

Abstract
In describing the DNA double helix, Watson and Crick suggested that “spontaneous mutation may be due to a base occasionally occurring in one of its less likely tautomeric forms.” Indeed, among many mispairing possibilities, either tautomerization or ionization of bases might allow a DNA polymerase to insert a mismatch with correct Watson–Crick geometry. However, despite substantial progress in understanding the structural basis of error prevention during polymerization, no DNA polymerase has yet been shown to form a natural base–base mismatch with Watson–Crick-like geometry. Here we provide such evidence, in the form of a crystal structure of a human DNA polymerase λ variant poised to misinsert dGTP opposite a template T. All atoms needed for catalysis are present at the active site and in positions that overlay with those for a correct base pair. The mismatch has Watson–Crick geometry consistent with a tautomeric or ionized base pair, with the pH dependence of misinsertion consistent with the latter. The results support the original idea that a base substitution can originate from a mismatch having Watson–Crick geometry, and they suggest a common catalytic mechanism for inserting a correct and an incorrect nucleotide. A second structure indicates that after misinsertion, the now primer-terminal G•T mismatch is also poised for catalysis but in the wobble conformation seen in other studies, indicating the dynamic nature of the pathway required to create a mismatch in fully duplex DNA.
Keywords: mutagenesis, replication fidelity, nascent base pair, mispair
In their work describing the structure of DNA (1, 2), Watson and Crick proposed that spontaneous base substitutions could be a consequence of bases spontaneously pairing in rare tautomeric forms (1). They suggested two possible transition mispairs, G•T and A•C, involving the enol form of guanine or thymine and the imino form of adenine or cytosine, respectively. Both mispairs fit well within the dimensions of the DNA double helix to preserve the geometry of a correct Watson–Crick base pair. Thus, the potential importance of mispair geometry to base substitution mutagenesis was implied even before the discovery of DNA polymerases (3). Since then, many other mispairing possibilities have been proposed (4, 5), some involving ionized mispairs with correct geometry (6). Moreover, many structural and biochemical studies of DNA polymerases have indicated that the fidelity of DNA synthesis heavily depends on base pair geometry (7, 8). While all DNA polymerases studied to date can occasionally incorporate an incorrect nucleotide, misincorporation is usually rare. This is because most DNA polymerases employ a series of prechemistry conformational transitions as checkpoints for exclusion of incorrect substrates (9–11). For this reason it is difficult to obtain structural support for Watson and Crick’s hypothesis on the origin of spontaneous base substitutions.
This difficulty is revealed by elegant structural studies of DNA polymerases in the process of preventing misincorporation. Multiple crystal structures of polymerases with mismatched base pairs at the active site have been solved. For example, a number of high-resolution structures are available of the large fragment of Bacillus stearothermophilus DNA polymerase bound to primer–terminal mispairs (12). They indicated that each of the 12 possible mispairs distorts the polymerase active site when present at the primer terminus, causing polymerase stalling. The nature and extent of the distortion, and the effect on further extension, depends on the composition of the mispair. Distorted polymerase active site geometry has also been observed in the structures of Dpo4 (13) and Pol β (14), members of family Y and X, respectively, in ternary complexes with DNA and an incorrect incoming nucleotide. In the Dpo4 structure, the incoming dGTP formed either a wobble base pair or was not coplanar with a template T, and the incoming dGTP did not stack well with the primer–terminal base. Structures of Pol β complexes with DNA and a nonhydrolyzable dAMPCPP opposite template G or C captured the enzyme in a closed conformation with the incoming incorrect nucleotide occupying a position similar to that of a correct incoming nucleotide. However, the templating nucleotide was shifted upstream, such that the incoming incorrect dAMPCPP was bound opposite an abasic pocket. Thus, structures of DNA polymerases bound to mismatched substrates have provided important insights into the mechanisms of polymerase error prevention. However, they leave open the question of how DNA polymerases actually do misinsert a natural (i.e., undamaged) incorrect dNTP.
