

Abstract
Bone loss due to metabolic or hormonal disorders and osteolytic tumor metastasis continues to be a costly health problem, but current therapeutics offer only modest efficacy. Unraveling of the critical role for the receptor activator of nuclear factor-kappa B (RANK) and its ligand, RANK ligand (RANKL), in osteoclast biology provides an opportunity to develop more effective antiresorptive drugs. The in vivo effectiveness of RANKL inhibitors demonstrates the potency of the RANKL/RANK system as a drug target. Here, we report the development of cell-based assays for high-throughput screening to identify compounds that inhibit signaling from two RANK cytoplasmic motifs (PVQEET559-564 and PVQEQG604-609), which play potent roles in osteoclast formation and function. Inhibitors of these motifs' signaling have the potential to be developed into new antiresorptive drugs that can complement current therapies. The cell-based assays consist of cell lines generated from RAW264.7 macrophages stably expressing a nuclear factor-kappa B-responsive luciferase reporter and a chimeric receptor containing the human Fas external domain linked to a murine RANK transmembrane and intracellular domain in which only one of the RANK motifs is functional. With these cells, specific RANK motif activation after chimeric receptor stimulation can be measured as an increase in luciferase activity. These assays demonstrated >300% increases in luciferase activity after RANK motif activation and Z ′-factor values over 0.55. Our assays will be used to screen compound libraries for molecules that exhibit inhibitory activity. Follow-up assays will refine hits to a smaller group of more specific inhibitors of RANK signaling.
Introduction
In normal physiology, bone homeostasis is maintained by the paired processes of bone resorption (carried out by osteoclasts) and bone formation (carried out by osteoblasts).1–3 This delicate homeostatic balance can be tipped in favor of the osteoclasts and bone resorption by several conditions. The chronic inflammation associated with rheumatoid arthritis leads to localized bone loss as does the osteolytic metastasis of some cancers.4,5 A more global loss of bone can be seen in osteoporosis, which is most commonly seen in postmenopausal women who have experienced a dramatic decrease in hormone (particularly estrogen) levels.6 Four major antiresorptive drugs (agents capable of inhibiting osteoclast formation and/or function) are currently available on the market: estrogen, selective estrogen receptor modulators, bisphosphonates, and calcitonin.7–11 Nonetheless, these drugs either offer only modest efficacy or may cause adverse side effects in clinical management of various bone disorders.11–14 Thus, there is a need for development of more efficacious and safer antiresorptive drugs.
Currently, the most attractive target for antiresorptive therapy is the receptor activator of nuclear factor-kappa B ligand (RANKL)/receptor activator of nuclear factor-kappa B (RANK) system. Together with the monocyte/macrophage colony stimulating factor (M-CSF), the interaction between RANK, located on the plasma membrane of bone marrow macrophages, and RANKL, present on the plasma membrane of bone stromal cells and osteoblasts and as an unbound, soluble variant, is both necessary and sufficient to induce differentiation into osteoclasts.15 In addition, the RANKL/RANK system also plays a potent role in the function and survival of differentiated osteoclasts.16 Notably, denosumab, an anti-RANKL antibody developed by Amgen that functions to block the RANKL-RANK interaction, has shown great therapeutic potential in clinical trials.17–19 As potent and clinically effective as such a protein-based approach would be in reducing bone loss, the cost of manufacturing and the means of delivery may stand as barriers to its widespread application. Further, since RANK has functions in biological processes beyond osteoclasts, global inhibition of the entirety of RANK's signaling via the blockage of the RANKL-RANK interaction is likely to be accompanied by side effects in other cells that utilize the RANKL/RANK system.20 As such, while targeting the RANKL-RANK interaction is a viable means for reducing bone resorption, a better approach would be to target individual RANK signaling pathways that are more specific to osteoclast formation and function.
RANK was identified as a member of the tumor necrosis factor receptor (TNFR) superfamily.21 As TNFR family members primarily employ TNFR-associated factors (TRAFs) to transmit downstream signaling, numerous studies have been performed to characterize RANK's TRAF-dependent signaling pathways, and these in vitro biochemical studies have collectively identified six TRAF binding motifs (Motifs 1, 2, 3, 4, 5, and 6) in the RANK cytoplasmic domain (Fig. 1).22–29 Our group has subsequently demonstrated that three of these TRAF-binding motifs (Motif 3: PFQEP,369-373 Motif 5: PVQEET,559-564 and Motif 6: PVQEQG604-609) play a functional role in osteoclast formation and function.30 Moreover, all three functional motifs activate the NF-κB signaling pathway in osteoclast precursors.30 Recently, we thoroughly evaluated the potential of these RANK functional motifs as therapeutic targets.31 Given that mutational inhibition of either Motif 5- or Motif 6-mediated signaling pathways in osteoclast precursors results in a dramatic reduction in osteoclast formation and function, Motif 5- and Motif 6-mediated signaling pathways can serve as effective antiresorptive targets.30,31 Moreover, mutation of any TRAF-binding motif other than Motif 5 or 6 does not greatly impact osteoclastogenesis, though motifs other than 5 and 6 may contribute to other aspects of RANK signaling. Thus, pharmacological blockage of Motif 5- and/or Motif 6-initiated signaling pathways should effectively inhibit osteoclast formation and reduce bone resorption when applied in a clinical setting.31
Fig. 1.

Diagrams showing the key components of the cell-based screening assays (hFas-W5 and hFas-W6) and two control assays (hFas-WT and hFas-P1-6). (A) hFas-W5 and hFas-W6 are designed to observe the effects of compounds on the signaling of individual RANK TRAF-binding motifs. (B) hFas-WT and hFas-P1-6 were used to determine maximum and minimum signaling activation for a given assay configuration. α-Fas induces an oligomerization of the chimeric receptor that fully activates the signaling of motifs that are not mutated. This leads to an increase in NF-κB translocation to the nucleus, and, as a consequence, an increase in luciferase gene activation. α-Fas, anti-human Fas activating antibody; hFas, human Fas; Luc, luciferase; NF-κB-RE, nuclear factor-kappa B responsive element; RANK, receptor activator of nuclear factor-kappa B; TRAF, TNFR associated factors.
