

Abstract
Aims
To study glycosidase activities of a Lactobacillus brevis strain and to isolate an intracellular β-glucosidase from this strain.
Methods and Results
Lactic acid bacteria isolated from a commercially available starter culture preparation for MLF were tested for β-glycosidase activities. A strain of L .brevis showing high intracellular β-d-glucosidase, β-d-xylosidase and α-l-arabinosidase activities was selected for purification and characterization of its β-glucosidase. The pure glucosidase from L. brevis has also side activites of xylosidase, arabinosidase and cellobiosidase. It is a homo-tetramer of 330 kDa and has an isoelectric point at pH 3.5.The Km for p-nitrophenyl-β-d-glucopyranoside and p-nitrophenyl-β-d-xylopyranoside is 0.22 mM and 1.14 mM, respectively. The β-glucosidase activity was strongly inhibited by gluconic acid δ-lactone, partially by glucose and gluconate, but not by fructose. Ethanol and methanol were found to increase the activity up to two fold. The free enzyme was stable at pH 7.0 (t1/2 = 50 d) but not at pH 4.0 (t1/2 = 4 d).
Conclusions
The β-glucosidase from L. brevis is widely different to that characterized from Lactobacillus casei (Coulon et al. 1998) and Lactobacillus plantarum (Sestelo et al. 2004). The high tolerance to fructose and ethanol, the low inhibitory effect of glucose on the enzyme activity and the good long-term stability could be of great interest for the release of aroma compounds during winemaking.
Significance and Impact of the study
Although the release of aroma compounds by LAB has been demonstrated by several authors, little information exists on the responsible enzymes. This study contains the first characterization of an intracellular β-glucosidase isolated from a wine related strain of L. brevis.
Keywords: glucosidase, xylosidase, arabinosidase, Lactobacillus brevis, Oenococcus oeni, lactic acid bacteria
INTRODUCTION
Although the majority of flavor and aroma compounds found in wine are produced during the alcoholic fermentation, the varietal bouquet is determined by volatile constituents derived from the grapes (e.g. terpenes, C13 norisoprenoids, benzene derivatives, etc) (Rapp 1992; Mateo and Jiménez 2000; Ribéreau-Gayon et al. 2006). Apart from the free flavor compounds present in the berries, a significant amount is present as odorless non-volatile glycosides (Günata et al. 1985, Maicas and Mateo 2005; Ribéreau-Gayon et al. 2006). These aroma compounds can be released from their glycosylated precursors by acid or enzyme catalyzed hydrolysis, thus altering the aroma profile of wines. Acid hydrolysis occurs very slowly during wine storage and can be accelerated by heat (Mateo and Jiménez 2000), but both processes may prompt a deterioration in wine quality. In contrast to this, enzymatic hydrolysis may improve the natural aroma spectrum of wine, but also fruit juices without having detrimental effects on the quality of the final product and is therefore of considerable interest in wine research (Günata et al. 1998; Mateo and Jiménez 2000).
Enzymatic hydrolysis of wine glycosides by grape glycosidases or yeast glycosidases is limited due to the very low activity of the enzymes under fermentation conditions (Mateo and Di Stefano 1998; Maicas and Mateo 2005). On the other hand, Aspergillus niger glycosidases present in commercial pectinase preparations, that are widely used in wine making, may possess considerable activity (Günata et al. 1993).
Relevant glycosidase activities have also been reported for lactic acid bacteria (LAB) responsible for malolactic fermentation (MLF). Numerous studies have investigated the glycosidase activity of Oenococcus oeni, which is usually the preferred species for MLF (Grimaldi et al. 2000; Boido et al. 2002; D’Incecco et al. 2004; Grimaldi et al. 2005b). But glycosidic activities have also been reported for strains of Lactobacillus and Pediococcus isolated from starter cultures for MLF (Grimaldi et al. 2005a). The β-glucosidase gene is probably widespread and highly conserved among wine LAB, its expression is regulated by a wide range of abiotic stresses (e.g. temperature, ethanol, pH) (Spano et al. 2005). However, previous studies on glycosidase activities of wine LAB mainly describe the hydrolysis of glycosides associated with the living cells. Only three β-glucosidases have been purified and characterized from Lactobacilli so far. Intracellular enzymes have been identified in Lactobacillus casei (Coulon et al. 1998) and Leuconostoc mesenteroides (Gueguen et al. 1997). Sestelo et al. (2004) purified an extracellular β-glucosidase from Lactobacillus plantarum. To our knowledge, this is the first study with the aim to investigate the properties of an intracellular β-glucosidase from a wine related Lactobacillus strain. Here, we describe the purification and basic characterization of a native β-glucosidase from a strain of Lactobacillus brevis isolated from a starter culture for MLF. Furthermore, we discuss its properties in contrast to the so far reported β-glucosidase activities of LAB.