Here we address this question by using DNA polymerase λ (Pol λ) as a structural model for misincorporation. Pol λ is a monomeric X family DNA polymerase that lacks a 3′ exonuclease activity (15, 16). Its cellular function is to fill short gaps in DNA generated during base excision repair and nonhomologous end joining of double strand breaks in DNA. We previously described several structures of Pol λ that provide information on how this polymerase incorporates a correct dNTP (17–19). In addition, we described structures of Pol λ containing single unpaired nucleotides in the template strand upstream of the active site (20, 21). In these structures, the active site geometry was consistent with catalysis, indicating that these structures are relevant to Pol λ’s ability to generate single nucleotide deletion errors at a relatively high rate (22). However, like many DNA polymerases, Pol λ is considerably more adept at preventing single base substitution errors that require misinsertion at the primer terminus followed by mismatch extension (7, 23). Consistent with this property, our previous attempts to obtain crystal structures of Pol λ with single base–base mismatches have met with limited success; i.e., the description of one mismatch containing structure with aberrant geometry (24). During investigation of this error prevention ability, we recently identified a derivative of Pol λ (25) that lacks five amino acids in a loop upstream of the polymerase active site (Pol λ DL). Deleting these residues did not reduce catalytic activity or alter polymerase active site geometry for correct incorporation. However, Pol λ DL generates single base substitution errors at rates that increased for all twelve mismatches. This property is consistent with the elimination of one (or more) of the kinetic checkpoints that prevent misincorporation (9). It is also consistent with quantum mechanical and molecular mechanical calculations of Pol β, a homolog of Pol λ, indicating that the major factor preventing incorrect nucleotide insertion is the free energy required to achieve active site geometry compatible with catalysis (26). We posited that this energy barrier might be reduced in Pol λ DL to the degree required to obtain structures of ternary Pol λ DL–DNA-incorrect dNTP complexes with active site geometry that is compatible with catalysis. We demonstrate here that this is indeed the case.
Results and Discussion
Structure of a dG•T Mispair at the Polymerase Active Site.
We were able to crystallize Pol λ DL in complex with a physiologically relevant one-nucleotide gapped DNA substrate and a nonhydrolyzable derivative of dGTP (dGMPCPP) paired with template T. The 2.6-Å structure (PDB ID code 3PML) contains two Pol λ DL ternary (Pol λ + primer-template + incoming dGMPCPP) complexes in the asymmetric unit, designated molecules A and B. The electron density at the active site in molecule B is consistent with multiple conformations for the template strand as well as for residues Phe506, Arg514, and Arg517. One set of conformations for Phe506, Arg514, Arg517, and the template strand has been modeled in a manner similar to that of molecule A (precatalytic conformation, see below) (Fig. S1). The other conformation set resembles the inactive form of the molecule, similar to what has been seen in the binary structures of polymerase λ, where Phe506 is rotated toward Tyr505, Arg517 occupies the position of the templating base in the nacent binding pocket, and Arg514 is also shifted (Fig. S1). We modeled the dGMPCPP with full occupancy, suggesting that it is binding in both conformations but only base pairing with the template strand in the precatalytic conformation. The multiple conformations of template DNA in molecule B create a blurring of the bases, rendering tenuous any discussion of the precise positions of atoms.
Fortunately, the DNA and the protein are in a single conformation in molecule A, and this conformation overlays well (the rmsd of 0.593 Å for 265 C-α atoms) with the structure of a ternary complex of Pol λ DL (25) containing correct incoming ddTTP paired opposite template A (Fig. 1). This structure also overlays well (Fig. 2A, the rmsd of 0.434 Å for 261 C-α atoms) with a ternary complex of wild-type Pol λ containing a correct A•dUMPNPP base pair in the binding pocket (19). The use of nonhydrolyzable nucleotides in these studies, as initially used in studies of pol β (27), allows visualization of the geometry of the polymerase active site in a precatalytic state, yet containing all the atoms required for catalysis (Fig. 2B and Fig. S2), including the attacking 3′-oxygen and two metal ions. In the mismatched structure, two metal ions are bound at the active site, and they, and the catalytic carboxylates with which they coordinate, overlay well with the equivalent atoms at the active site of the structure containing the correct base pair (Fig. 2B). The coordination spheres of both metals have octahedral geometry (Fig. 2B) consistent with magnesium ions occupying both metal sites. In the G•T-containing structure, the primer–terminal base pair occupies the same position as in the structure with the correct base pair, except for a slight twist in the position of the template strand base. The nascent mispair stacks with the primer–terminal correct base pair, just as observed in the structure with the correct base pair. The 3′-O of the primer–terminal nucleotide is in position for in line attack on the α-phosphate of the incoming nucleotide (Fig. 2B), and these atoms are separated by 3.9 Å. This distance is comparable to the separation between the 3′-O and the α-phosphate observed in the structures of ternary complexes of wild-type Pol λ (3.7 Å) (19), Pol β (3.4 Å) (28) and pol η (3.2 Å) (29), all of which contain a correct nascent base pair. Therefore, active site geometry with the G•T mismatch is similar to that for the correct base pair and is consistent with catalysis. This is particularly interesting because the incoming dG is paired with template T in a Watson–Crick conformation (Fig. 4A and compare with 4B). Thus, the conformation of the dGMPCPP•T mispair within the Pol λDL nascent base pair binding pocket and its position relative to the primer–terminal base pair differs significantly from that reported for the dGTP•T mispair in the structure of the Dpo4 ternary complex (13).