The potential for Motif 5 and Motif 6 to be highly effective antiresorptive targets prompted us to propose two cell-based assays (human Fas [hFas]-W5 and hFas-W6) for identifying compounds that block Motif 5- and/or Motif 6-mediated signaling pathways in osteoclast precursors (Fig. 1A). Each assay consists of a cell line generated from the RAW264.7 macrophage line stably expressing an nuclear factor-kappa B (NF-κB)-responsive luciferase reporter and one hFas-RANK chimeric receptor.31 The chimeric receptor allows for specific activation of targeted RANK motifs without interference from endogenous RANK; the chimeric receptor is activated not by RANKL, but by an anti-human Fas activating antibody (α-Fas) that specifically activates human (but not mouse) Fas (Fig. 2). hFas-W5 cells express an hFas-RANK chimeric receptor in which all motifs except Motif 5 are mutated (Fig. 1A). When hFas-W5 cells are treated with α-Fas, signaling through the chimeric receptor's Motif 5 is initiated, ultimately leading to an increase in NF-κB activation, which can be measured as an increase in luciferase expression and activity. If hFas-W5 is treated with a compound that can block the signaling of Motif 5, the induction of luciferase activity and, thus, luminescence after antibody treatment will be reduced. Inhibitors of Motif 6 signaling can likewise be identified using hFas-W6 cells (Fig. 1A). In addition, we have also generated two control assays (hFas-P1-6 and hFas-WT) (Fig. 1B).31 hFas-P1-6, in which all putative TRAF-binding motifs are mutated, serves as a negative control against which hFas-W5 and hFas-W6 baseline luciferase induction under different conditions can be measured. hFas-WT, in which all putative TRAF-binding motifs remain intact, can be used to assess the effects of varying assay conditions on unmodified RANK intracellular signaling.31
Fig. 2.

Rationale for use of a chimeric receptor consisting of hFas external domain linked to the transmembrane and intracellular domains of mouse RANK for the cell-based assays. Using a chimeric RANK receptor (left side) allows for specific activation of only the motifs of interest, whereas a mutated RANK (right side) would require RANKL treatment, which would also activate endogenous RANK. Endogenous RANK activation would make identification of motif-specific inhibitors difficult. ◊, α-Fas; Δ, RANK ligand (RANKL).
Here, we report the development of four cell-based assays (Fig. 1) for high-throughput screening (HTS) identification of small molecules that are capable of inhibiting TRAF-binding motif-specific RANK signaling.
Materials and Methods
Chemical and Reagents
Chemicals were purchased from Sigma unless indicated otherwise. NF-κB Activation Inhibitor II (Cat No. 481408) and Bay 11-7082 (Cat No. 196870) were purchased from EMD Chemicals. Dulbecco's modified Eagle's medium (DMEM) (Cat No. 10-013-CV), L-glutamine (Cat No. 25-005-CI), tetracycline (Cat No. 61-242-RG), G418 (Cat No. 61-234-RF), hygromycin B (Cat No. 30-240-CR), and puromycin (Cat No. 61-385-RA) were purchased from Mediatech. Fetal bovine serum was purchased from Invitrogen (Cat No. 26140-079). Recombinant GST-RANKL was purified as previously described.32 Mouse M-CSF was prepared from a M-CSF-producing cell line, CMG14-12, which was constructed and kindly provided by Dr. Sunao Takeshita.33
Culturing of RAW264.7 Cells
RAW264.7 (Cat No. TIB-71) cells were purchased from American Type Culture Collection and cultured at 37°C and 5% CO2 in treated tissue culture plates with DMEM containing 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, and 25 IU/mL penicillin/streptomycin. RAW264.7 cells were passaged by mechanical lifting with cell scrapers.
Stable Transfection of RAW264.7 Cells with NF-κB-Luciferase Reporter Plasmid
About 1 × 106 RAW264.7 cells were seeded into a 60 mm tissue culture-treated dish and allowed to attach and grow for 24 h. Eight micrograms of pGL4.32[luc2P/NF-κB-RE/Hygro] vector (Cat No. E8491) from Promega was transfected into seeded cells using Lipofectamine Plus™ from Invitrogen (Cat No. 15338-100) according to the manufacturer's instructions. Forty-eight hours after transfection, cells were selected with 300 μg/mL hygromycin B until resistant colonies appeared. Two colonies were isolated after emergence and the remaining colonies were allowed to expand into a mixed pool.
Construction of hFas-RANK Chimeric Receptor Retroviral Vectors
The TNFR external domain was cut out of five previously prepared TNFR-RANK chimeric receptor constructs using Xba I and Spe I.30 The differences between RANK intracellular region constructs were as follows: one in which only Motif 5 is fully functional (W5), one in which only Motif 6 is fully functional (W6), one in which no motif is fully functional (P1-6), and one in which all of the motifs are fully functional (WT). The hFas external region was cloned out of a previously prepared hFas-TNFR chimeric receptor construct using Xba I and Spe I and cloned into each of the previous plasmids between the Xba I and Spe I sites to generate the pBluescript-SK-hFas-(W5, W6, P1-6, and WT) constructs.34 The resulting chimeric hFas-RANK construct region was cloned out of pBluescript-SK using Xba I and BamH I and cloned between the Xba I and BamH I sites of pMX-Puro to generate the pMX-puro-hFas-(W5, W6, P1-6, and WT) retroviral vectors.