MATERIAL AND METHODS
Isolation and identification of Lactic acid bacteria
Lactic acid bacteria were isolated from the freeze-dried starter culture Biostart Bianco SK3 from Erbslöh, Germany. The enrichment media used were MRST pH 5.0 and MRST pH 5.0 with 10% (v/v) ethanol (MRST: glucose 10 g, fructose 5 g, yeast extract 5 g, tryptone 10 g, MgSO4·7 H2O 0.2 g, MnSO4·H2O 0.05 g, α-cycloheximide 0.1 g, cysteine hydrochloride 0.5 g, Tween 80 1 mL, plus filtered preservative free tomato juice 100 mL, in one liter). After incubation for 4 (MRST) or 14 days (MRS with ethanol) at 25°C the enrichment cultures were plated onto MRST 2% agar plates and incubated anaerobically at 25°C. Colonies were selected from the plates, the strains were then grown in MRST and stored in 30% glycerol at −70°C.
The single colony isolates were identified by amplified 16S-rDNA restriction analysis (16S-ARDRA) analysis following Rodas et al. (2003). The bacterial DNA was extracted with the GenElute™ Bacterial Genomic DNA Kit (Sigma). The primers used were 16SF: 5′ - AGAGTTTGATCCTGGCTCAG - 3′ and 16SR: 5′ - AAGGAGGTGATCCAGCCGCA - 3′. The PCR reactions were performed using the RedTaq Ready Mix (Sigma) in a Biometra T3 thermocycler. PCR conditions were: initial denaturation step 5 min at 94°C, followed by 35 cycles of denaturation at 94°C for 30s, annealing for 30s at 56°C, extension 1 min at 72°C and a final elongation step at 72°C for 5 min. Restriction analysis of the amplified 16S-rDNA fragment was done with the restriction enzymes MseI and BfaI (New England Biolabs) for 2 h at 37°C. The restriction fragments were separated on 2% agarose gels with ethidium bromide staining. The 50 bp and 100 bp ladders from New England Biolabs were used as molecular size marker, the reference strains used were O. oeni DSMZ 20252 and L. brevis DSMZ 20054. Additionally, the amplified 16S-rDNA fragments were purified with the Wizard SV Gel and PCR Clean-UP System (Promega) and sequenced (Agowa GmbH, Germany). The sequences were compared with the NCBI database using BLAST.
Culture conditions and preparation of crude extracts
The growth media used for glycosidase induction was MRSA pH 5.0 (glucose 5 g, fructose 2.5 g, d/l-malate 5 g, yeast extract 5 g, tryptone 10 g, MgSO4·7H2O 0.2 g, MnSO4·H2O 0.05 g, ammonium citrate dibasic 3.5 g, Tween 80 1 mL, preservative free apple juice 200 mL, in one liter). The apple juice was autoclaved separately and then added to the sterile MRS base. Precultures were prepared by inoculating 12 mL MRST and incubation at 25°C for 3 to 4 days. These precultures were used to inoculate 1L of MRSA in duplicate. After 4 days of growth the cells were harvested by centrifugation (6000 g, 30 min.), washed twice with 0.85% NaCl and resuspended in 0.85% NaCl for the determination of cell-bound glycosidase activities. After harvesting and for all the following steps the cells were kept at 4°C or on ice. For the determination of intracellular enzyme activities the harvested cells from the 1L cultures were resuspended in 20 mL cold 20 mM citrate phosphate buffer, pH 7.0 and kept on ice. Cell free extracts were prepared by disrupting the cell suspensions by passing them through a French Press 5 times at a pressure of 1200 psi (8.3 MPa). The cell debris were separated by ultracentrifugation in a Beckman L-70 ultracentrifuge with rotor 45Ti at 4°C, 105,000 g for 30 min.
Enzyme localisation studies
A protocol according to De Cort et al. (1994) was followed to determine the cellular localization of the enzymes. The washed cells were resuspended in 0.5 M sodium malate buffer pH 6 and treated with different concentrations of lysozyme (Sigma, ~93,000 U mg−1) for 2 h at 37°C. The remaining cells / spheroplasts were harvested (16,100 g, 15 min, Eppendorf Centrifuge 5415R) and washed with 0.5 M sodium malate buffer pH 6. The resulting supernatants and the remaining cells / spheroplasts were assayed for β-glucosidase activity. For hypotonic lysis, spheroplasts were suspended in 10 mM malate buffer, pH 6 and centrifuged after 30 minutes (16,100 g, 15 min). Again, the supernatant and the remaining fragments were tested for activity. A control without lysozyme- but otherwise the same treatments was included in these tests.
Glycosidase assays
The activity assays were performed according to Grimaldi et al. (2000). Citrate-phosphate buffers (CP) used for the enzyme assays were prepared following (McIlvaine 1921). A 200 mM citric acid solution was titrated with a 400 mM Na2HPO4 until the required pH value was achieved, and further diluted to give the required ion concentration defined as sum of citrate and phosphate ions.