Fig. 1.

Superposition of Pol λ DL ternary complexes. The complex with a dGMPCPP•T nascent mispair [the protein is green, the primer strand is yellow, the template strand is orange, the incoming dGMPCPP is magenta, and the active site metal ions (Me) are bright green] is overlaid with the ddTTP•A nascent base pair-containing complex, PDB ID code 3MGI (the protein is light cyan, the primer strand is beige, the template strand is light brown, and the incoming ddTTP is dark brown).
Fig. 2.

Watson–Crick conformation of a dGMPCPP•T mispair at the active site. (A) Superposition of the ternary complexes of Pol λ DL (green) in complex with DNA (primer strand is olive, template strand is orange) and incoming dGMPCPP (magenta) opposite template T (orange) and the WT Pol λ ternary complex (2PFO) (protein is gray, template strand is light orange, incoming dUMPNPP is light purple, and template adenine is brown. The metal ions A and B (Me) in the complex of Pol λ DL and WT Pol λ are green and purple, respectively. (B) Close up of the active site of the ternary complex of Pol λ DL wih the dGMPCPP•T nascent mispair. The magnesium ions are green, and active site H2O molecules are light red.
Fig. 4.

Geometry of G•T mispairs in Pol λ DL complexes. (A) Watson–Crick conformation of dGMPCPP•T nascent base pair from the ternary, precatalytic complex. (B) Correct dGMPCPP•C nascent base pair from the precatalytic complex with the G•T primer–terminal base pair. (C) G•T primer–terminal base pair in wobble conformation in the precatalytic, ternary complex. Simulated annealing Fobs-Fcalc omit maps contoured at 3.5σ are shown in dark blue. (D) Base pair parameters (derived using 3DNA software v.1.5, Lu and Olson), including H-bond information [atom, pair, and length (Å)], base pair width [C1′—C1′ distance (Å)], and lR and lY angles (in degrees) between the line joining the C1′—C1′ and the N9-C1′ (purine) and N1-C1′ (pyrimidine) glycosidic bonds. The positions of atoms in a G•C base pair are indicated.
pH Dependence of Single Nucleotide Misincorporation.
The dGMPCPP•T mispair in the Pol λ active site could conceivably contain either an ionized or rare tautomeric base. At alkaline pH, the N3 of thymine and the N1 of guanine deprotonize, increasing the ratio of ionized to tautomeric bases. Because the equilibrium between the keto and the rare enol tautomer is pH independent, the increase in ionized bases should concomitantly decrease the concentration of the rare enol tautomer that could mispair. Consequently, as discussed by Yu et al. (6), if an ionized base is involved, the efficiency of dGTP misinsertion opposite template T should increase with increasing pH, whereas if a rare tautomer is involved, the efficiency of dGTP misinsertion opposite template T should decrease with increasing pH. In an initial attempt to distinguish between the involvement of enol tautomers and ionized bases, we measured the efficiency of single nucleotide misinsertion of dGTP opposite template T by Pol λ DL as a function of pH. The results (Table 2 and Fig. S3) demonstrate that wild-type Pol λ and Pol λ DL misinsert dGTP opposite template T at substantially higher efficiencies in reactions performed at pH 9.0 as compared to pH 7. The relative misinsertion efficiency (fmis in Table 2), expressed as the ratio of catalytic efficiencies for incorrect dGTP relative to correct dATP, is increased by 90- and 13-fold, respectively, for wild-type pol λ and pol λ DL. These increases are in agreement with the results of Yu et al. (6) and are consistent with the possible involvement of an ionized base pair. On the other hand, the distance (2.7 Å), between the O6 atom of the guanine base and O4 of thymine in the dGMPCPP•T mispair at the pol λ active site suggests that a proton could be involved in the hydrogen bond between the two, which is consistent with a tautomeric base pair.