Preparation of Retrovirus
293GPG retrovirus packaging cells were kindly provided by Dr. Daniel S. Ory at Washington University and were cultured in DMEM containing 10% heat inactivated fetal bovine serum and supplemented with 500 μg/mL G418, 2 mM L-glutamine, 25 IU/mL penicillin/streptomycin, 2 μg/mL puromycin, and 1 μg/mL tetracycline as described.35 pMX-based retroviral vectors encoding the chimeric receptors were transiently transfected into 293GPG retrovirus packaging cells using the Lipofectamine Plus from Invitrogen according to the manufacturer's instructions. After the transient transfection, cells were cultured in DMEM supplemented with 10% heat inactivated fetal bovine serum and 2 mM L-glutamine. Virus-containing culture supernatant was collected at 48, 72, and 96 h after transfection and filtered through 0.45 μm low protein-binding filters. Supernatants were stored at −80°C.
Retroviral Infections of RAW264.7 Cells
RAW264.7 stable lines expressing the luciferase construct were seeded into 60 mm treated culture dishes at a density of 3 × 105 cells per dish. About 24 h later, the growth medium was aspirated from cells and cells were then infected with 1 mL prepared virus-containing supernatants and 1 mL growth medium in the presence of 8 μg/mL polybrene for 24 h. After the 24-h infection, virus-containing medium was removed and cells were cultured with growth medium for additional 24 h. Positively infected cells were then selected and maintained in growth medium containing 2 μg/mL puromycin.
Luciferase Assays
About 2 × 104 cells were seeded into the wells of clear (Costar® cell culture plate; Cat No. 07-200-90; Corning) or white (CulturPlate™-96; Cat No. 6005680; PerkinElmer) tissue culture-treated 96-well plates in 100 μL culture medium and allowed to attach and grow for 16 h. One hundred microliters of culture medium containing 2 × concentrations of either RANKL or α-Fas (CH11) (Millipore) (Cat No. 05-201) were added to cells cultured in 100 μL medium, and plates were incubated for varying times at 37°C. For experiments with NF-κB inhibitor, inhibitor was diluted to 500 × its working concentration in dimethyl sulfoxide (DMSO), diluted 1/250 into culture medium, and then added to cultured cells (under 100 μL culture medium) at a volume of 100 μL for a final 1 × concentration. When a flash-type substrate was used, the cells were washed once with phosphate-buffered saline and lysed in 30 μL passive lysis buffer (Cat No. E1941; Promega). Luciferase activity was measured by injecting 100 μL luciferase reagent (Cat No. E1483; Promega) directly into each well followed by a 1 s luminescence read using a microplate luminometer with automatic injector. When a glow-type substrate was used, 100 μL BriteLite™ Plus (Cat No. 50-904-9934; PerkinElmer) was added directly to each well with no washing, and plates were read at 1 s/well using a microplate luminometer. All treatments were performed in triplicate unless otherwise noted. The steps of the assay procedure are summarized in Table 1.
Table 1.
Assay protocol
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 |
Plate cells |
100 μL |
2 × 104 Assay cells |
| 2 |
Incubation time |
16 h |
37°C, 5% CO2 |
| 3 |
Reporter induction |
100 μL |
Induce NF-κB, Luciferase reporter |
| 4 |
Incubation time |
6 h |
37°C, 5% CO2 |
| 5 |
Remove medium |
N/A |
Remove via aspiration |
| 6 |
Wash |
100 μL |
Phosphate-buffered saline |
| 7 |
Remove PBS |
N/A |
Remove via aspiration |
| 8 |
Lyse cells |
30 μL |
Passive lysis buffer |
| 9 |
Reporter reagent |
100 μL |
Flash-type substrate |
| 10 | Assay readout | N/A | Microplate luminometer |
| Step Notes | |||
|---|---|---|---|
| 1. Solid white tissue culture plates. Tip dispense | |||
| 3. Tip dispense, 200 ng/mL α-hFas (final concentration 100 ng/mL) + 2× assay concentration library compound in 100 μL culture medium | |||
| 10. 1 s/well | |||
| NB: If a glow-type substrate is used, omit steps 5–8. Add 100 μL substrate directly to well. |
PBS, phosphate-buffered saline.
In Vitro Osteoclastogenesis
About 2 × 105 cells were seeded into the wells of a 12-well plate with 100 ng/mL RANKL. Medium with treatment was refreshed 48 h later. Untreated cells were maintained in culture medium without RANKL. The osteoclastogenesis cultures were stained for tartrate-resistant acid phosphatase (TRAP) expression with Leukacyte Acid Phosphatase Kit (Cat No. 387-A) from Sigma 96 h after seeding.
Data Analysis
Dose–response curves were drawn and IC50 values were calculated with a 4-parameter logistic fit using SigmaPlot 10. All other plots and statistical analyses were performed using Microsoft Excel 2007.
Results
Establishment and Characterization of RAW264.7 Stable Line Expressing an NF-κB-responsive Luciferase Reporter
RAW264.7 cells were chosen for the development of the cell-based assays because, among macrophage cell lines, RAW264.7 cells are unique in their ability to differentiate into osteoclast-like cells.36,37 This indicates that RAW264.7 cells retain all signaling components required for osteoclastogenic RANK signaling. Initially, multiple NF-κB reporters were examined, but ultimately Promega's pGL4.32 reporter was chosen for use due to its low background activation and rapid luciferase response time (Data not shown). We obtained two clones stably expressing Promega's pGL4.32 reporter (Clone 1 and Clone 2), which were derived from two isolated colonies that emerged from 3-weeks of hygromycin B selection after transfection. We also prepared a heterogeneous population of RAW264.7 cells stably expressing the reporter by expanding and pooling the remaining hygromycin B-resistant colonies (mixed pool).