Glycosidase activities were determined by use of p-nitrophenyl (pNP) -glycosides (Sigma) as substrates. The substrates used were pNP-β-d-glucopyranoside (pNPβGlc), pNP-β-d-xylopyranoside (pNPβXyl), pNP-α-l-arabinopyranoside (pNPαAra) and pNP-α-l-rhamnopyranoside (pNPαRha). The substrate stock solutions for the assays were prepared fresh for each assay. Substrates were dissolved in 10% dimethylsulfoxide and water to give concentrations of 40 mM. If not otherwise mentioned, all enzyme assays were performed at 37°C and pH 5.5 in duplicate. The enzyme solutions were stored in 20 mM CP, pH 7.0, and if necessary, further diluted in this buffer to give final absorbance readings between 0.3 and 1. The reaction mixture was made of 100 μL 200 mM CP pH 5.5, 50 μL sample and 50 μL substrate solution, to give substrate concentrations of 10 mM in the assay. After addition of the substrate the assays were incubated for 10 min at 37°C and then stopped with 400 μL 0.5 M Na2CO3. The absorbance of the yellow p-nitrophenolate anion was measured at 400 nm (ε400 = 18.300 M−1 cm−1 at pH 10.2) in a Beckman DU 800 spectrometer. Because of the spontaneous hydrolysis of the pNP-glycosides in alkaline solution the measurements were made immediately after the reaction. For the assays with intact biomass (cell-bound glycosidase activities) the cell suspensions were diluted to an OD600 of 1 for the glucosidase- or xylosidase-, and an OD600 between 3 and 4 for the arabinosidase- and rhamnosidase assays. The assays were incubated for 2 h at 37°C and stopped by the addition of 0.5 M Na2CO3. The cells were removed by centrifugation in an Eppendorf Centrifuge 5415R (6000 g, 5 min) and the absorbance of the supernatants was measured at 400 nm. The dry cell mass was estimated by measuring the absorbance of the cell suspension at 600 nm. For this, a calibration procedure was performed by drying 10 mL cell suspensions with known absorbances to constant weight at 105°C.
One unit (U) of glycosidase activity corresponds to 1 μmol of p-nitrophenol released per min. Cell-bound activities are expressed as U per gram of dry cell mass.
Temperature and pH activity profiles, inhibitory tests and long term stability
To determine the influence of pH on enzyme activity, the reaction mixture was made with CP buffers of pH values ranging from pH 3.0 to 7.0 in intervals of 0.5 units. Temperature influence on enzyme activity was determined between 15°C and 55°C. The assays were incubated in an Eppendorf thermomixer.
To determine the influence of possible inhibitors, the activity of the β-glucosidase from L. brevis was assayed in presence of varying concentrations of ethanol, methanol, glucose, fructose, sucrose, mannitol, sorbitol, d-malate, d-lactate, gluconic acid and gluconic acid δ-lactone in the standard glucosidase assay.
Enzyme stability was studied by storage of the purified enzyme at 4, 25, 37 and 45°C in 20 mM CP, pH 7.0 and at 4°C in 20 mM CP, pH 4.0.
Determination of Km-values
The standard test was modified such, that it was measured directly at pH 5.5 without the addition of the stop solution. Due to the low absorption of p-nitrophenol at the test pH, the addition of α-cyclodextrin was required to increase the absorption of p-nitrophenol at 400 nm. This was necessary to allow the use of lower enzyme concentrations and better Km estimates (Tate and Reynolds 2006). Extinction coefficients of the chromogenic complex of p-nitrophenol and α-cyclodextrin were determined for different concentrations of α-cyclodextrin at pH 5.5. Addition of 10 mM α-cyclodextrin was found to give the best results (ε400 = 3,300 M−1 cm−1 at pH 5.5). For actual tests substrate concentrations between 0 to 10 mM were used. The reaction mixture was maintained at 37°C and the reaction was started by addition of the pre-tempered enzyme solution. Measurements of absorbance change were taken continuously for 2 min using a Hitachi C-3000 spectrophotometer with the measurement cell tempered at 37°C. All assays were performed in triplicate. The initial reaction velocity was determined, and Km was calculated using the Hanes-Plot. The Km method was validated by measuring the Km value of the almond β-glucosidase (Sigma) at pH 5.5, for which a value of 7.15 mM at pH 6.5 was reported by Tate and Reynolds (2006). Our findings showed a Km of 3.15 mM pNPG at pH 5.5.
Protein assays
Protein concentrations were determined following Bradford (1976) using the Bio-Rad Protein Assay kit. Standard curves (0.1 to 1.0 mg mL−1) were prepared with bovine serum albumin (BSA).
Protein purification
A free β-glucosidase was purified from L. brevis. All chromatographic steps were performed on an ÄKTA Explorer system (GE Healthcare). Desalting and buffer exchange between the purification steps was accomplished by ultrafiltration with a Millipore filter (10,000 MWCO, at 4000 g). Ammonium sulfate was added to the crude cell extracts to give 30% saturation at 0°C. The precipitate was removed by centrifugation at 105,000 g for 30 min (Beckman L-70 Ultracentrifuge, rotor 45Ti). The supernatants were loaded directly onto a 60 mL Macro-Prep methyl HIC column (Biorad) at a flow rate of 0.2 cm min−1. A linear gradient, 10 column volumes (CV), from Buffer A (sodium phosphate buffer 50 mM, pH 7.0, 1.25 M (NH4)2SO4) to Buffer B (sodium phosphate buffer 50 mM, pH 7.0) was applied at a flow rate of 0.4 cm min−1. Fractions of 10 mL were collected and analysed for β-glucosidase activity and protein content. The active fractions were pooled and loaded onto a 21 mL Source 15 Q column (GE Healthcare). A linear gradient (7 CV, 1 cm min−1) from 0 M to 1 M NaCl in 20 mM Tris-HCl pH 7.0 was applied. The eluted enzyme was pooled and loaded onto a 190 mL Sephacryl S-300 column and eluted with 0.1 cm min −1 with 20 mM CP, pH 7.0, 150 mM NaCl. To determine the apparent molecular weight, the Sephacryl S-300 column was calibrated using the following molecular weight standard proteins (Sigma): cytochrome c 12 kDa, bovine serum albumin 67 kDa, alcohol dehydrogenase 141 kDa, β-amylase 200 kDa, apoferritin 443 kDa, thyroglobuline 669 kDa. The void volume of the column was determined with blue dextran (2,000 kDa). Molecular weight determination including column calibration was repeated with CP buffers pH 7.0; 100 and 500 mM, each containing 150 mM NaCl.