Table 2.
pH dependence of misinsertion
| dNTP | Km (μM) | kcat (1/s) | kcat/Km | fmis* |
| WT Pol λ | ||||
| pH 7.0 | ||||
| dATP | 0.30 ± 0.08 | 0.02 ± 0.008 | 6.7 × 10-2 ± 0.9 × 10-2 | |
| dGTP | 5.1 ± 2.5 | 0.0003 ± 6.7 × 10-5 | 5.4 × 10-5 ± 1.5 × 10-5 | 0.8 × 10-3 |
| pH 9.0 | ||||
| dATP | 1.0 ± 0.40 | 0.012 ± 0.0035 | 1.2 × 10-2 ± 0.25 × 10-2 | |
| dGTP | 1.2 ± 0.40 | 0.001 ± 0.0003 | 8.6 × 10-4 ± 3.1 × 10-4 | 72 × 10-3 |
| Pol λ DL | ||||
| pH 7.0 | ||||
| dATP | 0.3 ± 0.06 | 0.0055 ± 0.0013 | 1.9 × 10-2 ± 1.5 × 10-2 | |
| zdGTP | 13 ± 4.2 | 0.005 ± 0.0035 | 3.6 × 10-4 ± 1.2 × 10-4 | 1.9 × 10-2 |
| pH 9.0 | ||||
| dATP | 1.4 ± 0.40 | 0.013 ± 0.0035 | 1 × 10-2 ± 0.058 × 10-2 | |
| dGTP | 4.8 ± 1.09 | 0.012 ± 0.0042 | 2.5 × 10-3 ± 1.1 × 10-3 | 25 × 10-2 |
The kinetic constants (shown with standard deviations) are an average of 3 to 5 independent determinations.
*1fmis is the relative efficiency of misincorporation expressed as the ratio of [kcat/KmdGTP]/[kcat/KmdATP].
Structure of a Ternary Complex of Pol λ DL with Primer–Terminal dG•T Mispair.
Stable misincorporation to yield a single base substitution mutation requires that the G•T mismatch be extended by correct incorporation. To visualize this, we crystallized Pol λ DL in complex with a one-nucleotide gap DNA containing a G•T mispair at the 3′ terminus and correct dGMPCPP paired opposite template C. The crystals refined to 2 Å and contained one molecule of the ternary complex in the asymmetric unit (Table 1). The polymerase and the template strand were in the active conformation, but catalytic metal ion A did not have octahedral geometry, and the attacking 3′-O was not in line with the α-phosphorus of the incoming nucleotide (Table 1, Fig. S4). Therefore, to obtain a structure of a catalytically competent complex we soaked these crystals in a solution containing MnCl2, a procedure that previously induced active conformations with correct substrates bound to wild-type Pol λ (19) and Pol β (14). The resulting crystal diffracted to 2.2 Å (Table 1). Just as for the G•T mismatch in the nascent base pair binding pocket (Fig. 2), this structure overlays well with the precatalytic, wild-type Pol λ ternary complex (Fig. 3 A and B). Two metal ions are present (A = manganese, B = magnesium) and coordinated with octahedral geometry, all amino acid side chains are in their active conformations, and the 3′O on the primer–terminal mismatched dG is in line with the α phosphorus of the incoming nucleotide (3.7 Å apart) and the leaving group; i.e., this geometry is consistent with catalysis. However, unlike the G•T mismatch in the nascent base pair binding pocket, the template T of the primer–terminal mismatch is shifted toward the major groove, such that it is paired with the primer dG in a wobble conformation (Fig. 4A). This does not affect normal base pairing in the binding pocket, because the incoming correct dGMPCPP is paired with the templating C in the standard Watson–Crick conformation (Fig. 4A).
