a 2.5:1 ratio (Figure S5). Trypsin digestion of the acylated TMC TE protein followed by high-resolution MS confirmed that loading occurred at the Cys132 residue (Table S2). In contrast, no labeling of wild type TMC TE (Figure S4) nor the S132C or H255R single mutants was observed (not shown). Efforts to crystallize the acyl-enzyme species are ongoing.
After observation of an acyl-enzyme intermediate, we attempted to displace the acyl group with free CoA to create a new CoA thioester. This method would be useful for the formation of synthetically challenging CoA substrates and conversion of off-loaded biosynthetic intermediates into CoAs for use in chemoenzymatic synthesis. The reaction was attempted using two different methods, but formation of a new CoA species was only observed with method 2 (Supporting Information). However, this formation was nonenzymatic, as the new compound was observed in the presence and absence of enzyme (Figure S6), suggesting a chemical transthioesterification at high pH. While this result was surprising, we expect that this method will be generally applicable for the synthesis of small amounts of novel CoA compounds when the starting thioester is not sensitive to high pH. The TE Ser132Cys and His255Arg double mutant also provides a general strategy for affinity labeling of thioesterase enzymes with a range of natural and unnatural substrates.
In summary, the TMC TE is a polyketide hydrolase that exhibits a high degree of stereoselectivity at the β-hydroxy position. X-ray crystallography has provided the first high-resolution structure of a linear polyketide chain terminating TE, showing the enzyme to have a constrained substrate chamber relative to macrolactone-forming TEs. Although the basis for relatively low substrate tolerance remains unclear, it is now possible to assess the role of specific amino acid residues that might be important in the observed stereoselectivity toward acyl-(R)-β-OH. Moreover, construction of a double mutant form of select TMC TE active site residues has provided a new method for affinity labeling of the enzyme active site that should be applicable to other members of the β-hydrolase family of TEs. Finally, this work provides a path to explore further a process for polyketide termination that involves initial release of the polyketide chain followed by decarboxylative elimination. This represents a unique mechanism compared to the recently elucidated curacin TE that catalyzes concomitant hydrolysis and decarboxylative elimination of sulfate leading to a terminal olefin as a final step in the pathway.[21]
Supplementary Material
Footnotes
This work was supported by NIH grant GM076477 and the Hans W. Vahlteich Professorship (D.H.S.), UL1RR024986 (J.B.S), NIH grant DK042303 (J.L.S.) and by KOSEF grant MEST 2009-0078663 (E.-S.K.) GM/CA CAT is supported by the NIH National Institute of General Medical Sciences and the National Cancer Institute at the APS, which is supported by the US Dept. of Energy Office of Science
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Jamie B. Scaglione, Life Sciences Institute, University of Michigan, Ann Arbor, MI 48109 (USA)
David L. Akey, Life Sciences Institute, University of Michigan, Ann Arbor, MI 48109 (USA)
Rachel Sullivan, Life Sciences Institute, University of Michigan, Ann Arbor, MI 48109 (USA).
Jeffrey D. Kittendorf, Life Sciences Institute, University of Michigan, Ann Arbor, MI 48109 (USA)
Christopher M. Rath, Life Sciences Institute, University of Michigan, Ann Arbor, MI 48109 (USA)
Eung-Soo Kim, Department of Biological Engineering, Inha University, Incheon (Korea).
Janet L. Smith, Life Sciences Institute, University of Michigan, Ann Arbor, MI 48109 (USA). Department of Biological Chemistry, University of Michigan, Ann Arbor, MI 48109 (USA)
David H. Sherman, Life Sciences Institute, University of Michigan, Ann Arbor, MI 48109 (USA). Department of Medicinal Chemistry, Department of Chemistry, Department of Microbiology and Immunology, University of Michigan, Ann Arbor, MI 48109 (USA).