Electrophoresis
All electrophoretic steps including coomassie blue and silver staining were performed on the PHAST System (GE Healthcare) according to the suppliers instructions. For SDS and Native polyacrylamide gel electrophoresis (PAGE), precast gels (PhastGel Gradient 8-25) and the corresponding buffer strips (SDS / Native) were used. The molecular weight marker for SDS PAGE was High Precision Dual Colour (Biorad) 10 to 250 kDa range. For Native PAGE, the GE Healthcare HMW Native Marker Kit was used. SDS and Native Gels were stained for proteins with coomassie blue (PhastGel BlueR). Active staining of Native PAGE gels prior to coomassie blue staining was done by equilibrating the gel in CP 100 mM, pH 7.0 for 10 min and then for another 10 min in 1 mM 4-methylumbelliferyl-β-d-glucopyranoside (Sigma) in CP 100 mM, pH 7.0. The gel was photographed under UV light (Gel-Doc, Bio-Rad).
Isoelectric focusing (IEF) was done with PhastGel IEF - 3-9 gels and the IEF Mix 3.6 - 9.3 (Sigma) was used as pI marker. Both active and silver staining were applied to the IEF gels.
RESULTS
When the malolactic starter preparation SK3 was inoculated to MRST without ethanol, no strains of O. oeni could be detected. Nevertheless, the obtained isolates were screened for glucosidase activity, a strain with high cellular activity could be identified as L. brevis (“L. brevis SK3”). After incubating the preparation SK3 for 2 weeks in MRST with 10% ethanol, all examined isolates were identified as species of O. oeni and also tested for β-glucosidase activities. Fig. 1 shows the results of the 16S ARDRA analysis using the restriction enzymes MseI and BfaI compared with restriction patterns of reference strains. The identity of the strains was verified by comparing the sequences of the amplified 16S fragments with the NCBI database using the BLAST algorithm. The activities of the living biomass (cell-bound β-glucosidase activities) of the strains seen in table 1 were measured after 100 h of fermentation. Table 1 shows the cell-bound and the corresponding intracellular activities in the crude cell extracts for L. brevis SK3 and two isolates of O. oeni after growth in MRSA. Although both O. oeni strains exhibited considerable cell-bound activities, the yields of intracellular enzymes was low. α-l-rhamnosidase activity could not be detected and very low α-l-arabinosidase activities were recorded in accordance with the findings presented by Grimaldi et al. (2005 a; b). Due to the high yield of free glycosidase activities in L. brevis SK3, this strain was used for further characterization of the native enzymes. More than 80% (43 U per gram dry cell weight, table 1) of the total β-glucosidase activity determined after cell disruption (~50 U g−1) could be gained as soluble enzyme(s) in the supernatant after ultracentrifugation. One attempt was to further elucidate the cellular localization of the soluble β-glucosidase by degradation of the cell wall with lysozyme and subsequent hypotonic lysis of the resulting spheroplasts. Unfortunately, clear data could not be obtained by repeated experiments at varying conditions (table 2), even at high concentrations of lysozyme (5 mg mL−1 = 465,000 U mL−1) complete cell wall degradation could not be achieved. Incomplete spheroplast formation was also confirmed by phase contrast microscopy. As shown in table 2, the release of enzyme just by lysozyme treatment is low compared to the yield after hypotonic lysis of the lysozyme treated cells, therefore it could be deduced that the enzyme is cytoplasmic, rather than periplasmic. Fig. 2 (A, B) shows the pH and temperature dependence of the cell-bound activities. Glucosidase activity revealed optima at pH 6.0 and 45°C, xylosidase activity at pH between 5.5 and pH 6.0 and 45°C, and arabinosidase activity between pH 5.0 and pH 6.0 and 37°C. While the cell-bound activity of the glucosidase at pH 4.0 (37°C) is about 50% of the maximum, at low temperatures (i.e. 20°C) the cell activity was less than 10% of the maximum activity.
Figure 1.

16S ARDRA patterns on 2% Agarose. Digestion with MseI (A) and BfaI (B). Lanes 1 & 8: 50 bp ladder (A), 100 bp ladder (B). Lane 2: L. brevis DSMZ 20054. Lane 3: O. oeni DSMZ 20252. Lane 4: L. brevis SK3. Lane 5: Pediococcus acidilacti. Lane 6: O. oeni SK3Ba, Lane 7: O. oeni SK3Bb.
Table 1.