Table 1.
Crystallographic data and statistics
| Dataset | G•T nascent pocket (Mg, Mg) | G•T primer terminus; (Mn, Mg) | G•T primer terminus; (Na, Mg) |
| PDB ID code | 3PML | 3PMN | 3PNC |
| Wavelength (Å) | 1.000 | 1.5418 | 1.5418 |
| Unit cell (a,b,c) (Å) | 97.04, 191.57, 59.08 | 56.39, 62.42, 139.88 | 56.18, 62.27, 139.82 |
| Space Group | P21212 | P212121 | P212121 |
| Resolution (Å) | 25.0–2.6 | 50.0–2.2 | 50.0–2.0 |
| # of observations | 244,249 | 98,923 | 320,923 |
| Unique reflections | 34,636 | 25,329 | 33,148 |
| Redundancy | 7.1(7.0) | 3.9(2.5) | 9.7(5.7) |
| Rsym (%) *† | 7.0 (57.2) | 9.0 (43.1) | 8.2(53.7) |
| I/σI | 10.6 (3.5) | 17.6 (2.4) | 9.5(2.5) |
| Mosaicity range | 0.5–1.1 | 1.2–1.7 | 0.4–0.5 |
| Completeness (%) | 99.9 (99.8) | 99.2 (98.2) | 97.7(87.3) |
| Refinement statistics | |||
| Rcryst‡§, Rfree (%) | 23.3, 27.1 | 21.3, 24.3 | 20.8, 23.6 |
| No. of waters | 168 | 202 | 310 |
| Overall Wilson B value (Å2) | 53.1 | 41.1 | 38.3 |
| Average B for: | |||
| Protein atoms | 54.3 | 42.7 | 40.2 |
| DNA | 58.9 | 31.9 | 28.5 |
| Incoming nucleotide | 49.8 | 23.2 | 19.3 |
| Water | 43.0 | 40.1 | 42.4 |
| rmsd from ideal values | |||
| Bond length (Å) | 0.007 | 0.005 | 0.008 |
| Bond angle (°) | 1.2 | 1.0 | 1.3 |
| Dihedral angle (°) | 21.5 | 21.3 | 21.6 |
| Ramachandran statistics ¶ | |||
| Favored (98%) regions (%) | 93.9 | 96.0 | 96.5 |
| Allowed (> 99.8%) regions (%) | 99.8 | 99.7 | 100 |
* where Ii s the intensity of the ith observation and 〈I〉 is the mean intensity of the reflection.
where Ii s the intensity of the ith observation and 〈I〉 is the mean intensity of the reflection.
†Last resolution shell is in parentheses.
‡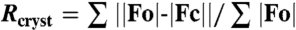 calculated from working dataset.
calculated from working dataset.
§Rfree was calculated from 5% of data randomly chosen not to be included in refinement.
¶Ramachandran results were determined by MolProbity.
Fig. 3.

G•T terminal mispair in the ternry complex of Pol λ DL. (A) Superposition of Pol λ DL ternary complex with a G•T terminal mispair and the WT Pol λ complex (2PFO). Pol λ DL is light gray, the template and primer strands are gray, and the G•T terminal mispair is bright green; the incoming dGMPCPP is magenta, and the active site metal ions A and B are purple and pea green, respectively. In the WT Pol λ complex, the protein is light blue, the DNA is light yellow, the incoming dUMPNPP is pink, and the metal ions A and B are light purple and dark green, respectively. (B) Close up of the active site of Pol λ DL ternary complex with a G•T terminal mispair.
Previous studies have described unusual, noncatalytic conformations of DNA polymerases bound to mismatched substrates (12–14, 27), including G•T mismatches in a wobble conformation (12, 13). Because these structures are inconsistent with catalysis, they have provided important insight into how base–base mismatches are prevented during DNA synthesis. The present study complements those studies by providing structures of a DNA polymerase with a G•T mismatch whose geometry appears to be consistent with misinsertion and with mismatch extension. The results suggest a catalytic mechanism for misinsertion and mismatch extension that is in common with correct incorporation, and they support Watson and Crick’s original idea that spontaneous base substitutions, in this case A•T to G•C transition mutations, may result from mismatches shaped like correct base pairs.