To determine the responsiveness of the NF-κB reporter, we treated the mixed pool, Clone 1, and Clone 2 with 100 μg/mL RANKL for 8 h before measuring their induced luciferase activity. While the level of luciferase activity from the RANKL-treated mixed pool was over 10-fold higher than that of untreated cells, the luminescence counts were markedly lower than those measured from clone 1 and clone 2 cells (Fig. 3). Further, clone 1 and clone 2 cells also demonstrated higher luciferase induction than the mixed population (Fig. 3B). Treatment of clone 1 and clone 2 cells with RANKL for 2, 4, 6, and 8 h revealed that a treatment time of 6 h is sufficient for maximum luciferase induction (Fig. 3C), and while Clone 2 cells appeared to demonstrate a higher induction fold than Clone 1 cells in a single time point experiment in the initial assay (Fig. 3B), the more thorough, multi-time point assessment revealed that Clone 1 consistently showed higher induction folds than Clone 2 (Fig. 3C). Because of this, Clone 1 was chosen for further assay development.
Fig. 3.

Responsiveness of NF-κB-luciferase reporter-expressing RAW264.7 cell to RANKL treatment. About 2 × 104 cells were seeded into the wells of clear tissue culture-treated 96-well plates in 100 μL culture medium and allowed to attach and grow for 24 h. One hundred microliters of culture medium containing 200 ng/mL RANKL was added to the cells for a final concentration of 100 ng/mL, and cells were incubated for 8 h at 37°C at 5% CO2. (A) Clone 1 and Clone 2 displayed greater luminescence intensity than the mixed pool (▪, untreated; □, 100 ng/mL RANKL). (B) Clone 1 and Clone 2 also displayed greater luciferase inductions than the mixed pool. (C) Time-dependant fold inductions for Clone 1 and Clone 2 (•, clone 1; ▪, clone 2). All treatments were performed in triplicate; error bars = standard deviation. Experiment was repeated three times; representative data are shown.
Construction and Initial Characterization of hFas-W5, hFas-W6, hFas-WT, and hFas-P1-6
Viruses encoding pMX-puro-hFas-(W5, W6, P1-6, and WT) were prepared by transiently transfecting the vectors into 293GPG packaging cells as described in the Materials and Methods section. Clone 1 cells were infected with these viruses and selected with puromycin. Cells infected with viruses encoding different chimeric receptors gave rise to hFas-W5, hFas-W6, hFas-WT, and hFas-P1-6 cells (Fig. 1). The infected cells were maintained in hygromycin and puromycin-containing medium to ensure continued presence of both the NF-κB reporter and chimeric receptor. To determine the responsiveness of each assay to α-Fas and to test whether the antibody stimulates a luciferase response through cross-reactions with receptors other than the chimeric receptor, parental Clone 1 cells and cells of hFas-W5, hFas-W6, hFas-WT, and hFas-P1-6 were treated with 50, 100, or 150 ng/mL α-Fas antibody. Assay cell lines demonstrated a dose-dependant response to the α-Fas antibody, with hFas-WT cells showing the strongest luciferase response and hFas-P1-6 the weakest (Fig. 4A). Although hFas-P1-6 cells demonstrated an increase in luciferase induction with increasing doses of antibody, hFas-W5, hFas-W6, and hFas-WT cells consistently exhibit higher inductions at all concentrations, with a marked increase in luciferase activity over untreated controls and hFas-P1-6 cells at 100 ng/mL (Fig. 4B). Because of this, we used 100 ng/mL α-Fas throughout the reminder of the assay development process. Clone 1 cells showed an apparent response to the antibody, but this response was not commensurate with antibody concentration. Further, when clone 1 cells were treated in a white plate, no increases in luciferase activation were observed (data not shown), indicating that the increases seen when the assay was previously performed were likely due to signal cross-talk from neighboring wells.
Fig. 4.

The responsiveness of parental Clone 1, hFas-WT, hFas-W5, hFas-W6, and hFas-P1-6 to α-Fas treatment. About 2 × 104 cells were seeded into the wells of clear 96-well plates in 100 μL culture medium and allowed to attach and grow for 24 h. One hundred microliters of culture medium containing various 2 × concentrations of α-Fas was added to the cells, and cells were incubated for 6 h at 37°C at 5% CO2. (A) Luminescence displayed by parental Clone 1, hFas-WT, hFas-W5, hFas-W6, and hFas-P1-6 cells in response to increasing concentrations of α-Fas (▪, 0 ng/mL; □, 50 ng/mL;  , 100 ng/mL;
, 100 ng/mL;  , 150 ng/mL). (B) Percent increase in luciferase activity of treated hFas-P1-6, hFas-W5, hFas-W6, and hFas-WT cells over that of their untreated counterparts (▪, hFas-P1-6; □, hFas-W5; , hFas-W6; , hFas-WT). All treatments were performed in triplicate; error bars = standard deviation. Experiment was repeated two times. Representative data are shown.
, 150 ng/mL). (B) Percent increase in luciferase activity of treated hFas-P1-6, hFas-W5, hFas-W6, and hFas-WT cells over that of their untreated counterparts (▪, hFas-P1-6; □, hFas-W5; , hFas-W6; , hFas-WT). All treatments were performed in triplicate; error bars = standard deviation. Experiment was repeated two times. Representative data are shown.
Assay Cell Lines Retain Capacity for RANKL-Induced Osteoclastogenesis
To determine whether the assay cell lines we have developed retain all the necessary components of RANK signaling and, thus, are appropriate for a RANK signaling inhibitor screen, we performed in vitro osteoclast formation assays with Clone 1, hFas-W5, hFas-W6, hFas-WT, and hFas-P1-6 cells. All cell lines were able to differentiate into large, multinuclear, TRAP-positive (TRAP+) osteoclast-like cells when treated with 100 ng/mL recombinant RANKL for 96 h, whereas untreated Clone 1 cell remained mononuclear and TRAP− (Fig. 5). This indicates that the cell line development process did not damage osteoclastogenic components of RANK signaling in any of the assay cell lines.
Fig. 5.