Cell-bound and intracellular glycosidase activities of selected strains of LAB.
| Cell-bound activity* (U g−1) |
Intracellular activity † (U g−1) |
|||
|---|---|---|---|---|
| pNPβGlc | pNPβXyl | pNPαAra | ||
| O. oeni SK3Ba | 7.4 | 2.3 | 1.5 | 0.3 |
| O. oeni SK3Bb | 5.8 | 2.0 | 1.5 | 0.4 |
| L. brevis SK3 | 10.6 | 43 | 16 | 1.6 |
Cellular β-glucosidase activities as units per gram of dry cell mass at pH 5.5 and 37°C.
Intracellular activities at 37°C as yield of soluble enzyme per gram dry cell mass.
Table 2.
Subcellular localization of the β-glucosidase in Lactobacillus brevis SK3. The enzyme activities are displayed as U per g of dry cell mass.
| Lysozyme concentration |
Supernatant A* after lysozyme treatment |
Cell-bound activity after lysozyme treatment |
Supernatant B† after hypotonic lysis of spheroplasts |
Cell-bound activity after hypotonic lysis |
|---|---|---|---|---|
| Control | 0.2 | 13 | 0.9 | 13 |
| 1 mg mL−1 | 0.3 | 17 | 0.9 | 16 |
| 2 mg mL−1 | 0.4 | 19 | 4.2 | 15 |
| 5 mg mL−1 | 1.6 | 21 | 12.5 | 13 |
soluble periplasmic fraction after lysozyme treatment.
soluble cytoplasmic fraction after hypotonic lysis.
Figure 2.

pH - and temperature dependence of glycosidase activities of L. brevis SK3. β-d-glucosidase (●), β-d-xylosidase (○), α-l-arabinosidase (▼). (A, B) Cell-bound activites displayed as U per g of dry cell mass. (C, D) Intracellular glycosidase activities of L. brevis SK3, 100% of relative activity refers to the enzyme activities measured at pH 5.5 and 37°C (see table 1).
For the free enzymes the temperature and pH dependence curves are shown in fig. 2 (C, D). The pH optimum for glucosidase, xylosidase and arabinosidase was pH 5.5. The temperatures at which the enzymes displayed maximum activities were: glucosidase 45°C, xylosidase 40°C and arabinosidase 37°C.
Subsequently, purification and further characterization of the native β-glucosidase was performed. A purification protocol for the β-glucosidase is shown in table 3. The enzyme was purified up to 244-fold to a specific activity of 70.9 U mg−1. The enzyme was electrophoretically pure and showed a single band at ~ 80 kDa on SDS PAGE (fig. 3A), gel filtration on Sephacryl S-300 suggested a molecular weight of 330 kDa, which implicates a homo-tetrameric structure. Although in Native PAGE (fig. 3B) coomassie staining revealed only one band resembling the tetramer, active staining detected three active bands which could represent the active tetramer, and to lesser degree, an active dimer and monomer as well. This was probably caused by Native PAGE conditions, when the enzyme was equilibrated and applied to the column again in buffers with higher ionic strength (CP 100 mM, 500 mM), no other than the tetrameric form could be observed. An isoelectric point at ~ pH 3.5 was determined by isoelectric focusing (fig. 3C). Results in table 4 represent the activities of the purified enzyme on several pNP-glycosides, indicating that the enzyme is specific for β-d- and α-l-linked glycosides. The relative activities of glucosidase, xylosidase and arabinosidase of the pure enzyme match the ratios found in the crude extract (table 1).
Table 3.
Purification protocol for the β-glucosidase from Lactobacillus brevis SK3.
| Purification step | Total activity (U) |
Total protein (mg) |
Specific activity (U mg−1) |
Purification factor |
Yield (%) |
|---|---|---|---|---|---|
| Crude extract | 172 | 575 | 0.29 | ||
| 30% AS * | 174 | 484 | 0.36 | 1.2 | 100 |
| Methyl HIC | 114 | 31.5 | 3.62 | 12.4 | 67 |
| Source 15 Q | 52 | 1.2 | 43.3 | 149 | 30 |
| Sephacryl S-300 | 29 | 0.41 | 70.9 | 244 | 17 |
supernatant of 30% ammonium sulfate precipitation.
Figure 3.

(A) SDS-PAGE with coomassie blue staining. Lane 1: High Precision Dual Colour molecular weight marker (kDa). Lane 2: ~ 2μg of β-glucosidase from L. brevis SK3 after purification (70.9 U mg−1). (B) Native PAGE. Lane 1: HMW Native Marker Kit (kDa). Lane 2: coomassie-staining of β-glucosidase. Lane 3: active staining with 4-methylumbelliferyl-β-d-glucopyranoside. (C) Isoelectric focusing with silver staining. Lane 1: Active staining. Lane 2: ~ 0.4 μg of β-glucosidase from L. brevis SK3, Lane 3: IEF Mix (Sigma).
Table 4.