Materials and Methods
Protein Expression and Purification.
Pol λ DL was expressed in Escherichia coli and purified as described (18).
Crystallization and Data Collection.
Crystals of the G•T basepair in the nascent binding pocket were obtained by the vapor diffusion sitting drop method by mixing 1 ul of preequilibrated protein/DNA/dGMPCPP solution (9.7 μg protein, 0.7 mM DNA, 1 mM dGMPCPP, 36 mM Tris, pH 7.5 and 10 mM MgCl2) with 1 ul of a reservoir solution consisting of 0.1 M NaCl, 0.1 M Hepes pH 7.5, and 10%PEG 4000. Crystals were transferred to a cryosolution consisting of 0.1 M Hepes pH 7.5, 0.1 M NaCl, 20% PEG 4000, 10 mM MgCl2, 1 mM dGMPCPP and 17.5% ethylene glycol, flash frozen in liquid nitrogen, and then placed in a stream of nitrogen gas cooled to -180 C for data collection. Data were collected at the SER-CAT beamline at the Advanced Photon Source. After the data had been processed and scaled, a model of polymerase lambda derived from the crystal structure of the binary complex of Pol λ DL (PDB ID code 3MGH) was refined against the data utilizing the same test reflections. The asymmetric unit consists of two molecules of polymerase lambda each binding DNA and an incoming nucleotide.
A crystal of the G•T base pair at the primer terminus site with a Na1+ and Mg2+ in the active site was obtained using the vapor diffusion sitting drop method by mixing 0.35 ul of preequilibrated protein/DNA/dGMPCPP solution (3.4 μg protein, 0.7 mM DNA, 1 mM dGMPCPP, 36 mM Tris pH 7.5, and 10 mM MgCl2) with 0.35 ul of 2 M formate. For data collection, a crystal was transferred directly to 0.1 M Tris pH 7.5, 100 mM NaCl, 1 mM dGMPCPP, 2.2 M Na formate and 17.5% ethylene glycol. Data were collected on an in-house MicroMax 007HF generator with VarimaxHF mirrors and a Saturn92 detector to a resolution of 2.0 Å. The ternary complex with the G•T basepair in the nascent binding pocket was used as the search model for molecular replacement using MOLREP (30). The asymmetric unit consists of one molecule of polymerase λ with DNA and an incoming nucleotide bound. The structure with two Mn2+ in the active site was obtained from a crystal from the same drop as the Na/Mg dataset but soaked in 2 M formate and 20 mM MnCl2 for 25 min before being transferred to the cryosolution consisting of 20 mM MnCl2, 0.1 M Tris pH 7.5, 100 mM NaCl, 1 mM dGMPCPP, 2.2 M Na formate and 17.5% ethylene glycol. The dataset was collected using the same in-house system at 2.2-Å resolution. Data were processed and the structure refined using the same test reflections as for the Na1+/Mg2+ dataset. All data were processed using HKL2000 (31) and refined using iterative cycles of refinement in CNS (32) and model building using O (33). The quality of the geometry for the structures was analyzed using Molprobity (34).
Kinetic Analysis of Nucleotide Insertion.
The steady state measurements of single nucleotide incorporation were performed as described in (25). DNA substrates were prepared by hybridizing a 32P-5′-end-labled 17-nucleotide primer (P17T, 5′GTACGACTGAGCAGTAC) and a 14-nucleotide downstream primer (DPT, 5′ GCCGGACGACGGAG) with a phosphate on the 5′ end to a 32-mer template (T32GT, 5′CTCCGTCGTCCGGCTGTACTGCTCAGTCGTAC) to create a one-nucleotide gap substrate. Reaction mixtures (10 μl) contained 50 mM Tris, pH 7 or pH 9, 1 mM dithiothreitol, 4% glycerol, 0.1 mg/ml bovine serum albumin, 2.5 mM MgCl2, 200 nM DNA, and 4 nM full-length WT pol λ or pol λ DL. Reactions were initiated by adding dATP at one of nine concentrations (0.05, 0.1, 0.2, 0.5, 1, 2, 5, 10, 20 μM) and incubated at 37 °C for 3 min. To measure misinsertion, reaction mixtures contained 50 or 100 nM WT pol λ or 5 or 7 nM pol λ DL. Reactions were initiated by adding dGTP at one of the concentrations (1, 2, 5, 10, 25, 50, 100, or 150 μM to reactions with WT pol λ and pol λ DL at pH 7 or 0.1, 0.3, 1, 2, 5, 8, 15, 20, 30 μM at pH 9) and incubated at 37 °C for 4 or 6 min at pH 9 and pH 7, respectively. After adding an equal volume of 99% formamide, 5 mM EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue, products were resolved on a 12% denaturing polyacrylamide gel and quantified by phosphor screen autoradiography. The data were fit to the Michaelis–Menten equation using nonlinear regression.