References
- 1.Cheng XC, Kihara T, Ying X, Uramoto M, Osada H, Kusakabe H, Wang BN, Kobayashi Y, Ko K, Yamaguchi I, et al. J Antibiot (Tokyo) 1989;42:141. [PubMed] [Google Scholar]
- 2.Shim JH, Lee HK, Chang EJ, Chae WJ, Han JH, Han DJ, Morio T, Yang JJ, Bothwell A, Lee SK. Proc Natl Acad Sci U S A. 2002;99:10617. doi: 10.1073/pnas.162522099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adler JT, Cook M, Luo Y, Pitt SC, Ju J, Li W, Shen B, Kunnimalaiyaan M, Chen H. Mol Cancer Ther. 2009;8:914. doi: 10.1158/1535-7163.MCT-08-0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitsuhashi S, Shima H, Li Y, Tanuma N, Okamoto T, Kikuchi K, Ubukata M. Int J Oncol. 2008;33:1027. [PubMed] [Google Scholar]
- 5.Lee JH, Lee JS, Kim SE, Moon BS, Kim YC, Lee SK, Choi KY. Mol Cancer Ther. 2006;5:3222. doi: 10.1158/1535-7163.MCT-06-0455. [DOI] [PubMed] [Google Scholar]
- 6.Wilkinson B, Micklefield J. Nat Chem Biol. 2007;3:379. doi: 10.1038/nchembio.2007.7. [DOI] [PubMed] [Google Scholar]
- 7.Giraldes JW, Akey DL, Kittendorf JD, Sherman DH, Smith JL, Fecik RA. Nat Chem Biol. 2006;2:531. doi: 10.1038/nchembio822. [DOI] [PubMed] [Google Scholar]
- 8.Akey DL, Kittendorf JD, Giraldes JW, Fecik RA, Sherman DH, Smith JL. Nat Chem Biol. 2006;2:537. doi: 10.1038/nchembio824. [DOI] [PubMed] [Google Scholar]
- 9.Walsh CT. Science. 2004;303:1805. doi: 10.1126/science.1094318. [DOI] [PubMed] [Google Scholar]
- 10.Cheng XC, Ubukata M, Isono K. J Antibiot (Tokyo) 1990;43:890. doi: 10.7164/antibiotics.43.890. [DOI] [PubMed] [Google Scholar]
- 11.Choi SS, Hur YA, Sherman DH, Kim ES. Microbiology. 2007;153:1095. doi: 10.1099/mic.0.2006/003194-0. [DOI] [PubMed] [Google Scholar]
- 12.Li W, Luo Y, Ju J, Rajski SR, Osada H, Shen B. J Nat Prod. 2009 doi: 10.1021/np8007478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagao Y, Dai WM, Ochiai M, Tsukagoshi S, Fujita E. J Org Chem. 1990;55:1148. [Google Scholar]
- 14.Hodge MB, Olivo HF. Tetrahedron. 2004;60:9397. [Google Scholar]
- 15.Tsai SC, Lu H, Cane DE, Khosla C, Stroud RM. Biochemistry. 2002;41:12598. doi: 10.1021/bi0260177. [DOI] [PubMed] [Google Scholar]
- 16.Kittendorf JD, Beck BJ, Buchholz TJ, Seufert W, Sherman DH. Chem Biol. 2007;14:944. doi: 10.1016/j.chembiol.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caffrey P. Chembiochem. 2003;4:654. doi: 10.1002/cbic.200300581. [DOI] [PubMed] [Google Scholar]
- 18.Kittendorf JD, Sherman DH. Bioorg Med Chem. 2009;17:2137. doi: 10.1016/j.bmc.2008.10.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsai SC, Miercke LJ, Krucinski J, Gokhale R, Chen JC, Foster PG, Cane DE, Khosla C, Stroud RM. Proc Natl Acad Sci U S A. 2001;98:14808. doi: 10.1073/pnas.011399198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Witkowski A, Witkowska H, Smith S. J Biol Chem. 1994;269:379. [PubMed] [Google Scholar]
- 21.Gu L, Wang B, Kulkarni A, Gehret JJ, Lloyd KR, Gerwick L, Gerwick WH, Wipf P, Håkansson K, Smith JL, Sherman DH. J Am Chem Soc. 2009;131:16033. doi: 10.1021/ja9071578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.