RANKL-induced osteoclastogenesis in parental Clone 1, hFas-WT, hFas-W5, hFas-W6, and hFas-P1-6 cells. About 2 × 105 cells seeded into the wells of a 12-well plate with 100 ng/mL RANKL. Medium was refreshed 48 h later. Cells were TRAP stained 96 h after seeding. All cell lines were capable of forming large, multinuclear, and TRAP+ osteoclasts, indicating that all assay cell lines retain osteoclast-critical RANK signaling components. Untreated Clone 1 cells were mononuclear and TRAP−. TRAP, tartrate-resistant acid phosphatase. Color images available online at www.liebertonline.com/adt.
NF-κB Inhibitors Reduce Induction of Luciferase by hFas-W5 and hFas-W6 Cells in a Dose-Dependant Manner
To further assess whether hFas-W5 and hFas-W6 are suitable for use in an HTS setting, we addressed whether the induced reporter activity of hFas-W5 and hFas-W6 cells can be suppressed by established NF-κB inhibitors. To this end, we determined the effect of two known small molecule NF-κB inhibitors (NF-κB Activation Inhibitor II and Bay 11-7082; Calbiochem) on α-Fas-induced luciferase induction. hFas-W5 and hFas-W6 cells were either untreated or treated with 100 ng/mL α-Fas plus vehicle (DMSO) or an equal volume of DMSO containing increasing concentrations of inhibitor, and their luminescence was measured. Using these data, percent inhibition was calculated for both cell lines and both inhibitors at each concentration using the following equation (μDMSO, mean luminescence with α-Fas and vehicle treatment; ν[Inhibitor], luminescence from individual well treated with α-Fas and inhibitor):
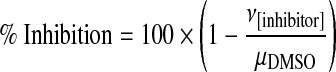 |
(1) |
Although α-Fas was still able to significantly activate expression of the reporter in the presence of DMSO in both hFas-W5 and hFas-W6 cells, addition of either NF-κB inhibitor reduced luminescence in a dose-dependant manner, and the inhibitory effect vs. concentration of both inhibitors followed a typical sigmoidal curve. On the basis of the dose–response curves (Fig. 6A, B), IC50 values for each inhibitor were calculated for both hFas-W5 and hFas-W6. These values are reported in Table 2. IC50 values for both inhibitors and both cell lines were acceptably similar to published values and those documented by the manufacturer.38,39 These data further support the suitability of hFas-W5 and hFas-W6 cells for use in HTS.
Fig. 6.

Effect of a known NF-κB inhibitors on induction of luciferase by hFas-W5 and hFas-W6 cells. About 2 × 104 cells were seeded into the wells of white tissue culture-treated 96-well plates in 100 μL culture medium and allowed to attach and grow for 24 h. About 100 μL of 200 ng/mL α-Fas and varying 2 × concentrations (dimethyl sulfoxide, 1.5625, 3.125, 6.25, 12.5, 25, 50, and 100 μM) of either NF-κB Activation Inhibitor II or Bay 11-7084 was added to cells and incubated for 6 h at 37°C at 5% CO2. Plots compare inhibitory effects of NF-κB Activation Inhibitor II (•) and Bay 11-7082 (○) on (A) hFas-W5 and (B) hFas-W6. IC50 values are reported in Table 2 alongside published values.38,39 Plotted data points are means ± standard deviation. All treatments were performed in triplicate.
Table 2.
IC50 Values
| Sample | NF-κB Activation Inhibitor II IC50 (μm) | Bay 11-7082 IC50(μm) |
|---|---|---|
| hFas-W5 |
18.69 |
6.74 |
| hFas-W6 |
26.47 |
7.11 |
| Published value | 7.1 | 2-10 |
Assessment of Well-to-Well Variability and Z′-Factor Calculation
To evaluate whether hFas-W5 and hFas-W6 are suitable for use in an HTS setting, we first determined the robustness of these two assays. The Z′-factor analysis has been developed as a useful tool to determine the robustness of HTS assays.40 The Z′-test was run by dividing a white 96-well plate into four equal quadrants. Two quadrants contained hFas-W5 or hFas-W6 cells treated with 100 ng/mL α-Fas (Treated), and the remaining two quadrants contained cells that were not treated with α-Fas (Untreated). After treatment, cells were lysed and assessed luminometrically with either the flash-type luciferase substrate that we used previously during assay development or a glow-type substrate that is more commonly used in HTS. After measuring the luciferase values of each well, the Z′-factor value was calculated using the following equation (σ, standard deviation; μ, mean luminescence):
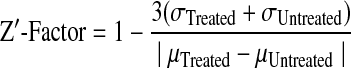 |
(2) |
This test quantifies the relationship between the assay's induced and baseline reporter activation, variation among measured baseline signals, and variation among induced signals.40 Both hFas-W5 and hFas-W6 cells exhibited induction levels similar to those seen in previous experiments. Z′-factor values for both flash-type and glow-type substrates are reported in Table 3. The calculated Z′-factor values indicate that our assays are suitable for HTS and standard glow-type substrates are appropriate for use with our assays.
Table 3.
Z′-Factor Values
| Assay | Z′-Factor: Flash Reagent | Z′-Factor: Glow Reagent |
|---|---|---|
| hFas-W5 |
0.56 |
0.64 |
| hFas-W6 | 0.61 | 0.61 |
Discussion
Despite continuous efforts to find new methods of clinical management, pathologic bone loss continues to present healthcare challenges. If a treatment is to be successful, it must meet three qualifications: (1) it must effectively provide relief from a pathologic state, (2) it must not have side effects severe enough to deter application, and (3) it must be simple, convenient, and affordable enough to ensure patient accessibility and compliance. No current antiresorptive therapy is capable of fulfilling all three requirements, highlighting a need for development of better antiresorptive drugs. Anti-RANKL antibodies have promise to display greater potency than bisphosphonates, but the problems of cost and safety remain to be addressed. Protein-based therapies are consistently several times higher in cost than small-molecule drugs. As such, there is a great impetus to develop new methods of discovering small molecule compounds that can be utilized in the development of affordable treatments that are not only effective, but also accessible. Indiscriminate inhibition of the totality of RANK signaling also presents a potential for side-effects. Given that RANK signaling has been shown to be an important regulator of dendritic cell survival and activation, T-cell activation, and B-cell differentiation, the possibility that total RANK inhibition may perturb immune responses may preclude such an approach.20,41,42 Our goal is to identify small molecule inhibitors of downstream RANK signaling that is essential to osteoclastogenesis. Such inhibitors could have the potency of total RANK inhibition, but because small molecules are relatively less expensive to produce than peptides, they would be more affordable than antibody therapy. More specific inhibition of RANK signaling by small molecules should also reduce the potential for side-effects on other RANK-utilizing cells.