Substrate specificity of β-glucosidase from Lactobacillus brevis SK3 using different p-nitrophenyl derivatives.
| Substrate | Specific activity* (U mg−1) |
Relative activity (%) |
|---|---|---|
| pNP-β-d-glucopyranoside | 71 | 100 |
| pNP-β-d-xylopyranoside | 33 | 46 |
| pNP-β-d-galactopyranoside | 0.1 | 0.1 |
| pNP-β-d-mannopyranoside | nd | |
| pNP-β-d-cellobioside | 9.4 | 13 |
| pNP-α-l-arabinopyranoside | 3.5 | 4.9 |
| pNP-α-l-rhamnopyranoside | nd | |
|
| ||
| pNP-α-d-glucopyranoside | <0.1 | <0.1 |
| pNP-α-d-xylopyranoside | nd | |
| pNP-α-d-galactopyranoside | nd | |
| pNP-α-d-mannopyranoside | nd | |
| pNP-β-l-arabinopyranoside | nd | |
nd: not detectable
The Km values found for the β-glucosidase were 0.22 mM with pNPβGlc and 1.14 mM with pNPβXyl. Table 5 shows the influence of several compounds on the β-glucosidase activity. The enzyme was partially inhibited by glucose (59% of initial activity at 0.2 M glucose) and gluconic acid (17% at 0.2 M), and strongly inhibited only by gluconic acid δ–lactone (14% at 10 mM). High concentrations (up to 0.2 M) of fructose, malate, lactate, mannitol and sorbitol had little or no effect on the enzyme activity. In the presence of 16% (v/v) ethanol and methanol the enzyme activity was increased more than two-fold.
Table 5.
Inhibition of β-glucosidase from Lactobacillus brevis SK3 by different compounds.
| Compound | Concentration (mM) |
Glucosidase activity (U mg−1) |
Relative activity (%) |
|---|---|---|---|
| Control | 42,6 | 100 | |
| Glucose | 10 | 41,0 | 96 |
| 200 | 25,3 | 59 | |
| Fructose | 200 | 41,6 | 98 |
| Gluconic acid | 10 | 29,2 | 69 |
| 200 | 7,3 | 17 | |
| Glucono-δ-lactone | 0,5 | 29,6 | 70 |
| 5 | 16,6 | 39 | |
| 10 | 5,8 | 14 | |
| l-Malate | 200 | 38,2 | 95 |
| l-Lactate | 200 | 37,8 | 89 |
| Mannitol | 200 | 47,4 | 111 |
| Sorbitol | 200 | 49,6 | 116 |
| Concentration (% [v/v]) |
|||
|---|---|---|---|
| Ethanol | 2 | 57,7 | 135 |
| 12 | 91,2 | 214 | |
| 16 | 95,6 | 224 | |
| Methanol | 16 | 100,4 | 236 |
Stability tests (fig. 4) in 20 mM CP, pH 7.0 showed that the enzyme displayed good stability when it was kept at 25°C and 37°C, with approximate half lifes of 160 h and 3.5 h, respectively. When the enzyme was kept at 45°C, less than 5% of initial activity was detectable after 30 min. For enzyme stored at 4°C in 20 mM CP, half lives of 4 days at pH 4.0 and 50 days at pH 7.0 were estimated.
Figure 4.
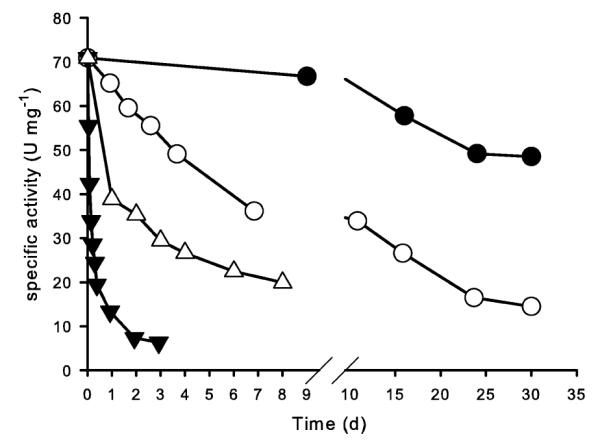
Temperature and long-term stability of the β-glucosidase from L. brevis SK3 at pH 7.0, 37°C (▼), 25°C (○), 4°C (●) and pH 4.0, 4°C (△).
DISCUSSION
The original intent of this study was to characterize free glycosidase enzymes from strains of O. oeni isolated from the commercially available MLF starter culture preparation Biostart Bianco SK3. However, by use of MRS with 10% tomato juice as enrichment media, the examined isolates belonged to the genus Lactobacillus. The isolation of Lactobacillus spp. has also been reported by Grimaldi et al. (2005a). Although these strains possibly overgrow O. oeni in rich media, they may play a minor role in MLF under wine conditions, after 14 days of incubation in media with 10% of ethanol no other species than O. oeni could be identified.
Although two O. oeni isolates exhibited considerable cell-bound activity, the yield of free enzyme(s) from these strains was poor. In contrast to this, the strain L. brevis SK3 showed high cell-bound and intracellular activities as well. This seems interesting, since L. brevis is not reported to be an important candidate for MLF in wine. Therefore, the strain L. brevis SK3 was selected for the purification and further characterization of its native β-glucosidase. The pure enzyme is a homo-tetramer with a molecular weight of 330 kDa and an isoelectric point at pH 3.5. The Km values for pNPβGlc and pNPβXyl were 0.22 and 1.14, respectively. Table 6 displays the properties of the L. brevis enzyme in comparison to other β-glucosidases reported in LAB so far. As judged from these data, the enzyme from L. brevis shows no resemblance to the β-glucosidases isolated from L. casei (Coulon et al. 1998) and L. plantarum (Sestelo et al. 2004). On the other hand, the L. brevis glucosidase could be related to the enzyme isolated from L. mesenteroides (Gueguen et al. 1997), the authors also reported inhibition by glucose and gluconolactone and activation of the enzyme by methanol and ethanol.