Supplementary Material
Acknowledgments.
We thank Lee Pedersen and William Beard for helpful discussions and critical reading of the manuscript. Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID (or 22-BM) beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract W-31-109-Eng-38. This work was supported by the Division of Intramural Research of the National Institutes of Health, National Institute of Environmental Health Sciences (Project Z01 ES065070 to T.A.K.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The structure factors have been deposited in the Protein Data Bank, www.pdb.org (crystal structure of a polymerase lambda variant with a dGTP analog opposite a templating T has been assigned the RCSB ID code rcsb062557 and PDB ID code 3PML; ternary crystal structure of polymerase lambda variant with a GT mispair at the primer terminus has been assigned the RCSB ID code rcsb062559 and PDB ID code 3PMN; ternary crystal structure of a polymerase lambda variant with a GT mispair at the primer terminus and sodium at catalytic metal site has been assigned the RCSB ID code rcsb062584 and PDB ID code 3PNC).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1012825108/-/DCSupplemental.
References
- 1.Watson JD, Crick FH. Genetical implications of the structure of deoxyribonucleic acid. Nature. 1953;171:964–967. doi: 10.1038/171964b0. [DOI] [PubMed] [Google Scholar]
- 2.Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 3.Lehman IR, Bessman MJ, Simms ES, Kornberg A. Enzymatic synthesis of deoxyribonucleic acid. I. Preparation of substrates and partial purification of an enzyme from Escherichia coli. J Biol Chem. 1958;233:163–170. [PubMed] [Google Scholar]
- 4.Drake JW, Baltz RH. The biochemistry of mutagenesis. Annu Rev Biochem. 1976;45:11–37. doi: 10.1146/annurev.bi.45.070176.000303. [DOI] [PubMed] [Google Scholar]
- 5.Topal MD, Fresco JR. Complementary base pairing and the origin of substitution mutations. Nature. 1976;263:285–289. doi: 10.1038/263285a0. [DOI] [PubMed] [Google Scholar]
- 6.Yu H, Eritja R, Bloom LB, Goodman MF. Ionization of bromouracil and fluorouracil stimulates base mispairing frequencies with guanine. J Biol Chem. 1993;268:15935–15943. [PubMed] [Google Scholar]
- 7.Echols H, Goodman MF. Fidelity mechanisms in DNA replication. Annu Rev Biochem. 1991;60:477–511. doi: 10.1146/annurev.bi.60.070191.002401. [DOI] [PubMed] [Google Scholar]
- 8.Kool ET. Active site tightness and substrate fit in DNA replication. Annu Rev Biochem. 2002;71:191–219. doi: 10.1146/annurev.biochem.71.110601.135453. [DOI] [PubMed] [Google Scholar]
- 9.Joyce CM, Benkovic SJ. DNA polymerase fidelity: Kinetics, structure, and checkpoints. Biochemistry. 2004;43:14317–14324. doi: 10.1021/bi048422z. [DOI] [PubMed] [Google Scholar]
- 10.Christian TD, Romano LJ, Rueda D. Single-molecule measurements of synthesis by DNA polymerase with base-pair resolution. Proc Natl Acad Sci USA. 2009;106:21109–21114. doi: 10.1073/pnas.0908640106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santoso Y, et al. Conformational transitions in DNA polymerase I revealed by single-molecule FRET. Proc Natl Acad Sci U S A. 2010;107:715–720. doi: 10.1073/pnas.0910909107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson SJ, Beese LS. Structures of mismatch replication errors observed in a DNA polymerase. Cell. 2004;116:803–816. doi: 10.1016/s0092-8674(04)00252-1. [DOI] [PubMed] [Google Scholar]