As previously described, the assay system we have developed to exploit the potential specificity of RANK motifs PVQEET559-564 and PVQEQG604-609 is based upon three components: the murine macrophage cell line RAW264.7, an NF-κB-responsive firefly luciferase reporter, and a chimeric receptor consisting of the extracellular region of the hFas receptor linked to the transmembrane and intracellular regions of murine RANK (Fig. 2).31 When a cell expressing both the luciferase reporter and chimeric receptor is treated with an activating antibody directed specifically against hFas, the chimeric receptors on the plasma membrane activate signaling through the unmutated TRAF-binding motif that ultimately promotes an increase in NF-κB signaling (Fig. 2, left side). The increased translocation of NF-κB to the nucleus stimulates the expression of luciferase via interaction with an NF-κB-responsive enhancer element. This increased expression level of luciferase can then be quantified via luminometric means. The use of a chimeric receptor allows us to eliminate NF-κB activation by endogenous RANK; for example, if we were to use a mutated RANK construct, we would be forced to use RANKL to activate signaling through the modified RANK's unmutated TRAF-binding motif. This RANKL treatment would also activate endogenous RANK and make interpretation of motif activity difficult (Fig. 2, right side). The chimeric receptor allows us to specifically activate only the mutant receptor with an antibody that reacts only with hFas, thus ensuring that NF-κB will be activated only by assay-specific motifs.
As was seen, assay cell lines demonstrate a different response to α-Fas treatment according to antibody concentration. It is not surprising that hFas-WT demonstrated the highest level of induction, where hFas-W5 and hFas-W6 showed lower levels of induction; the TRAF-binding motifs of hFas-WT are nonmutated and fully functional, where hFas-W5 and hFas-W6 bear inactivating mutations on all but a single motif. It is important to note, however, that the inactivating mutations do not result in the absolute ablation of signaling from the mutated motif. The point mutations applied to the different chimeric receptors were designed to reduce the signaling from specific motifs without significantly impacting the functionality of neighboring motifs. The requirement that mutations of one motif not interfere with the signaling of another compelled us to choose careful point mutations that cannot completely inactivate motif signaling. As a consequence, hFas-P1-6 cells retain some luciferase induction in response to the α-hFas antibody (Fig. 4). Nevertheless, hFas-W5 and hFas-W6 cells demonstrate a greater induction of luciferase than hFas-P1-6 cells, and, at 100 ng/mL, the difference in signal intensity between induced and noninduced hFas-W5 and hFas-W6 cells is high enough for screening purposes (Fig. 4B). In addition, at this concentration, hFas-W5 and hFas-W6 cells show increases over baseline that are more than double what is seen in hFas-P1-6 cells (Fig. 4B).
In comparison between hFas-W5, hFas-W6, and hFas-WT cells, it is clear that hFas-WT cells are capable of greater levels of luciferase induction than hFas-W5 or hFas-W6. This is not surprising, as hFas-WT retains full signaling functionality of all of its motifs. Although it would be preferable to increase the luciferase induction of hFas-W5 and hFas-W6 cells, the levels measured during the assay development process reflect the biology of the chimeric receptors. Further, for the purpose of screening compounds libraries, the inductions demonstrated by hFas-W5 and hFas-W6 coupled with their low well-to-well variability point to their sufficiency for identifying signaling inhibitors, and our experiments with NF-κB inhibitors indicate that the cells of hFas-W5 and hFas-W6 cells are sensitive enough to signaling inhibition that measurable decreases in signaling can be consistently detected.
The characterization of our cell-based assays supports their applicability toward HTS. It has been reported that if an assay's Z′-factor is ≥0.5, it is considered an excellent assay and generally a single test of a compound is sufficient.40 Given that the Z′-factor values for hFas-W5 and hFas-W6 are >0.5 regardless of luciferase substrate used, our assays are well suited for use in an HTS setting. Moreover, using known small molecule NF-κB inhibitors, we have also demonstrated that our assays are sensitive enough to identify signaling inhibitors (Fig. 6). Finally, the following factors support the cost effectiveness of our assays: (a) since RAW264.7 cells divide very rapidly and are easy to culture, large amounts of hFas-W5 and hFas-W6 cells for HTS assays can be obtained easily and rapidly; (b) the cell lysis/luciferase reagents that are widely used for HTS are inexpensive (<10 cents/well); (c) we have shown that 100 ng/mL is a reasonable dose for the cell-based assays, and, at a cost of $379 for 50 μg (enough for assays involving one hundred 96-well plates in which each well contains 100 μL medium), α-Fas will not contribute significantly to the cost of a screen.
In the early development of our assays we compared flash-type and glow-type luciferase substrate. In comparison with a glow-type luciferase substrate, we found that, while induction folds were similar between flash and glow substrates, raw luminescence counts were nearly 10-fold higher in both hFas-W5 and hFas-W6 when a flash-type reagent was used. For this reason, we used a flash-type reagent during much of the assay's development. Nevertheless, glow-type reagents are more commonly used in HTS due to the convenience of adding the substrates directly to cultured cells and the stability of their signals. Thus, after developing our assay, we validated the reproducibility of the luciferase response by performing Z′ analyses using both the flash-type reagent we used throughout development and a more common glow-type substrate. We found that both substrates give similar Z′-factor values, indicating that either a flash-type or glow-type reagent may be used according to the needs of the screening process.