Table 6.
Properties of β-glucosidases isolated from lactic acid bacteria.
The enzyme from L. brevis prefers β-d or α-l linked substrates which resemble the same stereochemical conformation, and could also hydrolyze pNP-β-d-cellobioside. Interestingly, for the glucosidase, xylosidase and arabinosidase of L. brevis the activity ratios found in the crude extract and the pure enzyme were comparable (tables 1 and 4). This leads to the interpretation that we are not dealing with three independent, but possibly only one enzyme. An interesting property is the activation of the enzyme by ethanol and methanol, an explanation might be the use of alcohols as acceptor for glycosylintermediates (Pemberton et al. 1979).
The reason for the high discrepancy in cell-bound / intracellular activities between O. oeni and L. brevis might be a predominace of membrane bound enzymes in O. oeni. On the other hand, the presence of deglycosylation systems other than β-glycosidases has been shown. Phosphoglycosylhydrolases (EC 3.2.1.86) may be part of a cellobiose-specific phosphotransferase system (PTS) (De Vos and Vaughan 1994; Nagaoka et al. 2008). Apart from characterizing the enzyme, one interesting issue was also to elucidate the subcellular localization of the glucosidase in L. brevis. Boido et al. (2002) indicated parietal localization of the glycolytic enzymes in O. oeni, since no intracellular aglycons could be found after hydrolysis of the glycosylated precursors. Furthermore, Spano et al. (2005) reported the presence of hydrophobic transmembrane domains in a β-glucosidase in L. plantarum and showed also, that the gene is widespread among LAB. The transmembrane localization may be true for glycosidases of O. oeni, since in the strains examined in this study the activities in the residual cell debris were always higher than the activities measured in the soluble fractions after cell disruption and ultracentrifugation (data not shown). Furthermore, a wild strain of Pediococcus pentosaceus isolated in our laboratory from Austrian wine displayed cell-bound glucosidase activity (7.8 U g−1), but no intracellular enzyme at all (data not shown). On the contrary, the isolated β-glucosidase from L. brevis turned out to be soluble. After thorough cell disruption (French pressure cell press) the remaining activity in the cell debris was less than 20% of the activity found in the cleared supernatant, but there might be a membrane bound enzyme as well. Our localization experiments had the aim to find out whether the free enzyme is periplasmic or cytoplasmic, but failed to give definite results. Nevertheless, from the results shown in table 2, it seems reasonable to propose a cytoplasmic localization, but due to the incomplete cell wall degradation and the high activities remaining in the cell fragments, it is invalid to draw coherent conclusions from these data. However inconclusive these results may seem, they have not been omitted from this report because the lysozyme treated cells were more active than the untreated cells, while the concomitant release of enzyme was insignificant. Since deglycosylation may also be performed by membrane bound proteins or by other mechanisms than glycosidases like EC 3.2.1.21, cells with perforated or at least partially degraded cell walls could play in interesting role as immobilized biocatalysts, which could overcome the detrimental effects on free enzymes presented by the harsh conditions as found in wine. Further studies with the aim to achieve a better understanding of the glycosidase mechanisms provided by wine lactic acid bacteria will be conducted in our division. The here presented glucosidase from L. brevis shows interesting characteristics. It’s good long-term and temperature stability and the tolerance to sugars and ethanol as well could be beneficial for some improvements in winemaking.
Furthermore, enzymes from different species of LAB, especially O. oeni, could display useful properties for the deglycosylation of aroma precursors, immobilization of these enzymes could further increase pH and temperature stability.
ACKNOWLEDGEMENTS
The authors appreciate the support given to KD Kulbe by the Austrian Science Fund (FWF) and the Research Centre Applied Biocatalysis Graz, Austria. This work is also dedicated to the memory of our technician Mr. Martin Schneider.