- 13.Vaisman A, Ling H, Woodgate R, Yang W. Fidelity of Dpo4: Effect of metal ions, nucleotide selection and pyrophosphorolysis. EMBO J. 2005;24:2957–2967. doi: 10.1038/sj.emboj.7600786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Batra VK, Beard WA, Shock DD, Pedersen LC, Wilson SH. Structures of DNA polymerase beta with active-site mismatches suggest a transient abasic site intermediate during misincorporation. Mol Cell. 2008;30:315–324. doi: 10.1016/j.molcel.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Diaz M, et al. DNA polymerase lambda (Pol lambda), a novel eukaryotic DNA polymerase with a potential role in meiosis. J Mol Biol. 2000;301:851–867. doi: 10.1006/jmbi.2000.4005. [DOI] [PubMed] [Google Scholar]
- 16.Moon AF, et al. The X family portrait: Structural insights into biological functions of X family polymerases. DNA Repair. 2007;6:1709–1725. doi: 10.1016/j.dnarep.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia-Diaz M, et al. A structural solution for the DNA polymerase lambda-dependent repair of DNA gaps with minimal homology. Mol Cell. 2004;13:561–572. doi: 10.1016/s1097-2765(04)00061-9. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Diaz M, Bebenek K, Krahn JM, Kunkel TA, Pedersen LC. A closed conformation for the Pol lambda catalytic cycle. Nat Struct Mol Biol. 2005;12:97–98. doi: 10.1038/nsmb876. [DOI] [PubMed] [Google Scholar]
- 19.Garcia-Diaz M, Bebenek K, Krahn JM, Pedersen LC, Kunkel TA. Role of the catalytic metal during polymerization by DNA polymerase lambda. DNA Repair. 2007;6:1333–1340. doi: 10.1016/j.dnarep.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia-Diaz M, Bebenek K, Krahn JM, Pedersen LC, Kunkel TA. Structural analysis of strand misalignment during DNA synthesis by a human DNA polymerase. Cell. 2006;124:331–342. doi: 10.1016/j.cell.2005.10.039. [DOI] [PubMed] [Google Scholar]
- 21.Bebenek K, et al. Substrate-induced DNA strand misalignment during catalytic cycling by DNA polymerase lambda. EMBO Rep. 2008;9:459–464. doi: 10.1038/embor.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bebenek K, Garcia-Diaz M, Blanco L, Kunkel TA. The frameshift infidelity of human DNA polymerase lambda. Implications for function. J Biol Chem. 2003;278:34685–34690. doi: 10.1074/jbc.M305705200. [DOI] [PubMed] [Google Scholar]
- 23.Kunkel TA, Bebenek K. DNA replication fidelity. Annu Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 24.Picher AJ, et al. Promiscuous mismatch extension by human DNA polymerase lambda. Nucleic Acids Res. 2006;34:3259–3266. doi: 10.1093/nar/gkl377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bebenek K, Garcia-Diaz M, Zhou R-Z, Povirk LF, Kunkel TA. Loop1 modulates the fidelity of DNA polymerase λ. Nucleic Acids Res. 2010;38:5419–5431. doi: 10.1093/nar/gkq261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin P, et al. Incorrect nucleotide insertion at the active site of a G:A mismatch catalyzed by DNA polymerase beta. Proc Natl Acad Sci USA. 2008;105:5670–5674. doi: 10.1073/pnas.0801257105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Batra VK, Beard WA, Shock DD, Pedersen LC, Wilson SH. Nucleotide-induced DNA polymerase active site motions accommodating a mutagenic DNA intermediate. Structure. 2005;13:1225–1233. doi: 10.1016/j.str.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 28.Batra VK, et al. Magnesium-induced assembly of a complete DNA polymerase catalytic complex. Structure. 2006;14:757–766. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Biertumpfel C, et al. Structure and mechanism of human DNA polymerase eta. Nature. 2010;465:1044–1048. doi: 10.1038/nature09196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vagin A, Teplyakov A. MOLREP: An automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 31.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 32.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 33.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 34.Lovell SC, et al. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.