In this report, we have described the development of two cell-based assays for identifying inhibitors of RANK signaling. In the process we have determined that assays should be conducted in HTS standard white plates, and either a flash-type or glow-type luciferase substrate is suitable. It is likely that the assay will be scaled down to a 384- or 1536-well format before screening, and we have confidence in the flexibility of our assays to accommodate this change.
In conclusion, throughout the development of these assays, we had three primary goals: to create an assay that was (1) capable of producing a robust, measurable response to RANK motif signaling activation, (2) simple and fast enough to allow its adoption into HTS systems, and (3) inexpensive enough as to not be prohibitively expensive to screen large chemical libraries. We believe that we have achieved these goals.
Abbreviations
- α-Fas
anti-human Fas activating antibody
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethyl sulfoxide
- hFas
human Fas
- HTS
high-throughput screening
- RANK
receptor activator of nuclear factor-kappa B
- NF-κB
nuclear factor-kappa B
- M-CSF
monocyte/macrophage-colony stimulating factor
- PBS
phosphate-buffered saline
- RANKL
RANK ligand
- TNF
tumor necrosis factor
- TNFR
TNF receptor
- TRAFs
TNFR associated factors
- TRAP
tartrate-resistant acid phosphatase
Acknowledgments
This work was supported by grant number NS061677 (to X.F.) from National Institute of Neurological Disorders and Stroke (NINDS), grant number AR47830 (to X.F.) from National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), the University of Alabama at Birmingham Marie and Emmett Carmichael Scholarship for Graduate Studies in Biosciences (to E.M.M.), and a training fellowship (to J.W.A.) from the University of Alabama at Birmingham's Center for Metabolic Bone Disease (T32 AR047512-09). We would like to thank Joanne Murphy-Ullrich, Ph.D., and Danny R. Welch, Ph.D., for use of equipment and the University of Alabama at Birmingham Center for AIDS Research sequencing core.
Disclosure Statement
No competing financial interests exist.
References
- 1.Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- 2.Teitelbaum SL. Ross FP. Genetic regulation of osteoclast development and function. [Review] Nat Rev Genet. 2003;4:638–649. doi: 10.1038/nrg1122. [DOI] [PubMed] [Google Scholar]
- 3.Lian J. Stein GS. Osteoblast biology. In: Marcus R, editor; Feldman D, editor; Nelson DA, editor; Rosen CJ, editor. Osteoporosis. Elsevier Academic Press; San Diego: 2008. pp. 93–150. [Google Scholar]
- 4.Goldring SR. Pathogenesis of bone and cartilage destruction in rheumatoid arthritis [Review] Rheumatology. 2003;42(Suppl 2):ii11–ii16. doi: 10.1093/rheumatology/keg327. [DOI] [PubMed] [Google Scholar]
- 5.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. [Review] Nat Rev Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 6.Raisz LG. Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J Clin Invest. 2005;115:3318–3325. doi: 10.1172/JCI27071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stefanick ML. Estrogens and progestins: background and history, trends in use, and guidelines and regimens approved by the US Food and Drug Administration. Am J Med. 2005;118(Suppl 12B):64–73. doi: 10.1016/j.amjmed.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 8.Prince R. Muchmore DB. Siris ES. Estrogen analogues: selective estrogen receptor modulators, phytoestrogens. In: Marcus R, editor; Feldman D, editor; Nelson DA, editor; Rosen CJ, editor. Osteoporosis. Academic Press; San Diego: 2008. pp. 1705–1723. [Google Scholar]
- 9.Miller P. Bisphosphonates: pharmacology, use in the treatment of osteoporosis. In: Marcus R, editor; Feldman D, editor; Nelson DA, editor; Rosen CJ, editor. Osteoporosis. Academic Press; San Diego: 2008. pp. 1725–1742. [Google Scholar]
- 10.Gruber HE. Ivey JL. Baylink DJ, et al. Long-term calcitonin therapy in postmenopausal osteoporosis. Metabolism. 1984;33:295–303. doi: 10.1016/0026-0495(84)90187-2. [DOI] [PubMed] [Google Scholar]
- 11.Miller PD. Derman RJ. What is the best balance of benefits and risks among anti-resorptive therapies for postmenopausal osteoporosis? Osteoporos Int. 2010. [Epub ahead of print]. [DOI] [PubMed]
- 12.Stepan JJ. Alenfeld F. Boivin G. Feyen JH. Lakatos P. Mechanisms of action of antiresorptive therapies of postmenopausal osteoporosis. [Review] Endocr Regul. 2003;37:225–238. [PubMed] [Google Scholar]
- 13.Lufkin EG. Sarkar S. Kulkarni PM, et al. Antiresorptive treatment of postmenopausal osteoporosis: review of randomized clinical studies and rationale for the Evista alendronate comparison (EVA) trial. [Review] Curr Med Res Opin. 2004;20:351–357. doi: 10.1185/030079904125003071. [DOI] [PubMed] [Google Scholar]
- 14.Marcus R. Wong M. Heath H., III Stock JL. Antiresorptive treatment of postmenopausal osteoporosis: comparison of study designs and outcomes in large clinical trials with fracture as an endpoint. [Review] Endocr Rev. 2002;23:16–37. doi: 10.1210/edrv.23.1.0453. [DOI] [PubMed] [Google Scholar]
- 15.Boyle WJ. Simonet WS. Lacey DL. Osteoclast differentiation and activation. [Review] Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 16.Lacey DL. Timms E. Tan HL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 17.Lewiecki EM. Current and emerging pharmacologic therapies for the management of postmenopausal osteoporosis. J Womens Health (Larchmt) 2009;18:1615–1626. doi: 10.1089/jwh.2008.1086. [DOI] [PubMed] [Google Scholar]