REFERENCES
- Boido E, Lloret A, Medina K, Carrau F, Dellacassa E. Effect of β-glycosidase activity of Oenococcus oeni on the glycosylated flavor precursors of Tannat wine during malolactic fermentation. J Agric Food Chem. 2002;50:2344–2349. doi: 10.1021/jf0109367. [DOI] [PubMed] [Google Scholar]
- Coulon S, Chemardin P, Gueguen Y, Arnaud A, Galzy P. Purification and characterization of an intracellular β-glucosidase from Lactobacillus casei ATCC 393. Appl Biochem Biotechnol. 1998;74:105–114. [Google Scholar]
- De Cort S, Kumara HMCS, Verachtert H. Localization and characterization of α-glucosidase activity in Lactobacillus brevis. Appl Environ Microbiol. 1994;60:3074–3078. doi: 10.1128/aem.60.9.3074-3078.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos WM, Vaughan EE. Genetics of lactose utilization in lactic acid bacteria. FEMS Microbiol Rev. 1994;15:217–237. doi: 10.1111/j.1574-6976.1994.tb00136.x. [DOI] [PubMed] [Google Scholar]
- D’Incecco N, Bartowsky E, Kassara S, Lante A, Spettoli P, Henschke P. Release of glycosidically bound flavour compounds of Chardonnay by Oenococcus oeni during malolactic fermentation. Food Microbiol. 2004;21:257–265. [Google Scholar]
- Grimaldi A, McLean H, Jiranek V. Identification and partial characterization of glycosidic activities of commercial strains of the lactic acid bacterium, Oenococcus oeni. Am J Enol Vitic. 2000;51:362–369. [Google Scholar]
- Grimaldi A, Bartowsky E, Jiranek V. Screening of Lactobacillus spp. and Pediococcus spp. for glycosidase activities that are important in oenology. J Appl Microbiol. 2005a;99:1061–1069. doi: 10.1111/j.1365-2672.2005.02707.x. [DOI] [PubMed] [Google Scholar]
- Grimaldi A, Bartowsky E, Jiranek V. A survey of glycosidase activities of commercial wine strains of Oenococcus oeni. Int J Food Microbiol. 2005b;105:233–244. doi: 10.1016/j.ijfoodmicro.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Gueguen Y, Chemardin P, Labrot P, Arnaud A, Galzy P. Purification and characterization of an intracellular β-glucosidase from a new strain of Leuconostoc mesenteroides isolated from cassava. J Appl Microbiol. 1997;82:469–476. [Google Scholar]
- Günata YZ, Bayonove CL, Baumes RL, Cordonnier RE. The aroma of grapes I. Extraction and determination of free and glycosidically bound fractions of some grape aroma components. J Chromatogr A. 1985;331:83–90. [Google Scholar]
- Günata Z, Dugelay I, Sapis JC, Baumes R, Bayonove C. Role of enzymes in the use of the flavour potential from grape glycosides in winemaking. In: Schreier P, Winterhalter P, editors. Progress in Flavour Precursor Studies. Allured Publishing Corporation; Carol Stream. IL, USA: 1993. pp. 219–234. [Google Scholar]
- Günata Z, Blondeel C, Vallier MJ, Lepoutre JP, Sapis JC, Watanabe N. An endoglycosidase from grape berry skin of Cv. M. Alexandria hydrolyzing potentially aromatic disaccharide glycosides. J Agric Food Chem. 1998;46:2748–2753. [Google Scholar]
- Maicas S, Mateo JJ. Hydrolysis of terpenyl glycosides in grape juice and other fruit juices: A review. Appl Microbiol Biotechnol. 2005;67:322–335. doi: 10.1007/s00253-004-1806-0. [DOI] [PubMed] [Google Scholar]
- Mateo JJ, Di Stefano R. Enological properties of β-glucosidase in wine yeasts. Food Microbiol. 1998;14:583–591. [Google Scholar]
- Mateo JJ, Jiménez M. Monoterpenes in grape juice and wines. J Chromatogr A. 2000;881:557–567. doi: 10.1016/s0021-9673(99)01342-4. [DOI] [PubMed] [Google Scholar]
- McIlvaine TC. A buffer solution for colorimetric comparison. J Biol Chem. 1921;49:183–186. [Google Scholar]
- Nagaoka S, Honda H, Ohshima S, Kawai Y, Kitazawa H, Tateno Y, Yamazaki Y, Saito T. Identification of five phospho-β-glycosidases from Lactobacillus gasseri ATCC33323T cultured in lactose medium. Biosci Biotechnol Biochem. 2008;72:1954–1957. doi: 10.1271/bbb.80089. [DOI] [PubMed] [Google Scholar]
- Pemberton MS, Brown RD, Emert GH. Role of beta -glucosidase in the bioconversion of cellulose to ethanol. Can J Chem Eng. 1979;58:723–729. [Google Scholar]
- Rapp A. Aromastoffe des Weines. Chemie in unserer Zeit. 1992;26:273–284. [Google Scholar]
- Ribéreau-Gayon P, Glories Y, Maujean A, Dubourdieu D. Handbook of Enology. Vol. 2. John Wiley & Sons, Ltd.; Chichester, England: 2006. The chemistry of wine stabilization and treatments; pp. 205–227. [Google Scholar]
- Rodas AM, Ferrer S, Pardo I. 16S-ARDRA, a tool for identification of lactic acid bacteria isolated from grape must and wine. Syst Appl Microbiol. 2003;26:412–422. doi: 10.1078/072320203322497446. [DOI] [PubMed] [Google Scholar]
- Sestelo ABF, Poza M, Villa TG. β-Glucosidase activity in a Lactobacillus plantarum wine strain. World J Microbiol Biotechnol. 2004;20:633–637. [Google Scholar]
- Spano G, Rinaldi A, Ugliano M, Moio L, Beneduce L, Massa S. A β-glucosidase gene isolated from wine Lactobacillus plantarum is regulated by abiotic stresses. J Appl Microbiol. 2005;98:855–861. doi: 10.1111/j.1365-2672.2004.02521.x. [DOI] [PubMed] [Google Scholar]
- Tate D, Reynolds AG. Validation of a rapid method for measuring β-glucosidase activity in fermenting muscat grape musts. Am J Enol Vitic. 2006;57:60–68. [Google Scholar]