- 18.Miller PD. Denosumab: anti-RANKL antibody. Curr Osteoporos Rep. 2009;7:18–22. doi: 10.1007/s11914-009-0004-5. [DOI] [PubMed] [Google Scholar]
- 19.Cummings SR. San Martin J. McClung MR, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361:756–765. doi: 10.1056/NEJMoa0809493. [DOI] [PubMed] [Google Scholar]
- 20.Fouque-Aubert A. Chapurlat R. Influence of RANKL inhibition on immune system in the treatment of bone diseases. Joint Bone Spine. 2008;75:5–10. doi: 10.1016/j.jbspin.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 21.Anderson DM. Maraskovsky E. Billingsley WL, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–179. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- 22.Locksley RM. Killeen N. Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. [Review] Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 23.Bodmer JL. Schneider P. Tschopp J. The molecular architecture of the TNF superfamily. [Review] Trends Biochem Sci. 2002;27:19–26. doi: 10.1016/s0968-0004(01)01995-8. [DOI] [PubMed] [Google Scholar]
- 24.Hsu H. Lacey DL. Dunstan CR, et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci USA. 1999;96:3540–3545. doi: 10.1073/pnas.96.7.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darnay BG. Haridas V. Ni J. Moore PA. Aggarwal BB. Characterization of the intracellular domain of receptor activator of NF-kappaB (RANK). Interaction with tumor necrosis factor receptor-associated factors and activation of NF-kappab and c-Jun N-terminal kinase. J Biol Chem. 1998;273:20551–20555. doi: 10.1074/jbc.273.32.20551. [DOI] [PubMed] [Google Scholar]
- 26.Wong BR. Josien R. Lee SY. Vologodskaia M. Steinman RM. Choi YW. The TRAF family of signal transducers mediates NF-KAPPA-B activation by the TRANCE receptor. J Biol Chem. 1998;273:28355–28359. doi: 10.1074/jbc.273.43.28355. [DOI] [PubMed] [Google Scholar]
- 27.Kim HH. Lee DE. Shin JN, et al. Receptor activator of NF-kappaB recruits multiple TRAF family adaptors and activates c-Jun N-terminal kinase. FEBS Lett. 1999;443:297–302. doi: 10.1016/s0014-5793(98)01731-1. [DOI] [PubMed] [Google Scholar]
- 28.Darnay BG. Ni J. Moore PA. Aggarwal BB. Activation of NF-kappaB by RANK requires tumor necrosis factor receptor-associated factor (TRAF) 6 and NF-kappaB-inducing kinase. Identification of a novel TRAF6 interaction motif. J Biol Chem. 1999;274:7724–7731. doi: 10.1074/jbc.274.12.7724. [DOI] [PubMed] [Google Scholar]
- 29.Galibert L. Tometsko ME. Anderson DM. Cosman D. Dougall WC. The involvement of multiple tumor necrosis factor receptor (TNFR)-associated factors in the signaling mechanisms of receptor activator of NF-kappaB, a member of the TNFR superfamily. J Biol Chem. 1998;273:34120–34127. doi: 10.1074/jbc.273.51.34120. [DOI] [PubMed] [Google Scholar]
- 30.Liu W. Xu D. Yang H, et al. Functional identification of three RANK cytoplasmic motifs mediating osteoclast differentiation and function. J Biol Chem. 2004;279:54759–54769. doi: 10.1074/jbc.M404687200. [DOI] [PubMed] [Google Scholar]
- 31.Chen T. Feng X. Cell-based assay strategy for identification of motif-specific RANK signaling pathway inhibitors. Assay Drug Dev Technol. 2006;4:473–482. doi: 10.1089/adt.2006.4.473. [DOI] [PubMed] [Google Scholar]
- 32.McHugh KP. Hodivala-Dilke K. Zheng MH, et al. Mice lacking b3 integrins are osteosclerotic due to dysfunctional osteoclasts. J Clin Invest. 2000;105:433–440. doi: 10.1172/JCI8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeshita S. Kaji K. Kudo A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res. 2000;15:1477–1488. doi: 10.1359/jbmr.2000.15.8.1477. [DOI] [PubMed] [Google Scholar]
- 34.Xu D. Shi Z. McDonald J, et al. Development of a chimaeric receptor approach to study signalling by tumour necrosis factor receptor family members. Biochem J. 2004;383:219–225. doi: 10.1042/BJ20040961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ory DS. Neugeboren BA. Mulligan RC. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc Natl Acad Sci USA. 1996;93:11400–11406. doi: 10.1073/pnas.93.21.11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shui C. Riggs BL. Khosla S. The immunosuppressant rapamycin, alone or with transforming growth factor-beta, enhances osteoclast differentiation of RAW264.7 monocyte-macrophage cells in the presence of RANK-ligand. Calcif Tissue Int. 2002;71:437–446. doi: 10.1007/s00223-001-1138-3. [DOI] [PubMed] [Google Scholar]
- 37.Battaglino R. Kim D. Fu B. Fu XY. Stashenko P. c-Myc is required for osteoclast differentiation. J Bone Miner Res. 2002;17:763–773. doi: 10.1359/jbmr.2002.17.5.763. [DOI] [PubMed] [Google Scholar]
- 38.Weldon CB. Burow ME. Rolfe KW. Clayton JL. Jaffe BM. Beckman BS. NF-kappa B-mediated chemoresistance in breast cancer cells. Surgery. 2001;130:143–150. doi: 10.1067/msy.2001.115512. [DOI] [PubMed] [Google Scholar]
- 39.Shin HM. Kim MH. Kim BH, et al. Inhibitory action of novel aromatic diamine compound on lipopolysaccharide-induced nuclear translocation of NF-kappaB without affecting IkappaB degradation. FEBS Lett. 2004;571:50–54. doi: 10.1016/j.febslet.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 40.Zhang JH. Chung TD. Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;10:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 41.Wong BR. Josien R. Lee SY, et al. TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J Exp Med. 1997;186:2075–2080. doi: 10.1084/jem.186.12.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kong YY. Yoshida H. Sarosi I, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]